Abstract
New 2-(3,4,5-triacetoxybenzoylamino)benzothiazoles (4a~5f) and 2-(galloylamino)benzothiazoles (6a~7f), were designed as topoisomerase-I inhibitors. Compare/fit studies between these molecules and the generated topoisomerase-I inhibitors hypothesis revealed that 4a~5f have higher fitting values than (6a~7f). Also, docking of 4a~7f with the topoisomerase-I enzyme prioritized the higher activity of (4a~5f) than (6a~7f). These molecules were synthesized and biologically evaluated for their in vitro cytotoxic activity against Hela and MCF7 human cancer cell lines in comparison to Camptothecin (topo-I inhibitor) and doxorubicin (topo-II inhibitors) as reference drugs. Such screening revealed that compounds 4d, 4e, 4h, 5b, 5c and 5e have comparable higher cytotoxic activity in both cultures than these reference drugs. The highest active molecule was 5f that gave 1.5 folds higher cytotoxic activity against Hela cell cultures and 1.9 folds higher activity against MCF7 cell lines than doxorubicin and 1.6 folds and 2.2 folds higher activity towards the two respective cultures than Camptothecin.
Introduction
The developments of anticancer agents became definite and selective for various targets. Among the important cellular targets which were reported to have a significant role in cancer treatment, is the inhibition of DNA topoisomerase enzymesCitation1. Topoisomerases are essential nuclear enzymes that catalyze the concerted breaking and rejoining of DNA strands. Also these enzymes are involved in the production of necessary topological and conformational changes in DNA which are critical to many cellular processes such as replications and transcriptionsCitation2–4. There are two human topoisomerases; ICitation5 and IICitation6, which have been isolated from mammalian cells. Topoismerase-I is an ATP independent and introduces transient break on a single strand of duplex DNA. It catalyzes the passage of another DNA strand through the broken part. On the other hand, topoisomerase II is an ATP dependent and introduces double strand breakdown of the DNA. It catalyzes the passage of another DNA double strand through this breakdown. Also, this enzyme plays important roles in chromosome assembly, condensation and segregation of chromosome as well as in completion of transcriptionCitation7–10.
Topoisomerases І and II have recently emerged as important cellular targets for chemical intervention in the development of anti-tumour drugs which were useful in preventing the actions of these enzymes, so they were used clinically in the treatment of different kinds of cancers. Many topoisomerases inhibitors are classified into three types. (i) Topoisomerases inhibitors that stabilize cleavable complex and intercalate into DNA (viz; DoxorubicinCitation11, AmsacrieCitation12 and EllipticinesCitation13). (ii) Topoisomerases inhibitors that stabilize the cleavable complex without intercalation with DNA (viz; CamptothecinCitation14, TopotecanCitation15 and EpipodophylotoxinCitation16). (iii) Cleavable complex non-forming type topoisomerase I and II inhibitors (viz; Epigallocatechin gallateCitation17, Peterselenic acidCitation18, ToposlationsCitation19–21, 2070-DTTCitation22, 2890-DTICitation23 and MacrostafinCitation24).
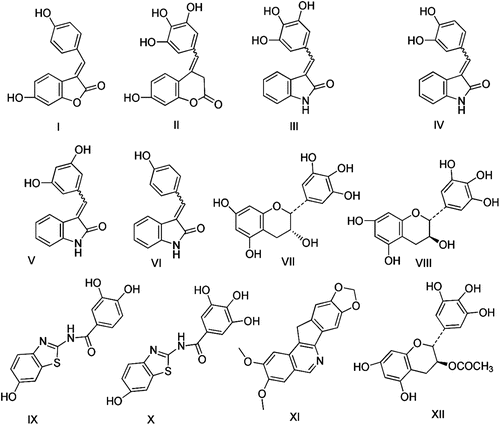
In 2002, Suzuki and coworkersCitation18 isolated Isoqurastatin (I) from Thermonospora alba and they reported that this compound have direct cleavable complex non-forming type topoisomerase I inhibitory activity with IC50 307 µM. Further modification in the structure of I (by the introduction of 1~3 hydroxy groups), into Comarinone (II) and Indolones (III, IV, V and VI), were found to increase its inhibitory activityCitation25. Meanwhile, it was found that Gallocatechin (VII) and Epigallocatechin (VIII) containing several hydroxyl groups on phenyl moiety had strong inhibitory activities against topoisomerasesCitation1. Moreover, new 2-(3,4-dihydroxybenzoyl-amino)-6-hydroxy-benzothiazole (IX) and 2-galloylamino-6-hydroxy-benzothiazoles (X), were evaluated to have potent topoisomerase-I (topo-I) inhibitory activity, with IC50 values = 8.8, 16.9 µM, respectivelyCitation26. More recently, several norindeneisoquinolines substituted with methoxy or methylenedioxy groups have been proven to have potent anticancer activityCitation27. Among these derivatives was the 2,3-dimethoxy-8,9-methylenedioxy-11H-indeno[1,2-c]isoquinoline (XI) molecule, that showed strong topo-I inhibitory activity with high cytotoxicity in the NCI cancer cell culture screening (mean graph midpoint of 50 µM)Citation27. Moreover, the 3-O-acyl-(-)epigallocatechin (XII) was reported to exhibit better anticancer activity than free hydroxy-epigallocatechin (VIII)Citation28. The structures of the topo-I inhibitors leads (I–XII) were given in .
Figure 1. Lead compounds of topoisomerase I inhibitors.
In this investigation, new topo-I inhibitors were designed by optimizing the structures of the leads (I–XII), followed by virtual screening of the proposed molecules by two integrated molecular modelling studies. The first was compare/fit of the proposed molecules with a generated common features pharmacophore (hypothesis) of topo-I inhibitors. The second was the docking studies of the same proposed molecules with the binding site of the same target.
The results of these two simulation studies (that will be discussed in details in the next part), predicted the higher affinity of the 2-(O-triacetyl-galloylamino)benzothiazoles (4a~5f) toward the topo-I enzyme than the lead molecule XI. However, the free hydroxyl galloyl analogues (6a~7f) were prioritized to have lower activity than the same lead ( and and Supplementary materials). Accordingly, we were motivated to synthesis these hit molecules (4a~7f), following the sequence outlined in to examine their cytotoxic activity against Hela and MCF7 cell cultures, hoping to develop new more potent anticancer topo-I inhibitors prototypes.
Table 1. Simulation fitting scores of the designed triacetyl (4a-h and 4a-f) and the free hydroxyl derivatives (6a-h and 7a-f) with the generated ideal hypothesis of topoisomerase I inhibitors in comparison to the leads compounds; I-XII.
Table 2. Docking energy and binding pattern of the docked triacetyl (4a-h and 5a-f) and the free hydroxyl derivatives (6a-h and 7a-f) with topo-I enzyme in comparison to the lead XI: (numbers of HB, numbers of Wan Der Wall attractions (pi-pi and sigma-pi) interactions and [pi-(+) interaction distance with Arg364 at the binding sites).
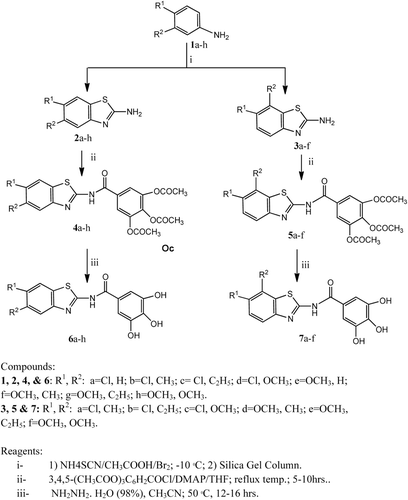
Scheme 1. Synthesis of the designed compounds (4a~7h).
Results and discussion
Synthesis
Treatment of 3,4-disubstituted-anilines (1b-d and 1f-h) with ammonium thiocyanate and bromine (NH4SCN/Br2) in acetic acid by a similar procedure for preparing compound 1aCitation29, resulted in the formation of a mixture of two products. TLC of such reaction products showed two main spots with distinguishable RF values, indicating the production of two main products in each reaction. Explanation of this may be attributed to the possible cyclization of the starting 3,4-disubstituted anilines (1b-d and 1f-h), during this reaction, at either the C2 or C6 (the two ortho positions) to give mixtures of the respective 2-amino-5,6-disubstituted-benzo[d]thiazoles (2b-h) and 2-amino-6,7-disubstituted-benzo[d]thiazoles (3a-f), in each case. The two components in each case were successfully separated on preparative silica-gel columns.
The structures of the isolated 5,6-disustituted-benzothiazole derivatives (2b-d and 2f-h) were confirmed from their 1H-NMR spectra, where they showed two aromatic singlets at δ = (7.08-7.93) and (7.25-8.02) of the two C-H aromatic at C4 and C7, respectively. But, the second isolated isomers; 6,7-disustituted-benzothiazole derivatives (3a-f), were also elucidated from their 1H-NMR spectra, where they gave two aromatic doublets at δ = (6.88-7.51) and (7.08-7.61) of the two adjacent aromatic hydrogens at C4 and C5, respectively.
Condensation of the crude products of 2a-h and 3a-f and 3,4,5-triacetoxy-benzoyl chlorideCitation30 was carried out in the presence of TEA in THF, where the 2-(O-triacetylgalloylamino)-5,6-disubstitutedbenzo-[d]thiazoles (4a-h) and 2-(O-triacetylgalloyl)amino-6,7-disubstituted-benzo[d]thiazoles (5a-f) were produced, respectively. Deacetylation of the respective triacetyl analogues (4a-h) and (5a-f), using hydrazine hydrate, gave the corresponding 2-(galloyl-amino)-5,6-disubstituted-benzothiazoles (6a-h) and 2-(galloylamino)-6,7-disubstituted-benzothiazoles (7a-f), respectively (). The structures of these new products were confirmed by spectral and analytical data.
Molecular modelling
Compare/fit studies of topo-I inhibitors hypothesis with 2-(triacetylgalloylamino)-5,6-disustituted benzo[d]thiazoles (4a~5f) and 2-(galloylamino)-6,7-disustituted-benzo[d]thiazoles (6a~7f)
The generation of the topo-I hypothesis was achieved by running common features pharmacophore generation, using the aforementioned leads (I-XII), where it generated 10 hypotheses. These hypotheses were assessed and the one ranked number 7 was proven to be the more ideal than the other hypotheses (1–6 and 8–10), due to the following; i) It has the highest fit values with the lead compounds I-XII (). ii) The data base searching, using ICI and Maybridge data bases revealed that such hypothesis map with only 0.01 % of the available data bases molecules, indicating its better selectivity. iii) Retrospectively, this hypothesis gave worst consistent fit values with the given in vitro biological activity of the test set molecules (4a~7f). Thus, this ideal hypothesis had reported here as the first time. It encompassed three features namely; 2 hydrogen bonds acceptors features (2HBA) and 1 aromatic vector feature (AR), with constraint distances and angels ( and ).
Figure 2. Constraint distances in the ideal Hypothesis of Topoisomerase-I inhibitors. (HBA = Hydrogen bond acceptor feature, AR =Ring Aromatic vector feature).
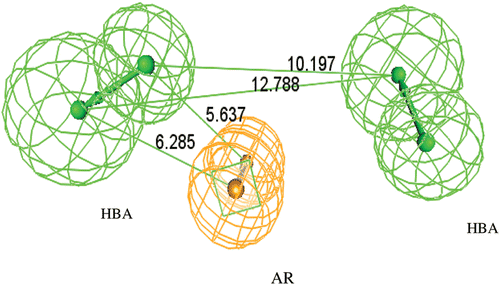
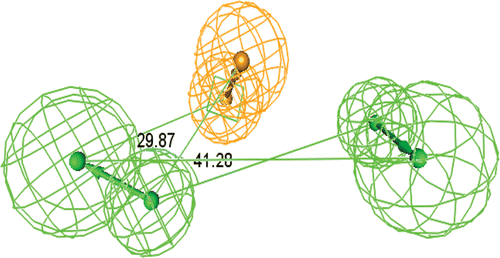
Figure 3. Constraint angles in the ideal hypothesis of topoisomerase-I inhibitors.
Comparative fit studies of the proposed compounds (4a~7f) with this ideal hypothesis showed that the 2-(O-triacetyl-galloyl) derivatives (4a~5f) had higher fit values than the leads І-XII, while the free hydroxyl galloyl derivatives (6a~7f) showed lower fit values than the same leads ( and and and Supplementary materials).
Figure 4. Hypothesis no. 7 and the top fitted compound no. 5f (Fit value = 2.78/3).
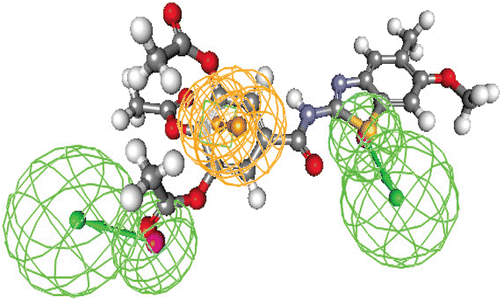
Figure 5. Hypothesis no. 7 and the top fitted compound no. 4h (Fit value = 2.79/3).
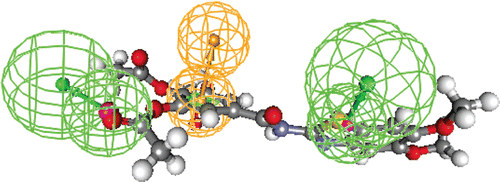
Docking of topo-I enzyme with 2-(O-triacetyl-galloylamino)-5,6-disustitutedbenzo[d]thiazoles (4a~5f) and 2-galloyl-amino-6,7-disustitutedbenzo[d]thiazoles (6a~7f)
This study was started by downloading the 3D crystal structure of topo-I enzyme complexed with both the combined DNA molecule and the newly developed inhibitor; 2,3-dimethoxy-8,9-methylenedioxy-11H-indeno[1,2-c]isoquinoline (XI)Citation27 (code; 1TL8)Citation1, from PBD web. Then this protein was docked with the test set compounds (4a~7f) by running Accelrys DS C-Docker protocol. Analysis of the docking diagrams of the test set compounds (4a~7h) in comparison to that of the lead XI in its crystal structure complexed with topo-I enzyme, were carefully studied, taking in consideration the reported SAR of the lead (XI) as inhibitor for this enzyme. Such reported SAR emphasized the existence of a crucial HB acceptor between the enzyme-Arg364 aminoacid and the ether bridge of XICitation27. Thus, the analyses of the 3D docking diagrams of the tested molecules, in this research, were monitored adopting Accelrys (DS) guidelines to search for the existence of such crucial HB with the enzyme-Arg364 aminoacid, to predict the fulfilling for the given SAR. Other details in the 3D docking diagrams were also automatically generated, in order to get the details of the binding patterns of the docked molecules. These details were found to have the following additional interactions: (i) The intermolecular H-bondings and their numbers including that with enzyme-Arg364 and DNA nucleotide structures at the binding site. (ii) The numbers of intermolecular lipophilic pi-pi interactions with the surrounding aminoacids and nucleotides. (iii) The distance of the intermolecular pi-cation [pi-(+)] interaction between the docked molecules and the Arg364 aminoacid.
Thus, monitoring of the 3D crystal structure of the lead (XI) with topo-I enzyme, showed one hydrogen bond with Arg364 (as reported)Citation27, 8 pi-pi interactions and pi-(+)interaction with distance equal 5.328 Å, between its structure and the enzyme-Arg364 and DNA nucleotide structures at the binding site. These binding interactions gave docking energy = −44.41 Kcal/mol (, ).
Figure 6. Publication quality 3D diagram of the X-ray crystal structure of lead (XI)complexed with topoisomerase-I enzyme (redocked with RMSD = 0.0023) and Docking value = −44.41. The binding pattern showed: [1HB with Arg364 (as reportedCitation27 and 8 lipophilic pi-pi interactions with the surrounded amino acids and DNA nucleotides, and a [ pi-(+)] interaction between it and Arg364 by distance = 6.95 Å.
![Figure 6. Publication quality 3D diagram of the X-ray crystal structure of lead (XI)complexed with topoisomerase-I enzyme (redocked with RMSD = 0.0023) and Docking value = −44.41. The binding pattern showed: [1HB with Arg364 (as reportedCitation27 and 8 lipophilic pi-pi interactions with the surrounded amino acids and DNA nucleotides, and a [ pi-(+)] interaction between it and Arg364 by distance = 6.95 Å.](/cms/asset/85f0f9ba-061d-4397-b843-824bfe8f5e7d/ienz_a_716835_f0007_b.gif)
Meanwhile, the O-triacetyl-galloyl-benzothiazoles test set compounds (4a~5f) demonstrated the same binding pattern like XI, but with more numbers of intermolecular hydrogen bonds (=1~6 HB) with different amino acids and DNA nucleotide structures at the binding site (including one HB of one of their triacetoxy functions with the enzyme-Arg364), more numbers of lipophilic pi-pi interactions (=6~11), and shorter [pi-(+)] interaction distances with Arg364 equal 4.09~4.88 Å, than that of the lead XI. Accordingly, the docking energies of these molecules gave higher values (=−62.34~ −67.55 Kcal/mol) than XI (, and Supplementary materials). In the contrarily, the docked free hydroxyl galloyl analogues (6a~7f) showed the same binding pattern like XI, but, with dramatically less number of intermolecular HB (=1~3) with the aminoacids and DNA nucleotide structures at the binding site (that included one HB with Arg364) and less numbers of lipophilic pi-pi interactions (=5~6), and longer [pi-(+)] interactions distances with Arg364 equal 5.48~5.58 Å, than both the lead XI and 4a~5f. These interactions with free hydroxyl analogues, gave lower docking values equal (−39.31~−45.10) than XI and 4a~5f (, and Supplementary materials).
Figure 7. Publication quality of the top docked compound 5f with topoisomerase-I enzyme with Docking value = −67.55. The binding pattern showed: [5 HB with the surrounded amino acids and DNA nucleotides including Arg364 (as crucial SAR), 7 lipophilic pi-pi interactions with the surrounded amino acids and DNA nucleotides, and a [pi-(+)] interaction between it and Arg364 by distance =4.49 Å].
![Figure 7. Publication quality of the top docked compound 5f with topoisomerase-I enzyme with Docking value = −67.55. The binding pattern showed: [5 HB with the surrounded amino acids and DNA nucleotides including Arg364 (as crucial SAR), 7 lipophilic pi-pi interactions with the surrounded amino acids and DNA nucleotides, and a [pi-(+)] interaction between it and Arg364 by distance =4.49 Å].](/cms/asset/3f3dcf3b-f7c9-40a8-95b4-47b8eae6caf9/ienz_a_716835_f0008_b.gif)
Figure 8. Publication quality of the lowest docked compound 6b with topoisomerase-I enzyme with Docking value = −45.10. The binding pattern showed: [3HB with the surrounded amino acids and DNA nucleotides including Arg364 (as a crucial SAR), 6 lipophilic pi-pi interactions with the surrounded amino acids and DNA nucleotides, and a [ pi-(+)] interaction between it and Arg364 by distance =5.69 Å].
![Figure 8. Publication quality of the lowest docked compound 6b with topoisomerase-I enzyme with Docking value = −45.10. The binding pattern showed: [3HB with the surrounded amino acids and DNA nucleotides including Arg364 (as a crucial SAR), 6 lipophilic pi-pi interactions with the surrounded amino acids and DNA nucleotides, and a [ pi-(+)] interaction between it and Arg364 by distance =5.69 Å].](/cms/asset/1ff90026-d09f-4768-bf32-9141fe92ff41/ienz_a_716835_f0009_b.gif)
Thus, the study concluded that the docking diagrams of the test set compounds (whether they were strongly or weakly docked with the enzyme), were complied with the same binding pattern like that of XI in its crystal structure with the topo-I enzyme, but with different rates (i.e. different docking scores, different numbers of HB, different numbers of pi-pi interactions and different [pi-(+)] distances with Arg364) ( and , , supplementary materials). This indicated that these molecules were predicted to exert their anticancer activity in a similar pattern like XI as topo-I inhibitors.
Biological evaluation (cell lines anticancer screening)
The designed and synthesized test set molecules in this research as topo-I inhibitors (4a~7f) were examined for their anticancer activities against human cervix carcinoma (Hela) and breast carcinoma (MCF7) cell lines, in comparison to two reference anticancer drugs; camptothecin (which is a selective topo-I inhibitor)Citation14 and doxorubicin (which is a selective topo-II inhibitor)Citation11. The comparative biological activities of most of the published works may compare the test set compounds with a reference drug that have the same biological activity and the same mode of actions or different mode of action. If the compared molecules have the same mode of action, they would be termed Me-Too drugs. But, if they have different mode of actions, they would be termed Break-Through drugs. Thus, our study, compared the cytotoxic activity of the test set compounds (4a~7f) (that were designed as topo-I inhibitors) as Me-Too drugs in comparison to Camptothecin, and as Break-Through drugs in comparison to doxorubicin.
Materials and methods
The frozen cultures of the above mentioned cell lines, were stored at (−80°C) in liquid nitrogenCitation31 and used for anticancer testing by serial sub-culturing. Different concentrations (20, 40, 60 and 80 µg/mL), of each of the test set compounds and the reference drugs (camptothecin and doxorubicin) were taken, separately. A control experiment was performed without adding any test or reference compound; i.e. 0.0 µg/mL dose. Subsequently, 100 µL calibrated culture cells of Hela and MCF7, in 800 µL Dulbecco’s Modified Eagle Medium (DMEM), were mixed with each concentration of each tested compounds and the reference drugs, separately, and the volumes were completed to 1 mL by adding DMEM medium. Cells were incubated for 72 hours and the produced suspensions were then counted by using Haemocytometer Tiefe Neubauer adopting a reported procedureCitation32. The cell cultures percentage growth inhibition (inhibition %) could be determined by calculating the mean number of the remaining survival cells after adding the tested compounds or reference drugs, at different doses in 3 × 3 groups of test sets (A) and calculating the mean number of survival cells of the control test (0.0 µg/mL), in 3 × 3 groups (B). Then, the inhibition % could be determined using the following reported equationCitation32:
Then, the IC50 values of the test compounds were calculated adopting the reported methodCitation32.
In this research, the comparison of the biological activities of the tested compounds towards each of the Hela and MCF7 cell lines, in comparison to the two reference drugs; doxorubicin and camptothecin were performed, separately, by determining their percentage-relative IC50 values (Equation 1), in comparison to each reference drug. Also, the numbers of folds (Equation 2) of higher activity than each of the reference drugs were determined for molecules with higher activity than these references.
Results of cytotoxicity tests
The results of in vitro anticancer testing for the test set compounds towards both Hela and MCF7 cell lines, which expressed in IC50 values and percentage-relative IC50 values in comparison to each of the reference drugs, for each cell cultures, were recorded in . The lower the IC50 values that inhibit the growth of the cancer cells the greater the activity of the tested compounds.
Table 3. Effects of test set compounds (4a~7f) and the reference drugs (doxorubicin and camptothecin) toward the growth of human cervix carcinoma (Hela) and human breast carcinoma (MCF7) cell lines expressed in IC50 values, % Relative IC50 and numbers of folds of more activity than the given reference drugs.
Comparative cytotoxicity in comparison to doxorubicin
The most prominent active molecules towards Hela cell line, in comparison to doxorubicin (the topo-II inhibitor), were four compounds; 5b, 5c, 5e and 5f, where they showed IC50 values equal 0.015, 0.012, 0.011 and 0.012 µM, respectively (IC50 value doxorubicin = 0.016 µM). These values indicated that they have percentage relative IC50 values more than the same reference equal 106% (=~1.1 folds), 133% (=~1,3 folds), 146% (=~1.5 folds) and 153% (=~1.5 folds), respectively.
In the other hand, the highest active hit molecules towards the MCF7 cell line, were; 4d, 4h, 5b, 5c and 5f. They showed the highest in vitro cytotoxic activity with IC50 values equal; 0.012, 0.015, 0.016, 0.012 and 0.009 µM, respectively, while doxorubicin have IC50 = 0.017 µM, that is they have % relative IC50 values equal; 142% (=~1.4 folds), 113% (=~1.1 folds), 106% (=~1.1 folds), 142% (=~1.4 folds) and 189% (=~1.9 folds), more activity than the same reference drug, respectively.
Meanwhile, the study demonstrated that compounds; 4c, 4e and 5d gave % relative IC50 activity within 56%~94% in comparison to the same reference drug, towards both the cell lines, while, the remaining five compounds; 4a, 4b, 4f, 4g and 5a exerted low % relative activity within range equal 20%~38%.
In the other hand, all the free hydroxyl-galloyl analogues (6a~7f) produced marginal cytotoxic activity within range equal 7%~39%, relative to the same reference drug, for both cell lines ().
Comparative cytotoxicity in comparison to camptothecin
The cytotoxic activities of all the tested compounds, in comparison to the selective topo-I inhibitor reference drug; Camptothecin, were found to show more % relative IC50 values (and hence more folds higher activity), towards both Hela and HCF7 cell lines, than that of doxorubicin (the topo-II inhibitor). Thus, compounds; 5b, 5c, 5e and 5f, showed percentage relative IC50, towards Hela cell lines, equal 126% (=~1.2 folds), 158% (=~1,6 folds), 172% (=~1.7 folds) and 158% (=~1.8 folds) higher than Camptothecin, respectively. While, compounds; 4d, 4e, 4h, 5b, 5c, 5e and 5f, showed relative % IC50 values, towards HCF7 cultures equal 167% (=~1.7 folds), 111% (=~1.1 folds), 133% (=~1.3 folds), 125% (=~1.2 folds), 167% (=~1.7 folds), 105% (=~1.1 folds) and 222% (=~2.2 folds), higher than Camptothecin, respectively.
In addition, the same comparative study towards Camptothecin, against the used cell cultures, demonstrated that the 4c, 4f, 4g and 5d compounds gave % relative IC50 activity within 40%~95%, while, the 4a, 4b, 5a and 5d) exerted low % relative activity within range equal 24%~33%. Meanwhile, the free hydroxyl-galloyl analogues (6a~7f) produced marginal cytotoxic activity within range equal 8%~46%, relative to the same reference drug, for both cell lines ().
Correlation of the in vitro cytotoxicity and the molecular modelling
Correlation of the above in vitro cytotoxic activity and the molecular modelling (compare fit and docking scores) for most of the synthesized molecules (4a~7f) were more or less matched and coincident with each other, while other molecules were not coincident. Examples of the matched molecules were: i) Compound 5f which showed the highest fitting scores (=2.78/3) and docking energy (−67.55 kcal/mol) with 5 HB (one of them with Arg364), 7 lipophilic pi-pi interactions and shorter [pi-(+)] distances = 4.48 Å with Arg364, was found to show 1.5 folds higher cytotoxic activity toward Hela cell line and 1.9 folds higher activity toward MCF7 cell culture more than the reference drug; doxorubicin. The same molecule (5f), showed 1.6 folds higher activity towards Hela and 2.2 folds higher activity towards HCF7 cultures than the topo-I inhibitor reference drug; Camptothecin. ii) The other active hits; 4d, 4e, 4h, 5b, 5c and 5e (that exhibited high docking and fitting scores but less than 5f), have comparable cytotoxic activity higher than both the reference drugs, but lower than 5f. iii) Compounds 4c, 4e, 4f, 5a and 5d, that have high fit and dock scores, but more less than 5f, showed % relative activities lower than both of the reference drugs within range = 40%~95% towards both cell cultures. iv) The free hydroxyl galloyl molecules (6a~7f), (that have low fitting and docking scores) than XI, produced marginal cytotoxic activity within range = 8%~46%, in comparison to both the two reference drugs, in both cell lines ().
Examples of molecules which showed non-correlated virtual scores with the cytotoxic activity were molecules; 4a, 4b, 4g, 5a and 5d. They exhibited high fit/dock scores, but, they exerted comparably low % relative IC50 values with range = 24%~33% towards both cell lines than the two reference drugs (). This deviation in the predictive power of the virtual scores and the in vitro experimental results in such integrated molecular modelling tools may be explained in terms of what had been reported that “despite the considerable success in molecular modelling scoring functions, yet, accurate and rapid prediction of protein–ligand interactions is still a challenge”Citation33.
Structure activity relationship of the designed molecules as topoisomerase-i inhibitors
The SAR of the molecules that showed high in vitro cytotoxic activity; 4d, 4e, 4h, 5b, 5c, 5e and 5f, towards Hela and MCF7 cultures, in comparison to the two mentioned reference drugs indicated that they have additional lipophilic methoxy or ethyl groups at the C5 or C7 positions of the benzo[d]thiazole ring system. This, besides the existence of other lipophilic groups like 6-chloro or 6-methoxy groups linked to the benzothiazole cycle as well as the triacetoxybenzoylamino functions. This could increase their lipophilic interactions with the binding site and hence increase their cytotoxic activity. Deacetylation of these molecules into (6a~7f) showed nearly 10 folds lower activity than the two reference drugs ().
The higher cytotoxic activity of the triacetyl-galloyl derivatives and the lower cytotoxic activity of the free-hydroxyl galloyl derivatives (4a~5f) was supported by the recently reported similar work in the literatureCitation28, where it was found that 3-O-alky and 3-O-acyl-(-)-epigallocatechin (XII) exhibited better anticancer activity than free hydroxy-gallate derivative (VIII). This could prove that acetylation of (-)-epigallocatechin into the 3-O-acyl-(-)-epigallocatechin (XII) could enhance its cytotoxic activity than the corresponding free hydroxyl analogues. Such reported results supported our approach of the higher activity of the triacetylgalloyl benzothiazole derivatives (4a~5f) than the deacetyl analogues (6a~7f).
The high activity of (4a~5f) than (6a~7f) may be explained in terms of the existence of: i) More numbers of HB with the amino-acids and DNA nucleotide structures at the binding cavity including Arg364, ii) More numbers of pi-pi interactions and iii) Shorter [pi-(+)] distances with Arg364 at the binding cavity ().
Experimental
Experiments of molecular modelling
During the compare/fit study, the training set topo-I leads (I-XI) were used to generate the ideal common feature hypothesis, followed by their fitting of the ideal one with the test set compounds (4a~7f), using Accelrys [Discovery Studio (DS) 2.55) modules, at Faculty of Pharmacy, Ain Shams University, Cairo, Egypt. The fitting scores of this study were recorded using best fit algorithm with all conformers of each of the test compounds, and the values of the highest fit conformers and their numbers for each molecule were recorded in .
The docking study was performed by running C-Docker protocol, between the binding cavity of the topo-I enzyme with the test set molecules (4a~7f) without deleting the complexed DNA structure, since it is assumed that the DNA shares in the bindings with the inhibitors. The lead XI was also docked with the same binding site in a parallel process as a reference molecule. The docking scores and the numbers of intermolecular hydrogen bonds of the test compounds with the amino acids and DNA nucleotides at the binding cavity including Arg364 were recorded. Also, the numbers of intermolecular pi-pi interactions and the distances of [pi-(+)] between the docked molecules and the Arg364 were measured by monitoring the 3D structures of the docking diagrams of the entire tested molecules.
Chemistry
All used reagents were supplied by Aldrich. The 3,4-disubstituted-anilines (1a-h) were commercially available from Chemexper, Chem-industry, ChimBlink, Sigma and Aldrich pharmaceutical companies. Also, the DMEM medium was commercially available from Gibco® Company. TLC was performed on silica gel 60 F254 plates (Merck). Preparative Column Chromatography was performed using Silica Gel 60 for separation of reaction mixture of the respective components 2b-d, f-h and 3a-f. Melting points were uncorrected and were determined on Stuart Scientific apparatus (capillary method). The FT-IR spectra were recorded on a Perkin-Elmer spectrum BX FT-IR spectrometer using KBr discs. Electron impact Mass spectrometry (EI-MS) was performed in The Microanalytical Centre, Cairo University, Egypt. The intermediates 2-amino-6-chloro-benzothiazole(2a)Citation28, 2-amino-6-methoxy-benzothi-azole(2e)Citation29 and 4,5-triacetoxybenzoyl chlorideCitation30 were prepared according to corresponding reported methods.
2-Amino-5,6-disubstitutedbenzo[d]thiazoles(2b-d and 2f-h) and 2-Amino-6,7-disubstituted-benzo[d]thiazoles(3a-f): General procedure
To an ice cooled and stirred mixture of 3,4–disubstituted-anilines [viz; 4-chloro-3-methyl-aniline(1b), 4-chloro-3-ethyl-aniline (1c), 4-chloro-3-methoxy-aniline(1d), 4-methoxy-3-methylaniline(1f), 3-ethyl-4-methoxy-aniline(1g), 3,4-dimethoxyaniline(1h)], (10 mmol) and ammonium thiocyanate (NH4SCN) (12 mmol), in acetic acid (20 mL) a solution of Br2 (12 mmol) in acetic acid (5 mL) was added dropwise within 60 minutes. Stirring was continued for another 60 minutes at 0°C. The reaction mixture was then diluted with H2O (100 mL) and the formed yellowish white precipitate was collected, air dried. The two components of the reaction mixtures (appeared in the TLC) were separated by their dissolving in acetone (0.5 mL), passing through a preparative silica gel column chromatography and elution with n-hexane:THF:methanol (9:1:0.2) mixture, where the 6,7-disubstituted derivatives (3a-f) were eluted at first followed by elution of the second components; 5,6-disubstituted derivatives (2b-d and 2f-h). Thus, the reaction products, upon using 4-chloro-3-methylaniline as a starting material in the above reaction, gave mixture of 2b and 3a. The TLC using n-hexane:THF:methanol (9:1:0.2) eluents, showed that 3a have RF values = 0.76, and 2b have RF values = 0.62. Accordingly, a preparative silica gel column was used to separate 2b and 3a using the same eluent mixture. The solvents of the eluted solution of 2b and 3a were evaporated under vacuum and the obtained crude solid products were crystallized from methylene chloride.
Physical and spectral data for compound 2b; It is separated as yellowish white crystals (30% yield), mp 201–2°C; IR 3210, 3190 cm−1(NH2); EIMS[m/z(%)]: M+ = 198(12.09), M++2 = 200(3.98); 1H-NMR(δ): 2.34 (s., CH3), 6.54(br., NH2), 7.8(s., 1H Ar-H), 8.02(s., 1H Ar-H); 13C-NMR(δ): 19.9, 121.6, 121.9, 129.5, 131.2, 134.0, 152.7, 166.1. Elemental analysis: Found: C, 47.99; H, 3.89; N, 13.82 for C8H7ClN2S (198.5); requires C, 48.36; H, 3.52; N, 14.10.
Physical and spectral data for compound 3a; It was separated as faint yellowish white crystals (18% yield), mp 187–8°C; IR: 3220, 3209 cm−1 (NH2); EIMS[m/z(%)]: M+ = 198(13.7), M++2 = 200(4.11); 1H-NMR(δ): 2.36 (s., CH3), 6.88(br., NH2), 7.54(s., 1H Ar-H), 7.59(s., 1H Ar-H); 13C-NMR(δ): 12.9, 120.0, 125.0, 126.3, 132.2, 134.0, 147.7, 166.1. Elemental analysis: found: C, 48.55; H, 3.41; N, 14.32 for C8H7ClN2S (198.5); requires C, 48.36; H, 3.52; N, 14.10.
Separation of reaction products 2c and 3b were performed as in case of 2b and 3a, where RF values for 3b = 0.77, and RF values of 2c = 0.64.
Physical and spectral data for compound 2c: It was separated as yellowish white crystals (28% yield), mp 198–9°C; IR 3215, 3191 cm−1(NH2); EIMS[m/z(%)]: M+ = 212(5.87), M++2 = 214(2.01); 1H-NMR(δ): 1.27 (t., CH3), 2.58(q., CH2), 6.95(br., NH2), 7.82(s., 1H Ar-H), 8.02(s., 1H Ar-H); 13C-NMR(δ): 14.01, 120.10, 121.03, 129.5, 130.2, 137.2, 151.27, 166.6. Elemental analysis: Found: C, 50.60; H, 4.55; N, 13.52 for C9H9ClN2S (212.5); requires C, 50.82; H, 4.23; N, 13.17.
Physical and spectral data for compound 3b: It was separated as faint yellowish white crystals (15% yield), mp 194–5°C; IR 3225, 3210 cm−1(NH2); EIMS[m/z(%)]: M+ = 212(8.8), M++2 = 214(2.98); 1H-NMR(δ): 1.23 (t., CH3), 2.63(q., CH2), 6.91(br., NH2), 7.50(s., 1H Ar-H), 7.51(s., 1H Ar-H); 13C-NMR(δ): 13.6, 19.54, 120.2, 123.6, 126.02, 130.2, 134.2, 148.20, 166.3. Elemental analysis: Found: C, 51.12; H, 4.47; N, 13.17 for C9H9ClN2S (212.5); requires C, 50.82; H, 4.23; N, 13.17.
Separation of reaction products 2d and 3c were performed as in case of 2b and 3a, where RF values for 3c = 0.81, and RF values of 2d = 0.69.
Physical and spectral data for compound 2d: It was separated as yellowish white crystals (28% yield), mp 198–9°C; IR 3219, 3201 cm−1(NH2); EIMS[m/z(%)]: M+ = 212(5.87), M++2 = 214(2.01); 1H-NMR(δ): 3.83 (s., CH3), 6.91(br., NH2), 7.93(s., 1H Ar-H), 8.06(s., 1H Ar-H); 13C-NMR(δ): 55.3, 106.6, 116.1, 122.20, 124.6, 148.1, 159.27, 166.6. Elemental analysis: Found: C, 44.52; H, 3.43; N, 12.79 for C8H7ClN2OS (214.5); requires C, 44.75; H, 3.23; N, 13.05.
Physical and spectral data for compound 3c: It was separated as faint yellowish white crystals (14% yield), mp 177–8°C; IR 3220, 3212 cm−1(NH2); EIMS[m/z(%)]: M+ = 214(18.32), M++2 = 216(5.78); 1H-NMR(δ): 3.83 (s., OCH3), 6.99(br., NH2), 7.25(s., 1H Ar-H), 7.45(s., 1H Ar-H); 13C-NMR(δ): 54.9, 109.6, 115.3, 116.6, 126.8, 148.10, 156.6, 166.3. Elemental analysis: Found: C, 45.01; H, 3.23; N, 13.05 for C8H7ClN2OS (214.5); requires C, 44.75; H, 3.23; N, 13.03.
Separation of reaction products 2f and 3d were performed as in case of 2b and 3a, where RF values for 3b = 0.82, and RF values of 2c = 0.70.
Physical and spectral data for compound 2f: It was separated as yellowish white crystals (34% yield), mp 138.9°C; IR 3210, 3201 cm−1(NH2); EIMS[m/z(%)]: M+ = 194(6.21), 1H-NMR(δ): 2.13 (s., CH3), 3.82(s., OCH3), 6.99(br., NH2), 7.42(s., 1H Ar-H), 7.78(s., 1H Ar-H); 13C-NMR(δ): 15.04, 56.1,104.8, 119.9, 121.3, 146.9, 128.8, 160.0, 166.6. Elemental analysis: Found: C, 55.34; H, 5.41; N, 14.12 for C9H9ClN2S (212.5); requires C, 55.67; H, 5.15; N, 14.43.
Physical and spectral data for compound 3d: It was separated as faint yellowish white crystals (16% yield), mp 161–2°C; IR 3232, 3219 cm−1(NH2); EIMS[m/z(%)]: M+ = 194(4.98), 1H-NMR(δ): 2.34(s., CH3), 3.83(s., OCH3), 6.99(br., NH2), 6.88(s., 1H Ar-H), 7.58(s., 1H Ar-H); 13C-NMR(δ): 11.8, 56.54, 113.2, 117.99, 119.6, 120.2, 124.3, 160.0, 166.3. Elemental analysis: Found: C, 56.01; H, 4.98; N, 14.71 for C9H10N2OS (194); requires C, 55.67; H, 5.15; N, 14.43.
Separation of reaction products 2g and 3e were performed as in case of 2b and 3a, where RF values for 3b = 0.79, and RF values of 2c = 0.66.
Physical and spectral data for compound 2g: It was separated as yellowish white crystals (29% yield), mp 138.9°C; IR 3218, 3211 cm−1(NH2); EIMS[m/z(%)]: M+ = 208 (5.10), 1H-NMR(δ): 1.27 (t., CH3), 2.68(q., CH2), 6.99(br., NH2), 7.47(s., 1H Ar-H), 7.84(s., 1H Ar-H); 13C-NMR(δ): 14.81, 22.3, 104.0, 120.0, 122.03, 129.8, 129.1, 147.2, 158.7, 166.6. Elemental analysis: Found: C, 57.63; H, 6.01; N, 13.32 for C10H12N2OS (208); requires C, 57.69; H, 5.76; N, 13.46.
Physical and spectral data for compound 3e: It was separated as faint yellowish white crystals (16% yield), mp 168–9°C; IR 3237, 3205 cm−1(NH2); EIMS[m/z(%)]: M+ = 208(11.91%); 1H-NMR(δ): 1.25 (t., CH3), 2.63(q., CH2), 3,83(s., OCH3), 6.91(br., NH2), 6.95(s., 1H Ar-H), 7.35(s., 1H Ar-H); 13C-NMR(δ): 14.6, 18.54, 56.1, 113.2, 119.8, 120.0, 123.0, 141.20, 158.7, 166.3. Elemental analysis: Found: C, 57.43; H, 5.42; N, 13.26 for C10H12N2OS (208); requires C, 57.69; H, 5.76; N, 13.46.
Separation of reaction products 2h and 3f were performed as in case of 2b and 3a, where RF values for 3b = 0.86, and RF values of 2c = 0.71.
Physical and spectral data for compound 2h: It was separated as faint yellowish white crystals (26% yield), mp 122–3°C; IR 3222, 3211 cm−1(NH2); EIMS[m/z(%)]: M+ = 210(6.87%); 1H-NMR(δ): 3.83 (s., 2OCH3), 6.91(br., NH2), 7.41(s., 1H Ar-H), 7.88(s., 1H Ar-H); 13C-NMR(δ): 56.10, 56.12, 105.9, 106.2, 124.2, 142.2, 144.20, 144.8, 166.3. Elemental analysis: Found: C, 51.11; H, 4.43; N, 13.10 for C9H10N2O2S (210); requires C, 51.42; H, 4.76; N, 13.33.
Physical and spectral data for compound 3f: It was separated as faint yellowish white crystals (16% yield), mp 144–5°C; IR 3231, 3215 cm−1(NH2); EIMS[m/z(%)]: M+ = 210(16.65%); 1H-NMR(δ): 1.23 (t., CH3), 2.63(q., CH2), 6.91(br., NH2), 7.50(s., 1H Ar-H), 7.51(s., 1H Ar-H); 13C-NMR(δ): 13.6, 19.54, 120.2, 123.6, 126.02, 130.2, 134.2, 148.20, 166.3. Elemental analysis: Found: C, 51.11; H, 4.43; N, 13.10 for C9H10N2O2S (210); requires C, 51.42; H, 4.76; N, 13.33.
2-[(3,4,5–(O-Triacetoxybenzoylamino)]-5,6-disubitituted-benzo[d]thiazoles (4a-h): General procedure
To a stirred solution of 2-amino-5,6-disubstituted-benzothiazoles (2a-h) (10 mmol) and DMAP (15 mmol), in THF (5 mL), was added 3,4,5-triacetoxybenzoyl chlorideCitation29 (15 mmol). The mixture was stirred for 6 hours at room temperature and the reaction was quenched by the addition of saturated NaCl (20 mL) and EtOAc (20 mL). The aqueous layer was extracted with EtOAc (2 × 20 mL). The combined EtOAc extracts were washed subsequently with 1 N HCl (10 mL), the (2 × 10 mL), NaHCO3 (10 mL), H2O (2 × 10 mL) and saturated NaCl (10 mL) and dried over MgSO4. After evaporation of the organic solvent using vacuum pump an oily crude product was formed which was purified by flash column chromatography using CH2Cl2: Acetone (10:1), as a mobile phase. The obtained oily products were then recrystallized from CH2Cl2: n-hexane to give 4a-h as faint yellowish white crystals of 4a-h.
Physical and spectral data for compound 4a; It was separated as pale yellowish white crystals (64% yield), mp 214–5°C; IR 3210 cm−1(NH); 1738, 1731, 1726 cm−1(3 COOCH3), 1661(CONH); EIMS[m/z(%)]: M+ = 462(24.2%), 464(M+2)(8.02%); 1H-NMR(δ): 2.82(s., 3CH3CO), 7.66(s., 2Ar-H), 7.56(d., 1H, Ar-H), 7.67(d., 1H Ar-H), 8.13(s., 1H, Ar-H), 9.11(br., NH); 13C-NMR(δ): 20.3, 118.3, 121.2, 124.3, 125.8, 127.4, 129.8, 132.3, 139.6, 146.2, 151.3, 155,7, 169.0, 174.5. Elemental analysis: Found: C, 51.65; H, 3.55; N, 6.40 for C20H15ClN2O7S (462.5); requires C, 51.89; H, 3.24; N, 6.05
Physical and spectral data for compound 4b; It was separated as pale yellowish white crystals (64% yield), mp: 208–9°C; IR 3214 cm−1(NH); 1742, 1736, 1722 cm−1 (3 COOCH3), 1669(CONH); EIMS[m/z(%)]: M+ = 476(16.11%), 478(M+2)(6.11%); 1H-NMR(δ): 2.28(s., 3CH3CO), 2.34(s., CH3-Ar), 7.66(s., 2Ar-H), 7.83(d., 1H, Ar-H), 8.01(d., 1H Ar-H), 9.15(br., NH); 13C-NMR(δ): 19.9, 20.3, 121.1, 121.7, 124.3, 127.4, 129.3, 131.8, 134.0, 139.6, 146.2, 152.7,165.7, 169.0, 174.5. Elemental analysis: Found: C, 53.00; H, 3.67; N, 6.00 for C21H17ClN2O7S (476.5); requires C, 52.89; H, 3.59; N, 5.87.
Physical and spectral data for compound 4c; It was separated as pale white crystals (81% yield), mp: 211–2°C; IR 3219 cm−1(NH); 1738, 1728, 1716 cm−1 (3 COOCH3), 1679(CONH); EIMS[m/z(%)]: M+ = 490(4.01%), 492(M+2)(1.41%); 1H-NMR(δ): 2.28(s., 3CH3CO), 1.25(t., CH3), 2.6(q., -CH2), 7.66(s., 2Ar-H), 7.89(s., 1H, Ar-H), 8.08(s., 1H Ar-H), 9.15(br., NH); 13C-NMR(δ): 19.9, 20.3, 121.1, 121.7, 124.3, 127.4, 129.3, 131.8, 134.0, 139.6, 146.2, 152.7, 165.7, 169.0, 174.5. Elemental analysis: Found: C, 53.83; H, 3.90; N, 5.71 for C22H19ClN2O7S (490.5); requires C, 53.83; H, 3.90; N, 5.71.
Physical and spectral data for compound 4d; It was separated as pale brownish white crystals (64% yield), mp: 184–5°C; IR 3210 cm−1(NH); 1734, 1726, 1719 cm−1 (3 COOCH3), 1651(CONH); EIMS[m/z(%)]: M+ = 492(6.8%), 494(M+2)(2.54%); 1H-NMR(δ): 2.27(s., 3CH3CO), 3.83(s., OCH3), 7.66(s., 2Ar-H), 7.83(s., 1H, Ar-H), 8.01(s., 1H Ar-H), 9.15(br., NH); 13C-NMR(δ): 20.3, 55.3, 106.6, 116.7, 122.2, 124.3, 124.6, 127.4, 139.6, 146.2, 148.1, 155.7, 159.0, 169.0, 174.5. Elemental analysis: Found: C, 51.44; H, 3.22; N, 5.45 for C21H17ClN2O8S (492.5); requires C, 51.17; H, 3.84; N, 5.68.
Physical and spectral data for compound 4e; It was separated as pale white crystals (53% yield), mp: 172–3°C; IR 3216 cm−1(NH); 1747, 1739, 1723 cm−1 (3 COOCH3), 1666(CONH); EIMS[m/z(%)]: M+ = 458(11.1%), 1H-NMR(δ): 2.28(s., 3CH3CO), 3.83(s., OCH3), 7.00(d., 1H Ar-H), 7.52(s., 1H, Ar-H), 7.53(d., 1H Ar-H), 7.66(s., 2Ar-H), 9.15(br., NH); 13C-NMR(δ): 20.3, 55.5, 104.9, 114.6, 118.2, 127.4, 124.3, 131.9, 139.6, 145.5, 146.7, 156.7, 165.7, 169.9, 174.5. Elemental analysis: Found: C, 54.89; 17H, 4.11; N, 6.42 for C21H18N2O8S (458); requires C, 55.02; H, 3.96; N, 6.11.
Physical and spectral data for compound 4f; It was separated as pale white crystals (44% yield), mp: 161–2°C; IR 3219 cm−1(NH); 1742, 1726, 1719 cm−1 (3 COOCH3), 1680(CONH); EIMS[m/z(%)]: M+ = 472(3.11%), 1H-NMR(δ): 2.15(s., CH3), 2.28(s., 3CH3CO), 3.83(s., OCH3), 7.4(s., 1H, Ar-H), 7.66(s., 2Ar-H), 7.78(s., 1H Ar-H), 9.15(br., NH); 13C-NMR(δ): 15.4, 20.3, 56.1, 104.8, 119.9, 121.3, 124.3, 128.9, 127.4, 139.6, 146.2, 146.9, 160.0, 165.7, 169.9, 174.5. Elemental analysis: Found: C, 56.26; H, 3.89; N, 5.65 for C22H20N2O8S (472); requires C, 56.93; H, 4.27; N, 5.93.
Physical and spectral data for compound 4g; It was separated as pale yellowish white crystals (53% yield), mp: 166–7°C; IR 3216 cm−1(NH); 1736, 1721, 1718 cm−1 (3 COOCH3), 1651(CONH); EIMS[m/z(%)]: M+ = 486(2.44%), 1H-NMR(δ): 1.25(t., CH3), 2.28(s., 3CH3CO), 2.8(q., CH2), 3.81(s., OCH3), 7.47(s., 1H, Ar-H), 7.66(s., 2Ar-H), 7.84(s., 1H Ar-H), 9.15(br., NH); 13C-NMR(δ): 14.8, 20.3, 22.3, 122.4, 56.1, 104.8, 120.4, 124.3, 127.4, 129.1, 139.6, 146.2, 146.9, 155.4, 158.7, 169.0, 174.5. Elemental analysis: Found: C, 57.22; H, 4.68; N, 6.00 for C23H22N2O8S (486); requires C, 56.79; H, 4.52; N, 5.76.
Physical and spectral data for compound 4h; It was separated as pale yellowish white crystals (39% yield), mp: 145–6°C; IR 3219 cm−1(NH); 1741, 1743, 1723 cm−1 (3 COOCH3), 1661(CONH); EIMS[m/z(%)]: M+ = 488(7.98%), 1H-NMR(δ): 2.28(s., 3CH3CO), 3.83(s., 2OCH3), 7.41(s., 1H, Ar-H), 7.66(s., 2Ar-H), 7.88(s., 1H Ar-H), 9.13(br., NH); 13C-NMR(δ): 20.3, 56.1, 105.9, 106.2, 124.2, 124.3, 127.4, 139.6, 142.3, 144.9, 146.2, 155.7, 169.0, 174.5. Elemental analysis: Found: C, 53.87; H, 3.88; N, 5.54 for C22H20N2O9S (488); requires C, 54.09; H, 4.13; N, 5.73.
2-[(3,4,5–Triacetoxybenzoyl]amino-6,7-disubitituted-benzo[d]thiazoles (5a-f): General procedure
To a stirred solution of crude 2-amino-6,7-substituted-benzothiazoles (3a-f) (10 mmol) and DMAP (15 mmol), in THF (5 mL), was added 3,4,5-triacetoxybenzoyl chlorideCitation30 (15 mmol) The mixture was stirred for 6 hours at room temperature and the products 5a-f of the reaction was isolated by similar procedure like that of 4a-h. The isolated crude product was purified by flash column chromatography using CH2Cl2: Acetone (10:2), as a mobile phase. The obtained semisolid products were then recrystallized from CH2Cl2: n-hexane to give 5a-f as faint yellowish white crystals.
Physical and spectral data for compound 5a; It was separated as pale yellowish white crystals (74% yield), mp: 192–3°C; IR 3222 cm−1(NH); 1734, 1721, 1712 cm−1 (3 COOCH3), 1673(CONH); EIMS[m/z(%)]: M+ = 476(22.9%), 478(M+2)(7.60%); 1H-NMR(δ): 2.28(s., 3CH3CO), 2.34(s., CH3-Ar), 7.44(d., 1H, Ar-H), 7.50(d., 1H Ar-H), 7.66(s., 2Ar-H), 9.15(br., NH); 13C-NMR(δ):12.6, 20.3, 120.0, 124.3, 124.7, 125.7, 127.4, 131.8, 132.3, 139.6, 146.2, 147.0, 155.7, 169.0, 174.5. Elemental analysis: Found: C, 52.56; H, 3.33; N, 6.02 for C21H17ClN2O7S (476.5); requires C, 52.89; H, 3.59; N, 5.87.
Physical and spectral data for compound 5b; It was separated as pale white crystals (46% yield), mp: 197.8°C; IR 3219 cm−1(NH); 1738, 1728, 1716 cm−1 (3 COOCH3), 1679(CONH); EIMS[m/z(%)]: M+ = 490(16.80%), 492(M++2)(5.21%); 1H-NMR(δ): 1.23(t., CH3), 2.28(s., 3CH3CO), 2.6(q., CH2), 7.66(s., 2Ar-H), 7.51(d., 1H, Ar-H), 7.52(d., 1H Ar-H), 9.15(br., NH); 13C-NMR(δ): 13.6, 19.9, 20.3, 120.7, 123.4, 124.3, 125.7, 127.3, 130.7, 134.8, 139.6, 146.2, 147.0, 155.7, 169.0, 174.5. Elemental analysis: Found: C, 53.55; H, 3.66; N, 6.00 for C22H19ClN2O7S (490.5); requires C, 53.83; H, 3.83; N, 5.71.
Physical and spectral data for compound 5c; It was separated as pale brownish white crystals (55% yield), mp: 192–3°C; IR 3210 cm−1(NH); 1734, 1726, 1719 cm−1 (3 COOCH3), 1651(CONH); EIMS[m/z(%)]: M+ = 492(11.3%), 494(M+2)(3.76%); 1H-NMR(δ): 2.28(s., 3CH3CO), 3.83(s., OCH3), 7.66(s., 2Ar-H), 7.25(d., 1H, Ar-H), 7.45(d., 1H Ar-H), 9.15(br., NH); 13C-NMR(δ): 20.3, 54.9, 109.6, 115.3, 116.7, 124.3, 126.8, 127.4, 139.6, 146.2, 148.1, 155.7, 156.6, 169.0, 174.5. Elemental analysis: Found: C, 51.45; H, 3.65; N, 6.11 for C21H17ClN2O8S (492.5); requires C, 51.17; H, 3.84; N, 5.87.
Physical and spectral data for compound 5d; It was separated as pale white crystals (30% yield), mp: 173–4°C; IR 3216 cm−1(NH); 1747, 1739, 1721 cm−1 (3 COOCH3), 1666(CONH); EIMS[m/z(%)]: M+ = 472(6.21%), 1H-NMR(δ):2.28(s., 3CH3CO), 2.34(s., CH3), 3.83(s., OCH3), 6.88(d., 1H, Ar-H), 7.34(d., 1H Ar-H), 7.66(s., 2Ar-H), 9.15(br., NH); 13C-NMR(δ): 11.8, 20.3, 56.1, 113.2, 117.5, 119.6, 124.30, 124.32, 127.4, 139.6, 141.2, 146.2, 160.0, 165.7, 169.0, 174.5. Elemental analysis: Found: C, 56.21; H, 4.56; N, 5.70 for C22H20N2O8S (472); requires C, 56.93; H, 4.27; N, 5.93.
Physical and spectral data for compound 5e; It was separated as pale yellowish white crystals (53% yield), mp: 169–70°C; IR 3219 cm−1(NH); 1742, 1726, 1720 cm−1 (3 COOCH3), 1680(CONH); EIMS[m/z(%)]: M+ = 486(4.12%), 1H-NMR(δ): 1.25(t., CH3), 2.28(s., 3CH3CO), 2.6(q., CH2), 3.83(s., OCH3), 6.95(d., 1H, Ar-H), 7.35(d., 1H Ar-H), 7.66(s., 2Ar-H), 9.15(br., NH); 13C-NMR(δ): 14.4, 18.7, 20.3, 56.1, 113.2, 119.8, 120.0, 123.0, 124.3, 127.7, 139.6, 141.2, 146.2, 158.7, 169.0, 174.5. Elemental analysis: Found: C, 57.01; H, 4.56; N, 5.76 for C23H22N2O8S (486); requires C, 56.79; H, 4.52; N, 5.76.
Physical and spectral data for compound 5f; It was separated as pale yellowish white crystals (48% yield), mp: 138–9°C; IR 3221 cm−1(NH); 1736, 1730, 1718 cm−1 (3 COOCH3), 1651(CONH); EIMS[m/z(%)]: M+ = 488(3.91%), 1H-NMR(δ): 2.28(s., 3CH3CO), 3.83(s., 2OCH3), 6.89(d., 1H, Ar-H), 7.09(d., 1H Ar-H), 7.66(s., 2Ar-H), 9.14(br., NH); 13C-NMR(δ): 20.3, 56.1, 60.9, 109.2, 114.3, 114.9, 124.3, 127.4, 139.6, 141.8, 142.3, 144.9, 146.2, 165.7, 169.0, 174.5. Elemental analysis: Found: C, 53.75; H, 3.90; N, 6.11 for C22H20N2O9S (488); requires C, 54.09; H, 4.13; N, 5.73.
2-(Galloylamino)-5,6-disubstitated-benzo[d]thiazoles (6a-h): General procedure
To a stirred solution of 4a-h (10 mmol) in acetonitrile (20 mL) was added hydrazine hydrate (15 mmol). The mixture was stirred for 90 minutes at room temperature, then 10 % aqueous KHSO4 was added until pH = 3. The total mixture was extracted with EtOAc (3 × 20 mL) and the combined EtOAc extracts were washed with saturated NaCl solution (10 mL) and H2O (2 × 10 mL) and then dried over MgSO4. Complete evaporation of the EtOAc was carried out using Vacuum pump to give a crude product which was recrystallized from pet. Ether (40–60) to give the titled compounds (6a-h) as faint yellowish white crystals in 60–65 % yield.
Physical and spectral data for compound 6a; It was separated as pale white crystals (64% yield), mp 142.3°C; IR: 3490, 3460, 3440 cm−1(3 OH); 3210 cm−1(NH), 1661 cm−1 (CON); EIMS[m/z(%)]: M+ = 336(12.11%), 338(M+2)(4.01%); 1H-NMR(δ): 5.3(br., 3OH), 6.93(s., 2Ar-H), 7.56(d., 1H, Ar-H), 7.69(d., 1H Ar-H), 8.13(s., 1H, Ar-H), 9.14(br., NH); 13C-NMR(δ): 108.9, 118.3, 121.2, 125.2, 129.8, 125.8, 132.3, 139.4, 146.3, 151.3, 165.0, 174.5. Elemental analysis: Found: C, 50.02; H, 3.00; N, 8.55 for C20H15ClN2O7S (336.5); requires C, 49.90; H, 2.67; N, 8.32.
Physical and spectral data for compound 6b; It was separated as pale white crystals (64% yield), mp 154–5°C; IR 3492, 3463, 3442 cm−1(3 OH); 3220 cm−1 (NH), 1675 cm−1 (CON); EIMS[m/z(%)]: M+ = 350(14.84%), 352(M+2) (4.94%); 1H-NMR(δ): 2.34(s., CH3), 5.3(br., 3OH), 6.93(s., 2Ar-H), 7.83(s., 1H Ar-H), 8.01(s., 1H, Ar-H), 9.15(br., NH); 13C-NMR(δ): 19.9, 108.9, 121.1, 121.7, 125.2, 129.3, 131.8, 134.7, 139.4, 146.3, 152.7, 165.7, 174.5. Elemental analysis: Found: C, 51.00; H, 3.29; N, 8.25 for C15H11ClN2O4S (350.5); requires C, 51.35; H, 3.13, N, 7.98.
Physical and spectral data for compound 6c; It was separated as pale white crystals (56% yield), mp 161–2°C; IR: 3496, 3473, 3454 cm−1(3 OH); 3232 cm−1 (NH), 1671 cm−1 (CON); EIMS[m/z(%)]: M+ = 364(21.81%), 366(M+2) (6.98%); 1H-NMR(δ): 1.25(t., CH3), 2.60(q., CH2), 5.35(br., 3OH), 6.93(s., 2Ar-H), 7.89(s., 1H Ar-H), 8.08(s., 1H, Ar-H), 9.15(br., NH); 13C-NMR(δ): 14.0, 23.1, 108.9, 120.4, 121.1, 125.2, 129.5, 130.5, 137.2, 139.4, 146.3, 152.7, 165.7, 174.5. Elemental analysis: Found: C, 53.00; H, 3.75; N, 8.00 for C16H13ClN2O4S (364.5); requires C, 52.67; H, 3.56, N, 7.68.
Physical and spectral data for compound 6d; It was separated as pale white crystals (64% yield), mp 141–2°C; IR: 3512, 3488, 3432 cm−1(3 OH); 3245 cm−1 (NH), 1679 cm−1 (CON); EIMS[m/z(%)]: M+ = 366(19.56%), 368(M+2) (6.71%); 1H-NMR(δ): 3.83(s., CH3), 5.35(br., 3OH), 6.93(s., 2Ar-H), 7.93(s., 1H Ar-H), 8.02(s., 1H, Ar-H), 9.15(br., NH); 13C-NMR(δ): 55.3, 106.6, 108.9, 116.7, 122.2, 124.6, 125.2, 139.4. 146.3, 148.1, 159.0, 165.7, 174.5. Elemental analysis: Found: C, 49.40; H, 3.25; N, 7.80 for C15H11ClN2O5S (366.5); requires C, 49.11; H, 3.00, N, 7.64.
Physical and spectral data for compound 6e; It was separated as pale white crystals (48% yield), mp 130–1°C; IR: 3497, 3473, 3449 cm−1(3 OH); 3210 cm−1 (NH), 1667 cm−1 (CON); EIMS[m/z(%)]: M+ = 332(21.11%), 1H-NMR(δ): 3.83(s., OCH3), 5.35(br., 3OH), 6.93(s., 2Ar-H), 7.00(d., 1H, Ar-H), 7.52(s., 1H Ar-H), 7.53(d., 1H, Ar-H), 9.15(br., NH); 13C-NMR(δ): 55.3, 104.9, 108.9, 114.6, 118.2, 125.5, 131.9, 139.4, 145.5, 146.3, 156.7, 165.7, 174.5. Elemental analysis: Found: C, 54.55; H, 3.64; N, 8.43 for C15H12N2O5S (332); requires C, 54.21; H, 3.00, N, 7.64.
Physical and spectral data for compound 6f; It was separated as pale white crystals (54% yield), mp 126–7°C; IR: 3492, 3453, 3440 cm−1(3 OH); 3220 cm−1 (NH), 1675 cm−1 (CON); EIMS[m/z(%)]: M+ = 346(16.80%), 1H-NMR(δ): 2.15(s., CH3), 3.82(s., OCH3), 5.37(br., 3OH), 6.91(s., 2Ar-H), 7.40(s., 1H, Ar-H), 7.78(s., 1H Ar-H), 9.15(br., NH); 13C-NMR(δ): 15.4, 56.1, 104.8, 108.9, 119.9, 121.3, 125.2, 128.9, 139.4, 146.3, 146.9, 160.0, 165.7, 174.5. Elemental analysis: Found: C, 55.21; H, 4.35; N, 8.22 for C16H14N2O5S (346); requires C, 55.48; H, 4.07, N, 8.09.
Physical and spectral data for compound 6g; It was separated as pale white crystals (38% yield), mp 111–2°C; IR: 3496, 3473, 3459 cm−1(3 OH); 3232 cm−1 (NH), 1667 cm−1 (CON); EIMS[m/z(%)]: M+ = 360(23.01%), 1H-NMR(δ): 1.25(t., CH3), 2.60(q., CH2), 3.81(s., OCH3), 5.37(br., 3OH), 6.91(s., 2Ar-H), 7.47(s., 1H, Ar-H), 7.84(s., 1H Ar-H), 9.15(br., NH); 13C-NMR(δ): 14.8, 22.3, 56.1, 104.8, 108.9, 120.0, 122.4, 125.2, 129.1, 139.4, 146.3, 146.9, 158.7, 165.7, 174.5. Elemental analysis: Found: C, 56.66; H, 4.47; N, 7.77 for C16H14N2O5S (360); requires C, 56.66; H, 4.47, N, 7.77.
Physical and spectral data for compound 6h; It was separated as pale white crystals (43% yield), mp 103–4°C; IR: 3512, 3478, 3402 cm−1(3 OH); 3245 cm−1 (NH), 1671 cm−1 (CON); EIMS[m/z(%)]: M+ = 362(14.02%), 1H-NMR(δ): 3.83(s., 2OCH3), 5.37(br., 3OH), 6.93(s., 2Ar-H), 7.41(s., 1H, Ar-H), 7.88(s., 1H Ar-H), 9.15(br., NH); 13C-NMR(δ): 56.1, 105.9, 106.2, 108.9, 124.2, 125.2, 139.4, 144.2, 144.9, 142.3, 143.6, 165.7, 174+. Elemental analysis: Found: C, 52.85; H, 3.65; N, 7.50 for C16H14N2O6S (362); requires C, 53.03; H, 3.86, N, 7.73.
2-(Galloylamino)-6,7-disubstitated-benzo[d]thiazoles (7a-f): General procedure
To a stirred solution of 5a-f (10 mmol) in acetonitrile (20 mL) was added hydrazine hydrate (15 mmol). The mixture was stirred for 2 h at room temperature. The products 7a-f were isolated in a similar procedure like those of 6a-h and crude products was crystallized from pet.ether 40–60 to give the titled compounds (7a-f) as faint yellowish white crystals, in 40–65 % yield.
Physical and spectral data for compound 7a; It was separated as pale white crystals (64% yield), mp 131–2°C; IR 3480, 3459, 3440 cm−1(3 OH); 3210 cm−1 (NH), 1661 cm−1 (CON); EIMS[m/z(%)]: M+ = 350(17.98%), 352(M+2) (5.65%); 1H-NMR(δ): 2.38(s., CH3), 5.35(br., 3OH), 6.93(s., 2Ar-H), 7.44(d., 1H Ar-H), 7.50(d., 1H, Ar-H), 9.15(br., NH); 13C-NMR(δ): 12.6, 108.9, 120.0, 125,7 124.7, 125.2, 131.8, 132.3, 139.4, 146.3, 147.0, 165.7, 174.5,. Elemental analysis: Found: C, 51.00; H, 3.29; N, 8.25 for C15H11ClN2O4S (350.5); requires C, 51.35; H, 3.13, N, 7.98.
Physical and spectral data for compound 7b; It was separated as pale white crystals (62% yield), mp 146–7°C; IR: 3489, 3473, 3442 cm−1(3 OH); 3220 cm−1 (NH), 1675 cm−1 (CON); EIMS[m/z(%)]: M+ = 364(8.21%), 366(M+2) (2.31%); 1H-NMR(δ): 1.25(t., CH3), 2.60(q., CH2), 5.35(br., 3OH), 6.93(s., 2Ar-H), 7.51(d., 1H Ar-H), 7.56(d., 1H, Ar-H), 9.16(br., NH); 13C-NMR(δ): 13.6, 19.5, 108.9, 120.2, 123.4, 125.2, 125.7, 130.5, 134.8, 139.4, 146.3, 147.0, 165.7, 174.5,. Elemental analysis: Found: C, 53.00; H, 3.90; N, 8.00 for C16H13ClN2O4S (364.5); requires C, 52.68; H, 3.59, N, 7.68.
Physical and spectral data for compound 7c; It was separated as pale white crystals (42% yield), mp 139–140°C; IR: 3488, 3472, 3443 cm−1(3 OH); 3230 cm−1 (NH), 1671 cm−1 (CON); EIMS[m/z(%)]: M+ = 366(6.87%), 368(M+2) (2.6%); 1H-NMR(δ): 3.83(s., CH3), 5.35(br., 3OH), 6.93(s., 2Ar-H), 7.25(d., 1H Ar-H), 7.45(d., 1H, Ar-H), 9.15(br., NH); 13C-NMR(δ): 54.9, 108.9, 109.6, 115.3116.7,, 125.2, 126.8, 139.4. 146.3, 148.1, 156.6, 165.7, 174.5. Elemental analysis: Found: C, 49.50; H, 3.50; N, 7.35 for C15H11ClN2O5S (366.5); requires C, 49.12; H, 3.02, N, 7.64.
Physical and spectral data for compound 7d; It was separated as pale white crystals (28% yield), mp 152–3°C; IR: 3511, 3465, 3439 cm−1(3 OH); 3254 cm−1 (NH), 1669 cm−1 (CON); EIMS[m/z(%)]: M+ = 346(4.11%), 1H-NMR(δ): 2.34(s., CH3), 3.83(s., OCH3), 5.35(br., 3OH), 6.93(s., 2Ar-H), 6.88(d., 1H, Ar-H), 7.54(d., 1H, Ar-H), 9.15(br., NH); 13C-NMR(δ): 11.8, 56.1, 108.9, 113.2, 117.5, 119.6, 124.3, 125.2, 139.4, 141.2, 146.3, 160.0, 165.7, 174.5. Elemental analysis: Found: C, 55.85; H, 4.25; N, 8.45 for C15H12N2O5S (346); requires C, 55.48; H, 4.07, N, 8.09, for compound 7e; Physical and spectral data for compound 6g; It was separated as pale white crystals (34% yield), mp 116–7°C; IR: 3497, 3481, 3449 cm−1(3 OH); 3217 cm−1 (NH), 1668 cm−1 (CON); EIMS[m/z(%)]: M+ = 360(12.03%), 1H-NMR(δ): 1.25(t., CH3), 2.60(q., CH2), 3.81(s., OCH3), 5.37(br., 3OH), 6.93(s., 2Ar-H), 6.95(d., 1H, Ar-H), 7.35(d., 1H Ar-H), 9.15(br., NH); 13C-NMR(δ): 14.8, 18.7, 56.1, 108.9, 113.2, 119.8, 120.0, 123.0, 125.2, 139.4, 141.2, 146.3, 158.7, 165.7, 174.5. Elemental analysis: Found: C, 57.00; H, 4.21; N, 7.45 for C16H14N2O5S (360); requires C, 56.66; H, 4.47, N, 7.77.
Physical and spectral data for compound 7f; It was separated as pale white crystals (26% yield), mp 98–9°C; IR: 3492, 3453, 3440 cm−1(3 OH); 3202 cm−1 (NH), 1664 cm−1 (CON); EIMS[m/z(%)]: M+ = 362(7.12%), 1H-NMR(δ): 3.83(s., 2OCH3), 5.39(br., 3OH), 6.98(s., 2Ar-H), 6.89(d., 1H, Ar-H), 7.09(d., 1H Ar-H), 9.15(br., NH); 13C-NMR(δ): 56.1, 60.9, 108.9, 109.2, 114.3, 114.9, 125.2, 139.4, 141.8, 142.3, 144.9, 146.3, 165.7, 174.5. Elemental analysis: Found: C, 52.85; H, 4.25; N, 7.85 for C16H14N2O6S (362); requires C, 53.03; H, 3.86, N, 7.73.
Experiments of anticancer activity
A 100 µL of Calibrated culture cells of Hela and MCF7, in 800 µL Dulbecco’s Modified Eagle Medium (DMEM), were mixed, separately, with each concentration for each tested compounds and the reference drugs (camptothecin and doxorubicin) (0, 20, 40, 60, 80 µg). The mixture was completed to 1 mL by DMEM medium. Cells were incubated for 72 h and cell suspensions were then counted by using Haemocytometer Tiefe Neubauer adopting a reported procedureCitation32. Each dose of the tested molecules was repeated 3 times. The mean number of survival cells with tested compounds in each test, at the different doses (µg/mL) in 3 × 3 groups of tests sets)/mean number of survival cells without drugs (in 3 × 3 groups) ×100 corresponding to each doses of the tested compounds [% mean survival cell numbers in relation to that of control test (0.0 µg/mL)], were calculated. The IC50 values for each of the tested compounds and the reference molecules (camptothecin and doxorubicin), were determined from the curve of the % of inhibition of cell numbers versus the doses (µg/mL) of the tested moleculesCitation32. The IC50 values were recorded in .
Conclusion
This research concluded that the 2-(triacetylgalloylamino)-benzo[d]thiazoles (4a~5f) have higher cytotoxicity than the corresponding deacetylated derivatives (6a~7f). The most active molecules against Hela culture cell line, in comparison to the topo-II inhibitor doxorubicin, among the prepared compounds were compounds 5b, 5c, 5e and 5f. They exhibited IC50 values; 0.011, 0.012, 0.012 and 0.015 µM, that means they have 106% (=~1.1 folds), 133% (=~1,3 folds), 146% (=~1.5 folds) and 153% (=~1.5 folds)] than the reference drug, respectively. Also, the most active molecules against MCF7 cell culture, in relation to the same reference were 4d, 4h, 5e, 5f, 5c and 5b. They showed IC50 values equal: 0.012, 0.015, 0.019, 0.009, 0.012 and 0.016 µM, that means they have 142% (=~1.4 folds), 113% (=~1.1 folds), 106% (=~1.1 folds), 142% (=~1.4 folds) and 189% (=~1.9 folds)], than the reference drug, respectively.
Meanwhile, The cytotoxic activity of all the tested compounds, in comparison to Camptothecin (the selective topo-I inhibitor reference drug), gave more folds higher activity towards the two cell cultures than doxorubicin. Thus, compounds; 5b, 5c, 5e and 5f, demonstrated 126% (=~1.2 folds), 158% (=~1.6 folds), 172% (=~1.7 folds) and 158% (=~1.8 folds) higher activity than Camptothecin, towards Hela cell lines, respectively. While, compounds; 4d, 4e, 4h, 5b, 5c, 5e and 5f, gave 167% (=~1.7 folds), 111% (=~1.1 folds), 133% (=~1.3 folds), 125% (=~1.2 folds), 167% (=~1.7 folds), 105% (=~1.1 folds) and 222% (=~2.2 folds), higher activity than Camptothecin, towards HCF7 cultures, respectively.
It is clear that the best result in this study is the achievement of the designed molecule; 2-[(3,4,5–triacetoxy-benzoyl]amino-6,7-dimethoxy-benzo[d]thiazoles (5f) that showed the highest cytotoxic activity among all the synthesized molecules. It exhibited 1.5 folds and 1.9 folds higher activity than doxorubicin, towards Hela and MCF7 cell cultures, respectively. Also, it demonstrated 1.6 folds and 2.2 folds higher activities than Camptothecin towards the same cell cultures, respectively. Thus, compound 5f was recommended to be a new prototype as anticancer agent.
Supplementary Material
Download PDF (1.9 MB)Acknowledgments
We deeply acknowledge the members of the Department of Tumor Pathology, National Cancer Institute of Egypt, for helping in performing the anticancer screening of the prepared molecules on Hela and MCF7 cell lines. Also, we deeply appreciate the cancer cells culture unit of USA cancer institute for delivering the frozen cancer cell cultures.
Declaration of Interest
All the authors have no interest and no personal relationships or financial with any peoples or organizations that could inappropriately influence (bias) their work directly or indirectly, during the subject matter or materials discussed in this manuscript.
References
- Liu LF. DNA topoisomerase poisons as antitumor drugs. Annu Rev Biochem 1989;58:351–375.
- Vilaivan T, Lowe G. A novel pyrrolidinyl PNA showing high sequence specificity and preferential binding to DNA over RNA. J Am Chem Soc 2002;124:9326–9327.
- D’Costa M, Kumar V, Ganesh KN. Synthesis of 4(S)-(N-Boc-amino)-2(S/R)-(thymin-1-ylmethyl)-pyrrolidine-N-1-acetic acid: A novel cyclic PNA with constrained flexibility. Tetrahedron Lett 2002;43:883–886.
- Sheigeyasu M, Kuwahara M, Sisido M, Ishikawa T. Synthesis and DNA binding property of a novel peptide nucleic acid that contains cis-4-adeninyl-L-prolinol unit. Chem Lett 2001;634–635.
- Ferro AM, Olivera BM. Poly(ADP-ribosylation) of DNA topoisomerase I from calf thymus. J Biol Chem 1984;259:547–554.
- Muller MT, Spitzner JR, DiDonato JA, Mehta VB, Tsutsui K, Tsutsui K. Single-strand DNA cleavages by eukaryotic topoisomerase II. Biochemistry 1988;27:8369–8379.
- Nitiss JL. Investigating the biological functions of DNA topoisomerases in eukaryotic cells. Biochim Biophys Acta 1998;1400:63–81.
- Tobey RA, Oishi N, Crissman HA. Cell cycle synchronization: Reversible induction of G2 synchrony in cultured rodent and human diploid fibroblasts. Proc Natl Acad Sci USA 1990;87:5104–5108.
- Woynarowski JM, McHugh M, Sigmund RD, Beerman TA. Modulation of topoisomerase II catalytic activity by DNA minor groove binding agents distamycin, Hoechst 33258, and 4’,6-diamidine-2-phenylindole. Mol Pharmacol 1989;35:177–182.
- McHugh MM, Woynarowski JM, Sigmund RD, Beerman TA. Effect of minor groove binding drugs on mammalian topoisomerase I activity. Biochem Pharmacol 1989;38:2323–2328.
- Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984;226:466–468.
- Nelson EM, Tewey KM, Liu LF. Mechanism of antitumor drug action: Poisoning of mammalian DNA topoisomerase II on DNA by 4’-(9-acridinylamino)-methanesulfon-m-anisidide. Proc Natl Acad Sci USA 1984;81:1361–1365.
- Tewey KM, Chen GL, Nelson EM, Liu LF. Intercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J Biol Chem 1984;259:9182–9187.
- Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem 1985;260:14873–14878.
- Yamashita Y, Kawada S, Fujii N, Nakano H. Induction of mammalian DNA topoisomerase I and II mediated DNA cleavage by saintopin, a new antitumor agent from fungus. Biochemistry 1991;30:5838–5845.
- Chow KC, Macdonald TL, Ross WE. DNA binding by epipodophyllotoxins and N-acyl anthracyclines: Implications for mechanism of topoisomerase II inhibition. Mol Pharmacol 1988;34:467–473.
- Suzuki K, Yahara S, Hashimoto F, Uyeda M. Inhibitory activities of (-)-epigallocatechin-3-O-gallate against topoisomerases I and II. Biol Pharm Bull 2001;24:1088–1090.
- Suzuki K, Shono F, Kai H, Uno T, Uyeda M. Inhibition of topoisomerases by fatty acids. J Enzym Inhib 2000;15:357–366.
- Suzuki K, Nagao K, Monnai Y, Yagi A, Uyeda M. Topostatin, a novel inhibitor of topoisomerases I and II produced by Thermomonospora alba strain No. 1520. I. Taxonomy, fermentation, isolation and biological activities. J Antibiot 1998;51:991–998.
- Suzuki K, Yahara S, Kido Y, Nagao K, Hatano Y, Uyeda M. Topostatin, a novel inhibitor of Topoisomerases I and II produced by Thermomonospora alba strain No. 1520. II. Physico-chemical properties and structure elucidation. J Antibiot 1998;51:999–1003.
- Suzuki K, Yamaizumi M, Tateishi S, Monnai Y, Uyeda M. Topostatin, a novel inhibitor of topoisomerases I and II produced by Thermomonospora alba strain No. 1520. III. Inhibitory properties. J Antibiot 1998;52:60–465.
- Suzuki K, Doi S, Yahara S, Uyeda M. 2070-DTI, a topoisomerase inhibitor produced by Streptomyces sp. strain No. 2070. J Enzyme Inhib Med Chem 2003;18:497–503.
- Suzuki K, Siddique T, Nishimura H, Sekimoto J, Uyeda M. Inhibition of DNA topoisomerases by microbial inhibitors. J Enzym Inhib 1998;13:41–55.
- Suzuki K, Sekimoto J, Siddique T, Kamiya A, Uyeda M. Macrostatin, a novel macromolecular inhibitor of topoisomerases produced by Streptomyces avermitilis no. C-127. J Enzym Inhib 1998;14:69–83.
- Suzuki K, Yahara S, Maehata K, Uyeda M. Isoaurostatin, a novel topoisomerase inhibitor produced by Thermomonospora alba. J Nat Prod 2001;64:204–207.
- Abdel-Aziz M, Matsuda K, Otsuka M, Uyeda M, Okawara T, Suzuki K. Inhibitory activities against topoisomerase I and II by polyhydroxybenzoyl amide derivatives and their structure-activity relationship. Bioorg Med Chem Lett 2004;14:1669–1672.
- Ioanoviciu A, Antony S, Pommier Y, Staker BL, Stewart L, Cushman M. Synthesis and mechanism of action studies of a series of norindenoisoquinoline topoisomerase I poisons reveal an inhibitor with a flipped orientation in the ternary DNA-enzyme-inhibitor complex as determined by X-ray crystallographic analysis. J Med Chem 2005;48:4803–4814.
- Park KD, Lee SG, Kim SU, Kim SH, Sun WS, Cho SJ et al. Anticancer activity of 3-O-acyl and alkyl-(-)-epicatechin derivatives. Bioorg Med Chem Lett 2004;14:5189–5192.
- El-Telbany F, Essaway M, Ismail MAH, El-Kersh T. A simple one-step synthesis of some new 4(6-substituted benzothiazol-2-yl)-1-thia-4-azaspiro[5:4]decan-3-one derivatives as potential antimicrobial agents. Egypt J Pharm Sci 1987;28:33–40.
- Ye J, Abiman P, Crossley A, Jones JH, Wildgoose GG, Compton RG. Building block syntheses of gallic acid monomers and tris-(O-gallyl)-gallic acid dendrimers chemically attached to graphite powder: A comparative study of their uptake of Al(III) ions. Langmuir 2010;26:1776–1785.
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990;82:1107–1112.
- Soeksmanto A, Subroto MA, Wijaya H, Simanjuntak P. Anticancer activity test for extracts of Sarang semut plant (Myrmecodya pendens) to HeLa and MCM-B2 cells. Pak J Biol Sci 2010;13:148–151.
- Sheng-You H, Sam ZG, Xiaoqin Z. Scoring functions and their evaluation methods for protein–ligand docking: Recent advances and future directions. Phys Chem Chem Phys 2010;12:12899–12908.