Abstract
The present study describes the synthesis of a series of new 4-aminoquinoline-derived thiazolidines and evaluation of their antimalarial activity against a NF-54 strain of Plasmodium falciparum in vitro and N-67 strain of Plasmodium yoelii in vivo. Among the series, two compounds, 2-(4-chloro-phenyl)-thiazolidine-4-carboxylic acid [2-(7-chloro-quinolin-4-ylamino)-ethyl]-amide hydrochloride (14) and 2-(2,6-dichloro-phenyl)-thiazolidine-4-carboxylic acid [2-(7-chloro-quinolin-4-ylamino)-ethyl]-amide hydrochloride (22) exhibited significant suppression of parasitaemia in the in vivo assay. All the analogues were found to form strong complex with haematin and inhibited the β-haematin formation in vitro. These results suggest that these compounds act on heme polymerization target.
Introduction
Malaria is one of the world’s most widespread infectious diseases and poses a great challenge to human health. Estimates range from 300 to 500 million clinical cases of malaria each year and approximately 1.5–2.5 million die due to non-availability of a proper therapeutic agentCitation1–3. During the past four decades, chloroquine (CQ) and other aminoquinolines have been the mainstays of malarial chemotherapy due to their therapeutic efficacyCitation4. However, development of resistance has severely limited the choice of available antimalarial drugs. As a result, there exists an urgent need of some novel chemotherapeutic agents for the treatment of malaria. Towards this objective, efforts are going on to develop either new chemical entities or to modify the existing therapeutic agents to overcome the drug resistance.
The structure–activity relationship studies on CQ analogues indicate that compounds synthesized by alteration of chain length are active against CQ-resistant parasite strains, strongly suggesting that the resistance mechanism does not involve any change in the target of this class of drugs but involves a compound-specific resistanceCitation4–6. Based on this affirmation, a number of groups have developed short chain analogues of 4-aminoquinoline derivatives, which are significantly more potent than CQ against a CQ-resistant strain of Plasmodium falciparum in in vitro studiesCitation6,Citation7. CQ and closely related 4-aminoquinolines form a complex with (Fe(III)FPIX) haematin, which is generated in the digestive vacuole of the intraerythrocytic malaria parasite as a result of proteolysis of host haemoglobinCitation7–9. Free haematin is cytotoxic for malaria parasite and it is sequestered in the form of haemozoin. The drug–haematin interactions inhibit the formation haemozoin crystals and the accumulation of significant concentrations of haematin which is toxic to parasite and it is believed to be responsible for killing the parasiteCitation9–12.
In our efforts to develop effective antimalarial agents, earlier we have reported design, synthesis and antimalarial activity of several side chain-modified 4-aminoquinolinesCitation13–16. We have demonstrated that 4-aminoquinoline-derived guanidine and tetramethylguanidine analogues () with altered chain length exhibit promising activity against CQ-sensitive strains of P. falciparum NF-54 in vitro and CQ-resistant N-67 strain of Plasmodium yoelii in vivoCitation13. Stocks et al. have shown that a series of short chain CQ derivatives, on replacement of the diethylamino function with a variety of heterocyclic ring substitutions including piperidinyl, pyrrolidinyl, morpholinyl and piperazinyl modifications, lead to a substantial increase in the antimalarial activityCitation7. Earlier we have also demonstrated that modification on 4-aminoquinoline lateral side chain with thiazolidine-4-one ring substitution () has led to compounds with improved antimalarial activity, and some of these compounds were indeed more effective than CQCitation14. Furthermore, lysine and ornithine conjugates were prepared by selectively modifying the pendant amino group of 4-aminoquinoline terminal side chain with amide bond (). Interestingly, these compounds also showed promising in vitro activityCitation15. Apart from these studies, we investigated quantitative structure–activity relationship studies of antimalarial activity of two distinct series of N1-(7-chloro-4-quinolyl)-1,4-bis(3-aminopropyl) piperazine analogues using the DRAGON descriptors in order to rationalize their activityCitation17. The study results suggested that amide moiety in the pharmacophore is favourable for the antimalarial activity of 4-aminoquinoline compoundsCitation17.
Figure 1. Some lead molecules of 4-aminoquinoline-derived antimalarials developed from this laboratory.
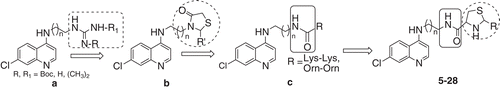
Based on these information, we thought it appropriate to introduce amide bond with heterocyclic ring system (4-thiazolidine) on lateral side chain of 4-aminoquinoline as it will provide lipophilic nature to molecules and could give compounds with improved antimalarial activity. The thiazolidine nucleus was selected based on the observation that it is a biologically privileged scaffold and well tolerated in human subjectsCitation18,Citation19. Due to the potential biological activity as well as our interest in the chemistry of thiazolidineCitation14,Citation20–22, we thought it would be appropriate to explore the antiplasmodial activity of derivatives having 4-thiazolidine nucleus at the lateral side chain and to the best of our knowledge such modifications have not been hitherto reported in the literature in the case of 4-aminoquinolines. The thiazolidine scaffold has basic nitrogen in the heterocyclic ring system and hydrophobicity is modulated by the substitution on C-2 position. It is therefore surmised that this will provide an optimal balance between basicity and hydrophobicity for their haematin binding and antimalarial activity. The present study describes synthesis, biophysical studies and antimalarial activity of these compounds.
Experimental section
General
Melting points (mp) were determined on a CompLab melting point apparatus and are uncorrected. Infrared (IR) spectra (cm−1) were recorded on Perkin-Elmer 621 spectrometer using the KBr disc technique. The 1H NMR spectra were recorded on a DPX-200 MHz Bruker FT-NMR spectrometer using CDCl3 and DMSO-d6 as solvent and 13C NMR spectra were recorded on Bruker DRX-300 FT-NMR spectrometer (75 MHz). Tetramethylsilane (δ 0.0 ppm) was used as an internal standard. Fast Atom Bombardment Mass Spectra (FAB-MS) were obtained on Jeol (Japan)/SX-102 spectrometer using glycerol or m-nitrobenzyl alcohol as matrix. Elemental analysis was performed on a Perkin-Elmer 2400 C, H, N analyzer and values were within the acceptable limits of the calculated values. The progress of the reaction was monitored on ready-made silica gel plates (Merck) using chloroform–methanol (9:1) as a solvent system. Iodine was used as developing agent or by spraying with Dragendorff’s reagent. Chromatographic purification was performed over silica gel (100–200 mesh). All chemicals and reagents were obtained from Aldrich (USA), Lancaster (UK) or Spectrochem Pvt. Ltd (India) and were used without further purification.
General synthetic procedure for the compounds (2a–2c)
l-Cysteine (0.5 g, 4.12 mmol) and appropriate aldehyde (1b–1c) (4.12 mmol) in methanol (15 mL) were stirred at 60°C for 5 h, and the reaction mixture was cooled and solid was collected, washed with anhydrous diethyl ether and dried to afford 2b–2c.
General synthetic procedure for the compounds (3a–3c)
Appropriate carboxylic acid (25.0 mmol) 2a–2c was dissolved in 100 mL of dioxane:water (1:1). To this, 25.0 mmol of 2N NaOH solution was added and the reaction mixture stirred at 0°C for 15 min. Then 25.0 mmol of Boc anhydride was added and reaction mixture was stirred for an hour at 0°C and the stirring was continued for 2–4 h at room temperature. Then reaction mixture was evaporated under reduced pressure and the residue obtained was acidified with the help 5% citric acid solution. This solution was extracted with the help of chloroform and then the chloroform layer was washed with brine. The organic layer was dried over anhydrous Na2SO4 and solvent was removed under reduced pressure and the crude product was purified by column chromatography over silica gel using chloroform–methanol as eluent.
General synthetic procedure for N-tert-butoxycarbonyl-thiazolidine-4-carboxylic acid-2-(substituted/unsubstituted)-[(7-chloro-quinolin-4-ylamino)-alkyl]-amide (5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 25, 27)
Compounds 3a–3c (2.0 mmol) were taken in anhydrous THF. To this, 1-hydroxy-benzotriazolehydrate (HOBt) (2.1 mmol), appropriate 4-aminoquinoline (4a–4d) (2.0 mmol) and N,N′-dicyclohexylcarbodiimide (DCC) (2.0 mmol) were added and resulting mixture was stirred for 30 min at 0°C and the stirring was continued for 2 h at room temperature. Dicyclohexylurea was removed by filtration and filtrate was evaporated to dryness in vacuo. The residue was taken in ethyl acetate and washed with 5% NaHCO3 and brine, dried on anhydrous Na2SO4. The crude product was purified by column chromatography over silica gel using chloroform–methanol as eluent.
General synthetic procedure for 2-(substituted/unsubstituted)-thiazolidine-4-carboxylic acid [2-(7-chloro-quinolin-4-ylamino)-alkyl]-amide hydrochloride (6, 8, 10, 12, 14, 16, 18, 20, 22, 24, 26, 28)
Compounds 5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 25, 27 were treated with 4N HCl/dioxane solution and kept for 1 h at room temperature. The solvent was evaporated under reduced pressure and the residue was precipitated with anhydrous ether. Filter and precipitate thoroughly were washed with ether and dried over anhydrous NaOH pellets under high vacuum.
2-(4-Chloro-phenyl)-thiazolidine-4-carboxylic acid [2-(7-chloro-quinolin-4-ylamino)-ethyl]-amide hydrochloride (14)
This compound was obtained as a yellowish gummy matter in 65% yield; IR (neat) 1636.5 cm−1; 1H NMR (200 MHz, DMSO-d6): δ 3.18–3.29 (m, 2H, CH2), 3.47–3.62 (m, 2H, CH2) 3.73–3.77 (m, 2H, CH-CH2-S), 4.26–4.32 (m, 1H, CO-CH-CH2), 5.70 (s, 1H, S-CH-N), 6.88–6.91 (d, J = 5.72 Hz, 1H, Ar-H quinoline), 7.54–7.60 (m, 2H, Ar-H), 7.86–7.90 (m, 2H, Ar-H), 8.06–8.07 (d, J = 1.88 Hz, 1H, Ar-H), 8.41–8.44 (d, J = 5.51 Hz, 1H, Ar-H quinoline), 8.71–8.89 (m, 2H, Ar-H quinoline), 8.93 (br s, 1H, NH), 9.31 (br s, 1H, NH), 9.67 (br s, 1H, NH); 13C NMR (75 MHz, CDCl3): δ 34.24, 41.46, 52.12, 65.54, 66.21, 97.61, 114.78, 118.14, 125.25, 125.88, 127.59, 128.28, 129.42, 129.68, 130.15, 131.22, 137.26, 137.68, 141.81, 148.72, 172.98; FAB-MS m/z 448 [M+H]+; Anal. Calcd for C21H21Cl3N4OS: C, 52.13; H, 4.37; N, 11.58; found: C, 52.17; H, 4.40; N, 11.56.
2-(2,6-Dichloro-phenyl)-thiazolidine-4-carboxylic acid [2-(7-chloro-quinolin-4-ylamino)-ethyl]-amide hydrochloride (22)
This compound was obtained as a yellowish white solid in 64% yield; mp 122–126°C; IR (KBr) 1627.9 cm−1; 1H NMR (200 MHz, DMSO-d6): δ 3.15–3.36 (m, 2H, CH2), 3.40–3.47 (m, 2H, CH2), 3.59–3.68 (m, 2H, CH2), 4.56–4.60 (m, 2H, CO-CH-CH2), 4.78–4.81 (m, 1H, CO-CH-CH2), 5.89 (d, J = 10.9 Hz, 1H, S-CH-N), 6.79–6.81 (d, J = 5.72 Hz, 1H, Ar-H quinoline), 7.40–7.71 (m, 4H, Ar-H), 8.00–8.05 (m, 2H, Ar-H), 8.43 (br s, 1H, NH), 8.80–8.83 (d, J = 5.96 Hz, 1H, Ar-H quinoline), 9.00 (br s, 1H, NH), 9.66 (br s, 1H, NH); 13C NMR (75 MHz, CDCl3): δ 36.57, 41.25, 49.45, 52.46, 69.71, 70.13, 97.26, 118.05, 125.88 (2C), 128.75 (2C), 129.16 (2C), 133.08 (2C), 134.83, 137.57, 141.26 (2C), 187.57; FAB-MS m/z 483 [M+H]+; Anal. Calcd for C21H20Cl4N4OS: C, 48.67; H, 3.89; N, 10.81; found: C, 48.71; H, 3.90; N, 10.84.
Biological and biophysical studies
Measurement of in vitro antimalarial activity
The in vitro antimalarial assay was carried out in 96-well microtitre platesCitation16,Citation23. The cultures of P. falciparum NF-54 strain are routinely maintained in medium RPMI 1640 supplemented with 25 mM HEPES, 1% d-glucose, 0.23% sodium bicarbonate and 10% heat inactivated human serumCitation24. The asynchronous parasites of P. falciparum were synchronized after 5% d-sorbitol treatment to obtain only the ring stage parasitized cells. For carrying out the assay, the initial ring stage parasitaemia of 0.8–1.5% at 3% haematocrit in a total volume of 200 µL medium RPMI 1640 was uniformly maintained. The test compound in 20 µL volume concentrations ranging between 0.5 and 50 µg/mL in duplicate well was incubated with parasitized cell preparation at 37°C in a candle jar. After 36–40-h incubation, the blood smears from each well were prepared and stained with Giemsa stainCitation23. The slides were microscopically observed to record maturation of ring stage parasites into trophozoites and schizonts in presence of different concentrations. The test concentrations, which inhibited the complete maturation into schizonts, were recorded as the minimum inhibitory concentration (MIC). CQ was used as the standard reference drug.
In vivo antimalarial efficacy test
The in vivo drug response was evaluated in Swiss mice infected with P. yoelii (N-67 strainCitation24). The mice (22 ± 2 g) were inoculated with 1 × 106 parasitized RBC on day 0 and treatment was administered to a group of five mice from day 0 to 3, once daily. The aqueous suspension of compounds was prepared with a few drops of Tween 80. The efficacy of test compounds was evaluated at 30.0 mg/kg/day and required daily dose was administered in 0.2 mL volume via intraperitoneal route. Parasitaemia levels were recorded from thin blood smears between days 4 and 28Citation24. The mean value determined for a group of five mice was used to calculate the percent suppression of parasitaemia with respect to the untreated control group. Mice treated with CQ served as positive controls.
Determination of haematin-4-aminoquinoline derivatives association constant
Association constant for haematin-4-aminoquinoline derivatives’ complex formation was determined by spectrometric titration procedure in aqueous dimethyl sulphoxide (DMSO) at pH 7.5Citation25. In this assay condition, haematin is strictly in monomeric state and interpretation of results is not complicated by the need to consider haematin disaggregation process. Association constant calculated in this technique is a good reflection of the interaction would occur in the acidic food vacuole. The pH 7.5 improves the stability of haematin solutions and quality of data.
In vitro inhibition of β-haematin polymerization
The ability of the 4-aminoquinoline derivatives to inhibit β-haematin polymerization was induced by 1-oleoyl-rac-glycerol using UV spectrophotometer and measurements were carried out at 405 nmCitation26. The triplicate values obtained from the assay are expressed as percent inhibition relative to haemozoin formation in a drug-free control. The 50% inhibitory concentration (IC50) values for the compounds were obtained from the sigmoidal dose–response curves using non-linear regression curve fitting analyses with GraphPad Prism v.3.00 softwareCitation27. Each IC50 value is the result of at least three separate experiments performed in duplicate.
Results and discussion
Chemistry
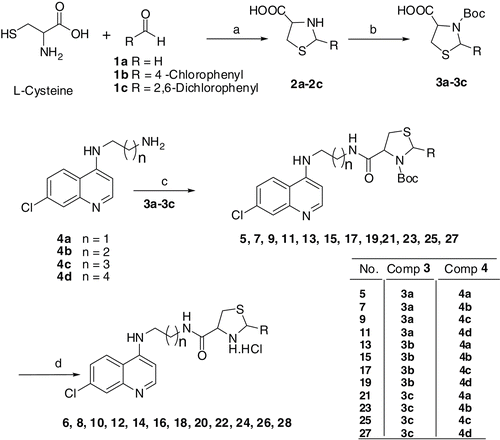
The desired 4-aminoquinoline-derived thiazolidines compounds (5–28) were synthesized as outlined in The synthesis of intermediate compounds of 4-aminoquinoline derivatives 4a–4d was carried out by the procedure reported earlier from this laboratoryCitation13. Boc-protected thiazolidine-4-carboxylic acid derivatives 3a–3c were synthesized by two-step process, in which thiazolidine-4-carboxylic acid 2a–2c was synthesized by condensation of l-cysteine with appropriate aldehyde 1a–1c in dry methanol. The 2-(substituted phenyl)thiazolidine-4-carboxylic acid derivatives (2b and 2c) were obtained as an inseparable diastereomeric mixture. This was further converted to Boc-protected derivatives by Boc anhydride in dioxane:water mixture (1:1). 4-Aminoquinolines 4a–4d were coupled with Boc-protected thiazolidine-4-carboxylic acid derivatives 3a–3c using HOBt/DCC in dry THF. The Boc group was removed by treatment with 4N HCl/dioxane to get the final compounds. The compounds reported in this study have been thoroughly characterized by elemental analyses and spectroscopic data.
Scheme 1 Synthesis of 4-aminoquinoline-derived thiazolidines.
Pharmacology
All the 4-aminoquinoline-derived thiazolidine compounds (5–28) were tested in vitro for antimalarial activity against NF-54 strain of P. falciparum. The MIC values were calculated from experiments carried out in triplicate and the results are presented in . These derivatives have shown significant antiplasmodial activity and among all the 24 compounds tested, two compounds 27 and 28 showed MIC range between 1.60 and 1.78 μM and 20 compounds showed MIC range between 3.28 and 4.98 μM. The remaining two compounds showed MIC range between 5.16 and 5.36 μM. The activity data highlight the importance of the number of carbon atoms in the lateral side chain and the nature of C-2 substitutions on the 4-thiazolidine ring systems. It may be inferred from the present results that increasing the carbon atoms in the side chain length moderately increase the antimalarial activity in this series. Furthermore, introduction of C-2 substitution by 4-chlorophenyl and 2,6-dichlorophenyl groups leads to a further increase in the antimalarial activity in comparison to unsubstituted analogues. This result clearly indicates that lipophilic character of these molecules is essential for the activity. Further, the data suggest that compounds with five carbon atoms 23–28 in the side chain are more active than the compounds with four 17–22, three 11–16 or two 5–10 carbon atoms in the side chain. The compounds having Boc-protecting group 5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 25 and 27 are relatively more active than their corresponding amino compounds 6, 8, 10, 12, 14, 16, 18, 20, 22, 24, 26 and 28. It is presumed that the protected derivatives are transformed to the corresponding free amino compounds, in the acidic milieu of the food vacuole, which is the site of action of 4-aminoquinoline class of compounds.
Table 1. In vitro antimalarial activity of synthesized compounds (5–28).
Selected compounds found active in vitro assay (14, 16, 18, 22, 24 and 26) were evaluated for in vivo activity in Swiss mice infected with N-67 strain of P. yoelli. The mice were treated with compounds (30 mg/kg) intraperitoneally once daily for 4 consecutive days, and their survival times and parasitaemia on day 4 were compared with those of control mice receiving saline (). These compounds showed moderate activity against P. yoelli infections in mice. Among these, compounds 14, 16 and 22 suppressed 99.70, 86.85 and 92.03% parasitaemia on day 4, respectively, compared to 100% suppression displayed by CQ.
Table 2. In vivo antimalarial activity data of selected compounds against N-67 strain of P. yoelii in Swiss mice.
To gain further insights into the potential of this new series of compounds as antimalarial agents, we assessed the mode of action of these analogues on heme polymerization target. The ability of the 4-aminoquinoline-derived thiazolidines to form association complex with haematin was investigated by UV spectrophotometric technique and the results are shown in . The data show that all the compounds bind to haematin with log K in the range of 5.24–6.25. It is appropriate to mention that higher log K value (i.e., >5.0) indicates tight binding of these analogues to haematin. The log K values of haematin association constant formation highlight the importance of C-2 substitution in the 4-thiazolidine system. In general, compounds that have 4-chlorophenyl and 2,6 dichlorophenyl (lipophilic) substitution at C-2 position of 4-thiazolidine system show very tight binding to haematin in comparison to unsubstituted analogues. It clearly indicates that all 4-aminoquinoline-derived thiazolidines might form complexes with haematin and the principal interaction might be involving π–π, stacking the quinoline ring with the porphyrin ring system of haematin. Similar experiments with Boc-protected derivatives could not be carried out because these compounds are not water-soluble.
Table 3. Biophysical data of 4-aminoquinoline-derived thiazolidines.
The ability of these compounds to inhibit haemozoin formation was studied to shed some light on the mode of action. The assay was performed according to the method described by Tripathi et al., which colorimetrically quantifies the ability of a compound to inhibit the conversion of haematin to β-haematin, the synthetic equivalent of haemozoinCitation26. The IC50 values represent the number of molar equivalents of the test compound, relative to haematin, that are required to induce 50% inhibition of the conversion of haematin to β-haematin. All the compounds were found to inhibit β-haematin formation () in a concentration-dependent manner. All the compounds were good inhibitors of β-haematin formation but some of them displayed moderate antimalarial activity. Compounds that have 4-chlorophenyl and 2,6-dichlorophenyl substitution at C-2 position of thiazolidine system show less µM concentration required for β-haematin formation inhibition in comparison to unsubstituted analogues. The study results suggest that this class of compounds binds to haematin and inhibits haematin polymerization by shifting haematin dimerization equilibrium to the µ-oxo dimer, thus reducing the availability of monomeric haematin for incorporation into β-haematin formation. These compounds may also inhibit β-haematin formation by blocking the growing face of crystal by a capping effect. It has been observed that there was no consistent correlation between haematin association constant, inhibition of haemozoin formation and in vitro antimalarial potency for this set of compounds, as the potent inhibitors of heme polymerization were not necessarily the more potent compounds in vitro. This indicates possible influence of other factors. One such factor could be the degree of accumulation within the parasite digestive vacuole, which is the definitive site of haemoglobin catabolism and, therefore, is the site of action of heme polymerization inhibitors.
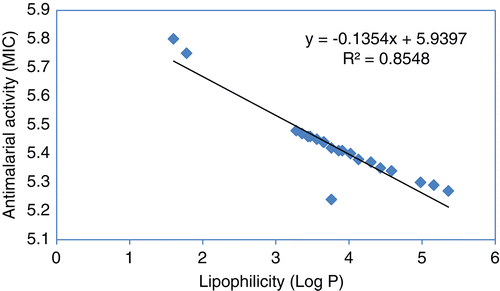
The modification carried out in the present study may affect the pKa values, which could have cascading effect on the antimalarial activity. Introduction of thiazolidine at 4-aminoquinoline lateral side chain amino group (pKa2) could render the ring nitrogen non-basic in nature. Therefore, we have calculated the pKa values () of these compounds using PallasCitation28. It clearly shows that introduction of thiazolidine on 4-aminoquinoline lateral side chain amino group leads to eliminate the pKa2 values and does not affect basicity of quinoline nitrogen atom (pKa1). However, thiazolidine skeleton itself generated one more pKa value (i.e., pKar) but these values are not correlated with antimalarial activity. The substituents at C-2 position of thiazolidine provide lipophilicity to the molecules as indicated by log p values (). This indicates a correlation between the antimalarial activity and log p (R2 = 0.85, p = 0.049; ). It clearly highlights the importance of lipophilicity in the antimalarial activity of the synthesized compoundsCitation14. The present study results are in accordance with our previous study inference and suggest that basicity of the lateral side chain nitrogen atom is not very essential for antiplasmodial activity of 4-aminoquinolinesCitation14,Citation15.
Figure 2. Correlation between log MIC and log p.
Conclusion
In summary, a new series of 4-aminoquinoline-derived thiazolidines has been synthesized and evaluated for their antimalarial activity. These derivatives have exhibited significant antimalarial activity against NF-54 strain of P. falciparum in the in vitro model and N-67 strain of P. yoelii in the in vivo model. Among the series, two compounds, 2-(4-chloro-phenyl)-thiazolidine-4-carboxylic acid [2-(7-chloro-quinolin-4-ylamino)-ethyl]-amide hydrochloride (14) and 2-(2,6-dichloro-phenyl)-thiazolidine-4-carboxylic acid [2-(7-chloro-quinolin-4-ylamino)-ethyl]-amide hydrochloride (22) exhibited significant suppression of parasitaemia in the in vivo assay. The present biophysical studies have suggested that this class of compounds forms association complex with haematin and thereby inhibits the haemozoin formation. The 4-aminoquinoline-derived thiazolidine analogue provided a promising lead for designing the antimalarial compounds targeting heme polymerization. The present findings are in accordance with our previous findings that the basicity of the side chain nitrogen is not an essential structural requirement for antiplasmodial activity of 4-aminoquinolines. The study provides a new opportunity for the development of new compounds for the treatment of malaria.
Supplementary material
The spectroscopic data for compounds 3a–3c and 5–28 (excluding 14 and 22) are available in supplementary material.
Acknowledgment
The authors thank the Director, CDRI for the support and the SAIF for the spectral data. One of the authors (V.R. Solomon) thanks the CSIR, New Delhi for Senior Research Fellowship. CDRI communication no. 49/2011/sbk.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S. Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov 2004;3:509–520.
- Wiesner J, Ortmann R, Jomaa H, Schlitzer M. New antimalarial drugs. Angew Chem Int Ed Engl 2003;42:5274–5293.
- Ridley RG. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature 2002;415:686–693.
- Kaur K, Jain M, Reddy RP, Jain R. Quinolines and structurally related heterocycles as antimalarials. Eur J Med Chem 2010;45:3245–3264.
- Ridley RG, Hofheinz W, Matile H, Jaquet C, Dorn A, Masciadri R et al. 4-aminoquinoline analogs of chloroquine with shortened side chains retain activity against chloroquine-resistant Plasmodium falciparum. Antimicrob Agents Chemother 1996;40:1846–1854.
- De D, Krogstad FM, Byers LD, Krogstad DJ. Structure-activity relationships for antiplasmodial activity among 7-substituted 4-aminoquinolines. J Med Chem 1998;41:4918–4926.
- Stocks PA, Raynes KJ, Bray PG, Park BK, O’Neill PM, Ward SA. Novel short chain chloroquine analogues retain activity against chloroquine resistant K1 Plasmodium falciparum. J Med Chem 2002;45:4975–4983.
- Pandey AV, Bisht H, Babbarwal VK, Srivastava J, Pandey KC, Chauhan VS. Mechanism of malarial haem detoxification inhibition by chloroquine. Biochem J 2001;355:333–338.
- Dorn A, Stoffel R, Matile H, Bubendorf A, Ridley RG. Malarial haemozoin/beta-haematin supports haem polymerization in the absence of protein. Nature 1995;374:269–271.
- Egan TJ, Marques HM. The role of haem in the activity of chloroquine and related antimalarial drugs. Coord Chem Rev 1999;190–192:493–517.
- Sullivan DJ Jr, Matile H, Ridley RG, Goldberg DE. A common mechanism for blockade of heme polymerization by antimalarial quinolines. J Biol Chem 1998;273:31103–31107.
- Ziegler J, Linck R, Wright DW. Heme Aggregation inhibitors: antimalarial drugs targeting an essential biomineralization process. Curr Med Chem 2001;8:171–189.
- Solomon VR, Puri SK, Srivastava K, Katti SB. Design and synthesis of new antimalarial agents from 4-aminoquinoline. Bioorg Med Chem 2005;13:2157–2165.
- Solomon VR, Haq W, Srivastava K, Puri SK, Katti SB. Synthesis and antimalarial activity of side chain modified 4-aminoquinoline derivatives. J Med Chem 2007;50:394–398.
- Solomon VR, Haq W, Smilkstein M, Srivastava K, Rajakumar S, Puri SK et al. Synthesis and antimalarial activity of novel side chain modified antimalarial agents derived from 4-aminoquinoline. Med Chem 2008;4:446–456.
- Solomon VR, Haq W, Smilkstein M, Srivastava K, Puri SK, Katti SB. 4-Aminoquinoline derived antimalarials: synthesis, antiplasmodial activity and heme polymerization inhibition studies. Eur J Med Chem 2010;45:4990–4996.
- Deshpande S, Solomon VR, Katti SB, Prabhakar YS. Topological descriptors in modelling antimalarial activity: N(1)-(7-chloro-4-quinolyl)-1,4-bis(3-aminopropyl)piperazine as prototype. J Enzyme Inhib Med Chem 2009;24:94–104.
- Prabhakar YS, Solomon VR, Gupta MK, Katti SB. QSAR studies on thiazolidines: A biologically privileged scaffold. In: Gupta SP ed Topics in Heterocyclic Chemistry. Berlin, Heidelberg, Germany: Springer, 2006, Vol. 4, pp. 161–249.
- Lesyk RB, Zimenkovsky BS. 4-Thiazolidones: Centenarian history, current status and perspectives for modern organic and medicinal chemistry. Curr Org Chem 2004;8:1547–1577.
- Prabhakar YS, Jain P, Khan ZK, Haq W, Katti SB. Synthesis and QSAR studies on the antifungal activity of 2,3,4-substituted thiazolidines. QSAR Comb Sci 2003;22:456–465.
- Rawal RK, Solomon VR, Prabhakar YS, Katti SB, De Clercq E. Synthesis and QSAR studies on thiazolidinones as anti-HIV agents. Comb Chem High Throughput Screen 2005;8:439–443.
- Srivastava T, Gaikwad AK, Haq W, Sinha S, Katti SB. Synthesis and biological evaluation of 4-thiazolidinone derivatives as potential antimycobacterial agents. ARKIVOC 2005; ii:120–130.
- Srivastava K, Puri SK. Plasmodium falciparum: modified medium composition supports continuous cultivation with foetal bovine serum. Exp Parasitol 2004;108:74–75.
- Puri SK, Singh N. Azithromycin: antimalarial profile against blood- and sporozoite-induced infections in mice and monkeys. Exp Parasitol 2000;94:8–14.
- Egan TJ, Mavuso WW, Ross DC, Marques HM. Thermodynamic factors controlling the interaction of quinoline antimalarial drugs with ferriprotoporphyrin IX. J Inorg Biochem 1997;68:137–145.
- Tripathi AK, Khan SI, Walker LA, Tekwani BL. Spectrophotometric determination of de novo hemozoin/beta-hematin formation in an in vitro assay. Anal Biochem 2004;325:85–91.
- GraphPad Prism, version 4.03. GraphPad Software Inc., 10855 Sorrento Valley Rd. #203, San Diego, CA 92121, 2005.
- Pallas 2.0. Compu-Drug Chemistry Ltd., San Francisco, CA, 1995.