Abstract
Famotidine was investigated as an inhibitor of glycogen synthase kinase-3β (GSK-3β) in an attempt to explain the molecular mechanism of its hypoglycemic side effects. The investigation included simulated docking experiments, in vitro enzyme inhibition assay, glycogen sparing studies using animal models and single dose oral glucose tolerance test (OGTT). Docking studies showed how famotidine is optimally fit within the binding pocket of GSK-3β via numerous attractive interactions with some specific amino acids. Experimentally, famotidine could inhibit GSK-3β (IC50 = 1.44 μM) and increased significantly liver glycogen spares in fasting animal models. Moreover, a single oral dose of famotidine was shown to decrease the glycemic response curve after 75 g OGTT
Introduction
Famotidine is a specific, long-acting H2-receptor antagonist which competes the binding of histamine to their receptors in the parietal cells. Famotidine decreases basal, nocturnal, pentagastrin and food-stimulated gastric acid productionCitation1. Famotidine is indicated for the management of peptic ulcer, esophageal refluxes, and the treatment of pathological hypersecretory conditions, like Zollinger–Ellison syndromeCitation2. Generally, famotidine is well tolerated in patients with various disorders like cardiovascular, renal and hepatic dysfunctions, moreover it is considered to have excellent safety profilesCitation3. Nevertheless, it is well known that H2-receptor inhibitors affect hormone metabolismCitation4; however, their effect on glucose metabolism is not well investigated. In fact several discrepancies are reported concerning the effects produced by these compounds on glycemic levelCitation4–7. In addition, most studies have investigated the effect of cimetidine and ranitidine and only few data are available for famotidine. In 2003, a study, carried out to investigate the effect of famotidine on some hematobiochemical parameters in mice, has revealed a glucose-lowering effect for famotidineCitation8.
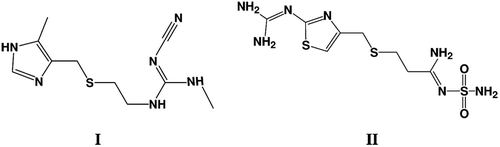
Recently, an extensive molecular modeling experiments, employing different molecular modeling techniques as pharmacophore modeling, QSAR predictions and docking investigations, were conducted to find out novel glycogen synthase kinase-3β (GSK-3β) inhibitorsCitation9. Cimetidine (I, ) was found to have a potent anti-GSK-3β bioactivity in vitro and illustrated a hypoglycemic activity and glycogen reservation capability in vivoCitation9. These findings were consistent with several previous studies which reported a significant cimetidine-induced hypoglycemiaCitation10. No studies were conducted to investigate the direct effect of famotidine on GSK-3β activity. The central role of GSK-3β in glucose metabolism and the structural similarities between cimetidine (I) and famotidine (II) () have encouraged us to investigate GSK-3β-inhibitory activity of famotidine as well as its effect on glycogen levels in animals and blood glucose level. To investigate if famotidine can bind to GSK-3β active site, an initial molecular docking was performed as a preliminary in-silico screening test.
Figure 1. Molecular structure of both Cimitidine (I) and Famotidine (II).
Materials and methods
Materials
Chemicals used in these experiments were of analytical grade and purchased from commercial suppliers: Recombinant GSK-3β (from Upstate Biotechnology, Waltham, MA); Tau [pS396] phosphoELISA kit) Biosource, Grand Island, NY); Assay buffer components (HEPES; MgCl2 and EDTA, Biosource); ATP (Sigma, Sigma, MO); heparin (Sandoz GmbH, Austria); Anthrone (Sigma); Thiourea (Sigma).
Molecular modeling
The 3D coordinates of GSK-3β were recovered from the Protein Data Bank (PDB code: 1Q5K, resolution = 1.94 Å)Citation11. The DS visualizer 2.0 templates for protein residues (Accelrys discovery studio (DS) visualizer 2.0, Accelrys Inc., San Diego, CA) was used to extract the co-crystallized ligand and to add hydrogen atoms to the protein. Famotidine chemical structure was sketched in ChemDraw Ultra (10.0), and saved in MDL molfile format (CS ChemDraw Ultra 10.0, Cambridge Soft Corp (Cambridgepark Drive, CA). (http://www.cambridgesoft.com) OMEGA software (Omega, Santa Fe, NY, USA, version 2.3.2, OpenEye Scientific Software (www. eyesopen.com)Citation12 was used to explore the conformational space of famotidine.
Famotidine was docked into the binding site of GSK-3β utilizeing docking capabilities of FRED software (FRED version 2.2.5, OpenEye Scientific Software (www.eyesopen.com)Citation13). This docking software considers a multiconformer database of multiple ligands, a target protein structure, a box defining the active site of the protein based on the co-crystallized ligand and other optional parameters as input.
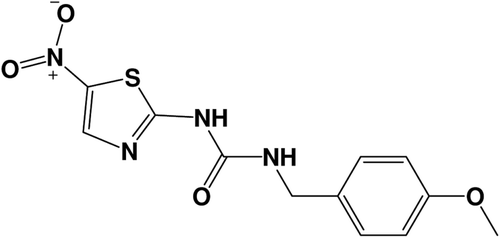
FRED’s docking process is used to thoroughly score all possible poses of each ligand in the active siteCitation13. The exhaustive search is based on rigid rotations and translations of each conformer. The best docking situations utilized were those succeeded to retrieve the pose of the co-crystallized ligand (AR-A014418, ). For more details about the docking experiment and docking parameters employed see referencesCitation14,Citation15.
Figure 2. Molecular structure of co-crystallized structure AR-A014418.
In vitro GSK-3β enzyme inhibitory assay
GSK-3β was dissolved in a buffer solution (pH 7.2) containing the following: 40 mM HEPES; 5 mM MgCl2; 5 mM EDTA; 100 μM ATP and 50 μg/ml heparin to get a concentration of 10 pg/ml. Subsequently, 50 μl aliquots of the enzymatic solution were transferred into 0.5 ml vials. Thereafter, appropriate volumes of famotidine stock solution were pipetted to the enzymatic solution to give final concentration of 0.1, 1, and 10 μM and the volumes were then completed upto 75 μl. Famotidine was incubated with the enzyme over 30 min at room temperature, then a proper volume of Tau solution (25 μl of 2000 pg/mlin HEPES) was added to get a final concentration of 500 pg/ml. This mixture was incubated over 1 h at room temperature.
Tau phosphorylation was detected as previously described by Bustanji et al.Citation14 A standard GSK-3β inhibitor (TDZD-8) was applied as positive controlCitation16 in this experiment.
In vivo evaluation
Preparation of famotidine doses
The effect of famotidine on liver glycogen content of fasting Balb/c mice was investigated. Famotidine suspension was suspended in 0.5% carboxy methylcellulose phosphate buffer saline (pH 7.2). For control animals, the vehicle without famotidine was employed. Three escalating doses of famotidine were used: 1.1, 2.2 and 4.4 mg/kg. These doses correspond to human dose, double the human dose and four times human dose, respectively. All famotidine doses were injected i.p. (ca. 0.20mL) to yield the desired dosage after normalization to mouse weight (ca. 20 g).
Determination of liver glycogen
All animal experiments comply with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. 10-week old male Balb/c mice of average weight of 20 g, obtained from Jordan University animal house, Jordan, were used for the investigations. The animals were randomly selected and fed ad libitum with standard feed and water. Animals were housed in the same conditions and separated randomly in four-mouse groups (5 animals each). The first group was injected i.p. with the vehicle as negative control. The other three groups were injected with the three escalating doses of famotidine. These doses are (i) human dose (1.1mg/Kg) (ii) double the normalized human dose, and (iii) four times the normalized human dose. All doses were normalized to mouse weight (ca. 20 g),
Animals were fasted for at least four hours before the doses administration. Three hours after the famotidine administration, the animals were sacrificed by cervical dislocation and their livers were removed immediately for glycogen determination. Liver glycogen content was determined quantitatively using anthrone reagent methodCitation17. For further procedure details see referencesCitation14,Citation15.
Oral glucose tolerance test (OGTT)
The study was placebo-controlled crossover study. Six healthy human volunteers (28–40 years old) were fasted for at least 10 h before the starting point of experiments in which they received single oral doses of famotidine 80 mg or placebo, separated by at least a 7-d washout interval. Thirty minutes after the administration, the participants were perorally administered with 225 ml containing 75 g glucose (consumed within 5 min). Plasma was obtained by centrifugation (3000 rpm) and plasma glucose determination were measured before and 30, 60, and 120 min after the administration with glucose by the hexokinase method. The study was conducted in accordance with the guidelines in the Declaration of Helsinki. Written informed consent was obtained after the purpose, nature and potential risks had been explained to the subjects. All subjects were within 10% of ideal body weight, and none had undergone abdominal surgery or were taking any medications. All the subjects enrolled in this study were ethnic Caucasian. Subjects who are smoking and/or alcoholic were excluded.
Data are presented as means ± SEM. Statistical comparisons were performed using Student’s t-test. In all cases, p < 0.05 is considered statistically significant.
Results and discussion
Efforts to investigate the inhibitory effect of famotidine on GSK-3β were started by using in-silico docking technique to investigate its possibility of binding within GSK-3β binding pocket. The docking simulations were conducted using FRED docking engine. The binding pocket was defined from the crystallographic structure of AR-A014418Citation11, which is a known GSK-3β ligand with high affinity ().
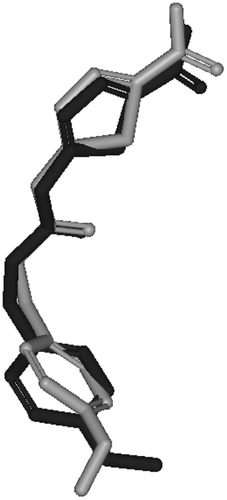
The molecular modeling study of famotidine- GSK-3β binding has shown that famotidine can be successfully docked within the binding pocket of GSK-3β making several significant interactions with key hot spots within the GSK-3β binding pocket. shows the solvent accessible surface as well as the key residues of GSK-3β binding site with the co-crystallized inhibitor AR-A014418 () and the highest ranking docked pose of famotidine (). However, to validate our docking-scoring procedure, we employed the same docking conditions to dock AR-A014418 into the binding pocket of GSK-3β. Docking simulations created close models to the crystallographic structure (RMSD = 0.84 Å, ), which supported the docking results of famotidine into GSK-3β binding site, as shown in , and supports our conclusions regarding famotidine/GSK-3β binding.
Figure 3. (A) Detailed view of the co-crystallized structure (AR-A014418) and the corresponding interacting amino-acids within the binding site of GSK-3β. (B) Detailed view of the docked famotidine structure and the corresponding interacting amino-acid moieties within the binding site of GSK-3β.
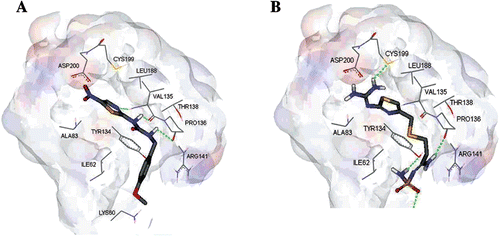
Figure 4. Comparison between the docked conformer/pose of inhibitor AR-A014418 (dark black) as produced by the docking simulation and the crystallographic structure of this inhibitor within GSK-3β (gray, PDB code: 1Q5K).
Comparison of the highest ranking docked pose of famotidine () with the co-crystallized ligand within the binding site of GSK-3β () illustrates similarities in their binding profile. Both have strong hydrogen bond interactions with the key amino acids PRO-136 and VAL-135 and potential hydrophobic interaction with LEU-188. However, famotidine showed more several important interactions which might contribute to its high GSK-3β binding affinity and inhibitory activity. The guanidine group of famotidine interacts with the sulfahydryl moiety of CYS-199 and carboxyl group of ASP-200 through hydrogen bonding (2.2 Å) and electrostatic interactions (3.2 Å), respectively. Moreover, the terminal NH2 group makes a strong hydrogen bond with the OH of the TYR-143 (2.2 Å), while the sulfonyl group is hydrogen bonded to LYS-60 (2.5 Å). Furthermore, the sulfur atom in the thioether flexible chain in famotidine has a potential hydrophobic interaction with ILE-62. Overall, these attractive interactions cooperate in stabilizing the proposed complex. This preliminary docking simulation finding has encouraged us to further evaluate the inhibitory actions of famotidine in vitro and to investigate the effects of famotidine on animal’s liver glycogen levels and OGTT.
The proposed inhibitory action of famotidine was experimentally validated against recombinant GSK-3β. The in vitro inhibitory activity was expressed as the concentration of famotidine that inhibits the enzymatic activity by 50% (IC50) when is compared to the un-inhibited enzymatic reaction performed under the same conditionsCitation14. The inhibitory effects of three escalating concentrations; (0.1, 1, and 10 μM) of famotidine on GSK-3β activity was performed. Results have shown that these concentrations could inhibit GSK-3β activity in a dose dependent manner showing 29.4, 35.5, 71.2% inhibition, respectively. Linear regression of these results revealed (R2 = 0.918) an estimated IC50 value of 1.44 μM TDZD-8, a standard GSK-3β inhibitor showed an IC50 value of 1.5 µM that is comparable to the published valueCitation16.
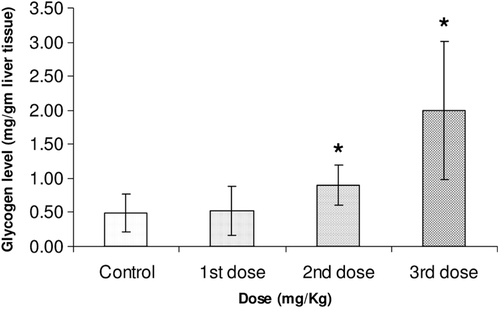
Moreover, famotidine significantly increased liver glycogen reserves in fasting Balb/c mice in a dose-dependent manner. shows the responses of liver glycogen reserves to i.p. administered famotidine at doses 1.1, 2.2, 4.4 mg/kg normalized to mice weight. It is obvious in this figure that there is a significant elevation of liver glycogen (fourfold increase) with the use of the highest dose of famotidine (4.4 mg/kg).
Figure 5. Liver glycogen reserves in fasting 10-week old male Balb/c mice three hours after i.p. administration of vehicle control, 1.1, 2.2 and 4.4 mg/kg of famotidine normalized to mice weights (20 g). Each result represents the average glycogen level from 5 mice. Error bars indicate the standard deviations of the measurements. (*) p value < 0.05 versus control.
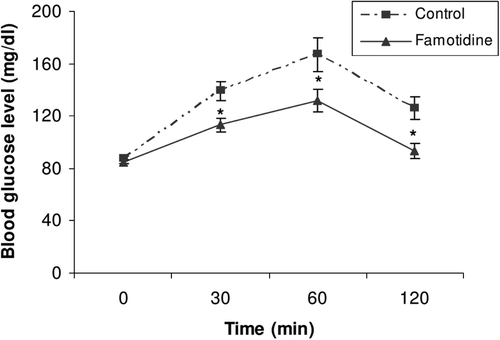
Finally, we have also investigated whether oral glucose tolerance is improved in healthy volunteers by famotidine. Blood glucose levels were measured before and 30, 60, and 120 min after famotidine administration. Serum glucose levels were observed to be significantly decreased 30 min after oral glucose load following famotidine administration (113.0 ± 14 mg/dl compared to the control value of 139.0 ± 17 mg/dl; p < 0.05). Similarly, blood glucose level significantly decreased 60 min after glucose load from 167.0 ± 32 to 132.0 ± 21 mg/dl (p < 0.05). At 120 min, serum glucose levels decreased to less than 120 mg/dl (93.0 ± 14 mg/dl compared to the control value of 126.0 ± 21 mg/dl; p < 0.05). Average area under the serum glucose-time curve also was decreased significantly (13415 compared to the control value of 17020; p < 0.05), as shown in . This is the first report indicating that famotidine improves the glycemic curve in OGTT.
Figure 6. Glycemic response during a 75 g OGTT (n = 6) in healthy humans with (▴) or without (▪) famotidine (80 mg p.o.). Asterisks denote a significant difference compared to the control values. Data are expressed as geometric mean ± SEM.
Glycogen metabolism is tightly regulated metabolic process and is under the control of various metabolic signalsCitation18. GSK-3β plays crucial role in the regulation of glycogen synthesis through phosphorylation and thus inhibition of the activity of glycogen synthase which is a key metabolic enzyme in the glycogen synthesis pathway. Consequently, inhibitors of GSK-3β could improve glucose disposal via increasing the rate of glycogen synthesis in the liver and skeletal muscles. In our study, the significant increase in liver glycogen reserves caused by famotidine may be attributed, at least partially, to its inhibitory action of GSK-3β, a hypothesis that was supported by the initial docking studies.
Conclusion
In summary, our studies provide evidence that famotidine could inhibit GSK-3β in vitro. Moreover, we provide evidence showing that famotidine promotes glucose metabolism in healthy humans. A single oral dose of famotidine was shown to lessen the glycemic response curve after 75 g OGTT. These effects were paralleled by the enhancement in liver glycogen synthesis in vivo.
Acknowledgements
This project was sponsored by the Deanship of Scientific Research at the University of Jordan. The authors wish to thank the Deanship of Scientific Research. The authors would like to thank also the OpenEye Scientific Software Corporation for providing free license of FRED software.
Declaration of interest
The authors report no declarations of interest.
References
- Fit KE, Williams PC. Use of histamine2-antagonists for the treatment of verruca vulgaris. Ann Pharmacother 2007;41:1222–1226.
- Campoli-Richards DM, Clissold SP. Famotidine. Pharmacodynamic and pharmacokinetic properties and a preliminary review of its therapeutic use in peptic ulcer disease and Zollinger-Ellison syndrome. Drugs 1986;32:197–221.
- Howden CW, Tytgat GN. The tolerability and safety profile of famotidine. Clin Ther 1996;18:36–54; discussion 35.
- Stubbs WA, Delitala G, Besser GM, Edwards CR, Labrooy S, Taylor R et al. The endocrine and metabolic effects of cimetidine. Clin Endocrinol (Oxf) 1983;18:167–178.
- Verdy M, Bolte E, Verdy I. Cimetidine and glucose tolerance (oral and intravenous). Int J Clin Pharmacol Ther Toxicol 1983;21:487–488.
- Gentile S, Marmo R, Costume A, Orlando C, D’Alessandro R, De Bellis G et al. The role of ranitidine infusion on glucose, insulin and C-peptide serum levels induced by oral glucose tolerance test in healthy subjects. Acta Diabetol Lat 1986;23:165–170.
- Scarpignato C, Tirelli F, Starcich R, Bertaccini G. Effect of acute and chronic cimetidine administration on glucose tolerance and insulin secretion in man. Horm Res 1981;15:228–236.
- Das PK, Mostofa M, Hasan Q, Shil GC, Islam MR. Effects of ranitidine, famotidine and omeprazole on some haematobiochemical parameters in mice. J Anim Vet Adv 2003 2:321–326.
- Taha MO, Bustanji Y, Al-Ghussein MA, Mohammad M, Zalloum H, Al-Masri IM et al. Pharmacophore modeling, quantitative structure-activity relationship analysis, and in silico screening reveal potent glycogen synthase kinase-3beta inhibitory activities for cimetidine, hydroxychloroquine, and gemifloxacin. J Med Chem 2008;51:2062–2077.
- Støa-Birketvedt G, Paus PN, Ganss R, Ingebretsen OC, Florholmen J. Cimetidine reduces weight and improves metabolic control in overweight patients with type 2 diabetes. Int J Obes Relat Metab Disord 1998;22:1041–1045.
- Bhat R, Xue Y, Berg S, Hellberg S, Ormö M, Nilsson Y et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem 2003;278:45937–45945.
- OMEGA VUM, OpenEye Scientific Software Inc., Santa Fe, New Mexico, Jul., 2006
- FRED VUM, 2006. OpenEye Scientific Software, (www.eyesopen.com), USA.
- Bustanji Y, Taha MO, Almasri IM, Al-Ghussein MA, Mohammad MK, Alkhatib HS. Inhibition of glycogen synthase kinase by curcumin: Investigation by simulated molecular docking and subsequent in vitro/in vivoevaluation. J Enzyme Inhib Med Chem 2009;24:771–778.
- Mohammad MK, Al-Masri IM, Taha MO, Al-Ghussein MA, Alkhatib HS, Najjar S et al. Olanzapine inhibits glycogen synthase kinase-3beta: an investigation by docking simulation and experimental validation. Eur J Pharmacol 2008;584:185–191.
- Martinez A, Alonso M, Castro A, Pérez C, Moreno FJ. First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J Med Chem 2002;45:1292–1299.
- Carroll NV, Longley RW, Roe JH. The determination of glycogen in liver and muscle by use of anthrone reagent. J Biol Chem 1956;220:583–593.
- Carbohydrate biosynthesis. In: Lehninger principles of biochemistry. 3rd ed. New York: Worth Publishers; 1999. p 735–739.