Abstract
The α-glucosidase inhibitory activities of bergenin derivatives were evaluated. Bergenin derivatives were synthesized from bergenin which is a characteristic compound of B. ligulata. A new bergenin derivative, 11-O-(3′,4′-dimethoxybenzoyl)-bergenin showed the highest potent inhibitory activity among those of bergenin derivatives. The presence of substituents at 3′,4′-position in bergenin derivatives altered the α-glucosidase inhibitory activity. 11-O-(3′,4′-dimethoxybenzoyl)-bergenin was noncompetitive inhibitor for α-glucosidase. The present study reveals that bergenin derivatives could be classified as a new group of α-glucosidase inhibitors.
Introduction
Diabetes mellitus is a worldwide health problem that affects increasingly people every year. It is also a lifestyle-related disease known to trigger many complications, nephropathy, retinopathy, neuropathy, cardiovascular disease and so on. Diabetes mellitus type 2, which is caused by a secretory decrease in insulin from pancreatic Langerhans β cells or a lowering of the insulin resistanceCitation1,Citation2, is a serious problem in developed countries. One of the most beneficial therapies for diabetes mellitus type 2 is said to be one that achieves optimal blood glucose control after a meal, and α-glucosidase inhibitors perform a very useful activity by delaying glucose absorptionCitation3–5. Glucosidase inhibition is applicable to the treatment of numerous diseases including diabetes mellitus type 2Citation6, cancerCitation7 and HIVCitation8. This wide rage of uses for glucosidase inhibitors exemplifies the pervasive importance of glycosylation/deconjugation of sugars in such varied processes as cell signalling/recognition, cell cycle regulation and metabolism.
Interest in glucosidase inhibitors is growing because the number of diabetes mellitus cases has been increasing worldwide in recent years. Glucosidase inhibitors have been very interesting biological activitiesCitation9,Citation10, thus, there might be value in searching for glucosidase inhibitors derived from food and plant in the view of safety.
In humans, four enzymes are causative for the complete degradation of starch into glucose, which is then able to enter the bloodstream by a specific transport systemCitation11,Citation12. Firstly, salivary and pancreatic α-amylases (EC 3.2.1.1) are endohydrolases that cleave the internal α-1, 4 bonds of starch into shorter linear and branched dextrin chains. The dextrins are then further hydrolyzed at the nonreducing ends into glucose by two small-intestinal brush-border exohydrolases: maltase-glucoamylase (EC 3.2.1.20 and 3.2.1.3) and sucraseisomaltase (EC 3.2.148 and 3.2.10)Citation13.
In previous studies, human pancreatic α-amylase has been widely known as family 13 of the glycoside hydrolases (GH13), which gathers a large variety of enzymes with varying substrate and product specificities as reflected by the 26 different EC numbers found in this family of enzymes: glycoside hydrolases (EC 3.2.1.X, the most abundant), glycoside transferases (EC 2.4.1.X) and even isomerases (EC 5.4.99.15 and EC 5.4.99.16)Citation14–16. Regardless of the low amino acid sequence similarity between the evolutionarily distant members of this family, some highly conserved residues are still present. Consequently, it has been established by structural studies that all GH13 members share similar foldingCitation17.
The α-glucosidase (EC 3.2.1.20) from Saccharomyces cerevisiae (baker’s yeast), coded by the MAL12 gene, is also one of the GH13 family. It can catalyze the hydrolysis of terminal, nonreducing 1,4-linked α-d-glucose residues with release of α-d-glucose molecule and retention of configuration at the anomeric carbon, characteristic of GH13 enzymesCitation15,Citation18. Because of its ready availability and ease of handling, baker’s yeast α-glucosidase (EC 3.2.1.20; GH13 family) has been widely used for enzymatic assays to find novel α-glucosidase inhibitors with different biological activitiesCitation19–24.
Bergenia ligulata (Wall.) Engl. (syn. Saxifraga) is a perennial herb that grows wild in North India and particularly between the stones and rocks. This plant has been used in the Ayurvedic formulations for various ailments. B. ligulata contained β-sitosterolCitation25, a number of phenolic compounds like (+)-afzelechinCitation26 and catechin and other compoundsCitation27–29. Among these compounds, bergenin were identified as the major unique phenolic compound of B. ligulata. It has been reported that bergenin has such as anti-inflammatoryCitation30, antioxidantCitation31, hepatoprotectiveCitation32, neuroprotectiveCitation33, anti-HIVCitation34, antiarrhythmicCitation35, antifungalCitation36 and gastroprotectiveCitation37 activities. Furthermore, bergenin and its esterified derivatives occur widely in a number of plants and have been found as ingredients in plant extractsCitation38–42. Bergenin is modified by coupling with a variety of fatty acids on hydroxyl groups through chemical catalysis, Takahashi et al. synthesized a series of esterified derivatives of bergenin with greatly enhanced biological activityCitation33. Some other chemical modifications of bergenin derivatives were also reportedCitation43.
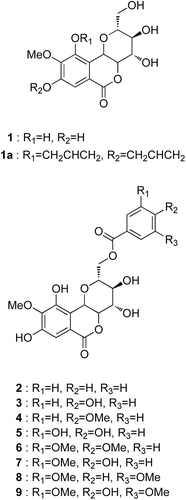
In present study, bergenin derivatives were prepared from the naturally occurring bergenin and evaluated to the inhibitory effects of the bergenin derivatives on α-glucosidase from baker’s yeast (E.C. 3.2.1.20), to study the structure-activity relationships and kinetics of inhibitory activity ().
Figure 1. Chemical structures of bergenin (1) and its derivatives (2–9).
Materials and methods
Chemistry
All reagents were purchased from Wako Pure Chemistry (Osaka, Japan). All solvents were purchased from Kanto Chemical (Tokyo, Japan). Roots of Bergenia ligulata (Wall.) Engl. were provided by Koei Kogyo Co., Ltd. (Tokyo, Japan). Thin layer chromatography (TLC) was performed on precoated plates (silica gel 60 F254, 0.25 mm, Merk, Darmstadt, Germany). Column chromatography was carried out using 70–230 mesh silica gel (Kieselgel 60, Merk, Germany). Optical rotations were measured on a Japan Spectroscopic Co. LTDDIP-1000. Infrared (IR) spectra were recorded with a JASCO FT/IR-470 plus Fourier transform infrared spectrometer (JASCO Co., Ltd, Japan) with an ordinate scale in the region 4000–450. Nuclear magnetic resonance (NMR) spectra were recorded at 400 MHz for 1H and 100 MHz for 13C on a JEOL ECA 400 spectrometer in DMSO or CDCl3 with TMS as internal standard (chemical shift in d, ppm). J values are reported in Hertz. EI-MS spectra were obtained on a JEOL the Tandem MS station JMS-700 (Japan Electron Optics Laboratory Co., Ltd., Tokyo, Japan). The absorbance was measured with a MTP-800Lab microplate reader. α-glucosidase from baker’s yeast (EC 3.2.1.20; GH13 family) was purchased from Oriental Yeast Co., Ltd (Tokyo, Japan). p-nitrophenyl α-d-glucopyranoside was purchased from Wako Pure Chemistry (Osaka, Japan).
Extraction and isolation
The barks (5.0 kg) of B. ligilata were extracted once with MeOH. The solution was evaporated to dryness in vaccuo to give a methanol extract (480 g). The residue was re-extracted successively with hexane, dichloromethane, ethyl acetate, butanol and water. Each fraction was concentrated to dryness in vaccuo to give hexane, dichloromethane, ethyl acetate, butanol and water fractions respectively.
The butanol extract was fractionated to fraction 1–3 by silica gel column chromatography with dichloromethane and methanol as eluants. Fraction 2 was recrystallized, thereby bergenin (1) (3.7 g) was isolated. The structure of 1 was identified by the comparison of its physical and spectral data with those described in the literatureCitation39,Citation44.
Synthesis
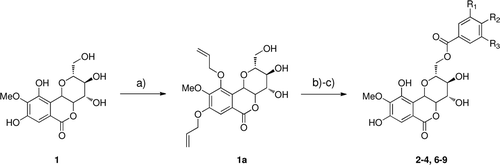
General procedure for the synthesis of 8,10-diallyloxy-bergenin (1a)
First, the phenolic hydroxy groups in bergenin (1) (700 mg, 2.13 mmol) were selectively allylated. Allyl bromide and NaI were added to a solution bergenin and K2CO3 in anhydr. DMF (5 mL). After the mixture was stirred 2 h at 55°C under nitrogen, it was concentrated in vacuo. The residue was treated with water (15 mL) and extracted with ethyl acetate (3 × 10 mL). The organic extract was washed successively with brine (3 × 15 mL). The extract was dried over Na2SO4 and concentrated in vacuo. The residue was chromatographed on silica gel to afford 1a (554 mg, 64%) as a white powder.
General procedure for the synthesis of bergenin derivatives (2–4, 6–9)
Firstly, to a solution of 1a in anhydr. THF (2 mL/mmol), various benzoic acids and Ph3P (2 equiv) were dissolved. DIAD (1.7 equiv) was added dropwise under 0°C. The mixture was stirred for 2 h under nitrogen. It was concentrated in vacuo. The residue was chromatographed on silica gel to afford benzoic acid ester of 1a (yield 79–92%) as a white powder.
Secondly, the allyl protected esters and Pd(PPh3)4 (1 mol%, freshly prepared) were dissolved in degassed anhydrous. THF (2 mL/mmol) and morpholine (10 equiv per allylgroup to be cleaved) was added dropwise. The mixture was stirred at r.t and concentrated in vacuo. The residue was taken up in EtOAc. The organic layer was washed several times with small amounts of 1 N HCl, dried and concentrated. The crude material was purified by silica gel column chromatography. The purified material was bergenin derivatives (2–4, 6–9) in yield 28–59% ().
Scheme 1. Reagents and conditions: (a) allylbromide, K2CO3, NaI, DMF; (b) DIAD, P(Ph)3, RCOOH, THF; (c) 10% Pd(PPh3)4, 20 equiv., morpholine, THF.
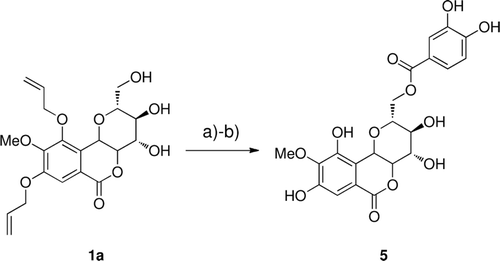
Synthesis of 11-O-protocatechuoylbergenin (5)
Firstly, the phenolic hydroxy groups in protocatechualdehyde were selectively allylated. Secondly, 3,4-diallyloxy-protocatechuic acid was accomplished according to previous studyCitation45. Then, the same procedure as for 2–4, 6–9 was used by starting from 1a ().
Scheme 2. Reagents and conditions: (a) DIAD, P(Ph)3, 3,4-diallyloxy-protocatechuic acid, THF; (b) 10% Pd(PPh3)4, 20 equiv., morpholine, THF.
8,10-diallyloxy-bergenin (1a)
Yield 64%, white powder. 1H NMR (CDCl3, 400 MHz) δ 3.54–3.59 (m, 1H), 3.54–3.59 (m, 1H), 3.68–3.73 (m, 1H), 3.83–4.01 (m, 1H), 3.91 (s, 3H), 3.98 (d, J = 9.2 Hz, 1H), 4.41–4.55 (m, 4H), 4.72 (d, J = 10.4 Hz, 1H), 5.26 (dd, J1 = 1.8 Hz, J2 = 10.8 Hz, 2H), 5.40 (dd, J1 = 1.8 Hz, J2 = 17.2 Hz, 2H), 5.97–6.12 (m, 2H), 7.34 (s, 1H).
11-O-benzoylbergenin (2)
Yield 25%, amorphous powder; [α]25D +66.8° (c = 0.15, MeOH); IR (KBr) νmax cm−1: 3474 (OH), 1722 (ester), 1610 (arom. C=C); 1H NMR (DMSO-d6, 400 MHz) δ 3.41 (dd, J1 = 9.2 Hz, J2 = 8.8 Hz, 1H), 3.69 (m, 1H), 3.74 (s, 3H), 3.93 (ddd, J1 = 9.2 Hz, J2 = 7.2 Hz, J3 = 2.0 Hz, 1H), 4.03 (t, J = 10.0 Hz, 1H), 4.34 (dd, J1 = 12.4 Hz, J2 = 7.2 Hz, 1H), 4.78 (dd, J1 = 12.4 Hz, J2 = 2.0 Hz, 1H), 5.03 (d, J = 10.8 Hz, 1H), 7.00 (s, 1H), 7.54 (m, 2H), 7.67 (m, 1H), 8.01 (m, 2H); 13C NMR (DMSO-d6, 100 MHz): ; EI-MS, m/z (rel. intensity) 432 [M]+ (2), 370 (4), 256 (6), 122 (53), 105 (100), 77 (56).
Table 1. 13C NMR spectral data for compounds 2–9 (δ in ppm).
11-O-p-hydroxybenzoylbergenin (3)
Yield 38%, amorphous powder; [α]25D +18.3° (c = 0.15, MeOH); IR (KBr) νmax cm−1: 3380 (OH), 1712 (ester), 1608 (arom. C=C), 1238 (C-O); 1H NMR (DMSO-d6, 400 MHz) δ 3.40 (m, 1H), 3.69 (t, J = 9.2 Hz, 1H), 3.73 (s, 3H), 3.87 (ddd, J1 = 9.2 Hz, J2 = 6.4 Hz, J3 = 1.6 Hz, 1H), 4.03 (t, J = 9.8 Hz, 1H), 4.26 (dd, J1 = 12.0 Hz, J2 = 6.4 Hz, 1H), 4.71 (dd, J1 = 12.0 Hz, J2 = 1.6 Hz, 1H), 5.02 (d, J = 10.0 Hz, 1H), 6.86 (d, J = 8.8 Hz, 2H), 7.00 (s, 1H), 7.85 (d, J = 8.8 Hz, 2H); 13C NMR (DMSO-d6, 100 MHz): ; EI-MS, m/z (rel. intensity) 448 [M]+ (2), 328 (1), 208 (24), 121 (100), 81 (13), 65 (28).
11-O-p-methoxybenzoylbergenin (4)
Yield 24%, amorphous powder; [α]25D +44.4° (c = 0.15, MeOH); IR (KBr) νmax cm−1: 3373 (OH), 2959 (C-H), 1720 (ester), 1609 (arom. C=C), 1237 (C-O); 1H NMR (DMSO-d6, 400 MHz) δ 3.40 (t, J = 8.8 Hz, 1H), 3.69 (t, J = 8.8 Hz, 1H), 3.74 (s, 3H), 3.89 (m, 1H), 3.83 (s, 3H), 4.04 (t, J = 10.0 Hz, 1H), 4.29 (dd, J1 = 12.0 Hz, J2 = 6.8 Hz, 1H), 4.74 (dd, J1 = 12.0 Hz, J2 =2.1Hz, 1H), 5.03 (d, J = 10.0 Hz, 1H), 7.00 (s, 1H), 7.05 (d, J = 8.8 Hz, 2H), 7.95 (d, J = 8.8 Hz, 2H); 13C NMR (DMSO-d6, 100 MHz): ; EI-MS, m/z (rel. intensity) 462 [M]+ (17), 250 (11), 208 (50), 152 (93), 135 (100), 92 (34), 77 (17).
11-O-protocatechuoylbergenin (5)
Yield 16%, amorphous powder; [α]25D +25.2° (c = 0.25, MeOH); IR (KBr) νmax cm−1: 3364 (OH), 1709 (ester), 1610 (arom. C=C), 1227 (C-O); 1H NMR (DMSO-d6, 400 MHz) δ 3.39 (t, J = 9.2 Hz, 1H), 3.69 (t, J = 9.2 Hz, 1H), 3.74 (s, 3H), 3.86 (m, 1H), 4.01 (t, J = 10.0 Hz, 1H), 4.26 (dd, J1 = 11.6 Hz, J2 = 6.0 Hz, 1H), 4.67 (brd, J = 11.6 Hz, 1H), 5.05 (d, J = 10.4 Hz, 1H), 6.81 (d, J = 8.4 Hz, 1H), 7.00 (s, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.38 (d, J = 1.6 Hz, 1H); 13C NMR (DMSO-d6, 100 MHz): ; EI-MS, m/z (rel. intensity) 464 [M]+ (11), 328 (23), 262 (15), 137 (100), 77 (54), 55 (32).
11-O-(3′,4′-dimethoxybenzoyl)-bergenin (6)
Yield 28%, amorphous powder; [α]25D +32.6° (c = 0.025, MeOH); IR (KBr) νmax cm−1: 3395 (OH), 1716 (ester), 1598 (arom. C=C), 1225 (C-O); 1H NMR (DMSO-d6, 400 MHz) δ 3.38 (m, 1H), 3.70 (t, J = 9.6 Hz, 1H), 3.73 (s, 3H), 3.83 (s, 3H), 3.84 (s, 3H), 3.91 (m, 1H), 4.04 (dd, J1 = 10.0 Hz, J2 = 9.6 Hz, 1H), 4.21 (dd, J1 = 12.0 Hz, J2 = 7.2 Hz, 1H), 4.83 (brd, J = 12.0 Hz, 1H), 5.05 (d, J = 10.0 Hz, 1H), 7.00 (s, 1H), 7.09 (d, J = 8.4 Hz, 1H), 7.48 (d, J = 1.6 Hz, 1H), 7.63 (dd, J1 = 8.4 Hz, J2 = 1.6Hz, 1H); 13C NMR (DMSO-d6, 100 MHz): ; EI-MS, m/z (rel. intensity) 492 [M]+ (9), 208 (20), 182 (50), 165 (100), 152 (9), 79 (9).
11-O-vanilloylbergenin (7)
Yield 30%, amorphous powder; [α]25D +56.4° (c = 0.1, MeOH); IR (KBr) νmax cm−1: 3365 (OH), 1717 (ester), 1599 (arom. C=C), 1222 (C-O); 1H NMR (DMSO-d6, 400 MHz) δ 3.39 (m, 1H), 3.69 (dd, J1 = 9.6 Hz, J2 = 8.8 Hz, 1H), 3.73 (s, 3H), 3.85 (s, 3H), 3.89 (ddd, J1 = 8.0 Hz, J2 = 7.6 Hz, J3 = 2.0 Hz, 1H), 4.04 (dd, J1 = 10.4 Hz, J2 = 9.6 Hz, 1H), 4.19 (dd, J1 = 11.6 Hz, J2 = 8.0Hz, 1H), 4.81 (dd, J1 = 11.6 Hz, J2 = 2.0 Hz, 1H), 5.05 (d, J = 10.8 Hz, 1H), 6.88 (d, J = 8.4 Hz, 1H), 7.00 (s, 1H), 7.48 (d, J = 2.0 Hz, 1H), 7.51 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H); 13C NMR (DMSO-d6, 100 MHz): ; EI-MS, m/z (rel. intensity) 478 [M]+ (7), 328 (16), 208 (53), 180 (17), 151 (100), 97 (13), 43 (18).
11-O-(3′,5′-dimethoxybenzoyl)-bergenin (8)
Yield 26%, amorphous powder; [α]25D +157.9° (c = 0.025, MeOH); IR (KBr) νmax cm−1: 3355 (OH), 1717 (ester), 1602 (arom. C=C), 1237 (C-O); 1H NMR (DMSO-d6, 400 MHz) δ 3.38 (m, 1H), 3.69 (m, 1H), 3.73 (s, 3H), 3.81 (s, 6H), 3.91 (ddd, J1 = 9.2 Hz, J2 = 7.6 Hz, J3 = 1.6 Hz, 1H), 4.04 (dd, J1 = 10.4 Hz, J2 = 9.6 Hz, 1H), 4.23 (dd, J1 = 12.0 Hz, J2 = 7.6 Hz, 1H), 4.90 (dd, J1 = 12.0 Hz, J2 = 1.6 Hz, 1H), 5.05 (d, J = 10.4 Hz, 1H), 6.79 (t, J = 2.4 Hz, 1H), 7.00 (s, 1H), 7.19 (d, J = 2.4 Hz, 2H); 13C NMR (DMSO-d6, 100 MHz): ; EI-MS, m/z (rel. intensity) 492 [M]+ (12), 328 (5), 208 (31), 182 (78), 165 (100), 137 (18), 122 (17), 55 (15).
11-O-syringylbergenin (9)
Yield 20%, amorphous powder; [α]25D +24.5° (c = 0.1, MeOH); IR (KBr) νmax cm−1: 3371 (OH), 1718 (ester), 1610 (arom. C=C), 1235 (C-O); 1H NMR (DMSO-d6, 400 MHz) δ 3.38 (m, 1H), 3.69 (m, 1H), 3.72 (s, 3H), 3.85 (s, 6H), 3.92 (ddd, J1 = 10.0 Hz, J2 = 8.1 Hz, J2 = 2.4 Hz, 1H), 4.06 (t, J = 10.0 Hz, 1H), 4.12 (dd, J1 = 11.6 Hz, J2 = 8.8 Hz, 1H), 4.89 (dd, J1 = 11.6 Hz, J2 = 2.4 Hz, 1H), 5.07 (d, J = 10.8 Hz, 1H), 7.00 (s, 1H), 7.26 (s, 2H); 13C NMR (DMSO-d6, 100 MHz): ; EI-MS, m/z (rel. intensity) 508 [M]+ (2), 328 (8), 237 (16), 208 (100), 182 (50), 165 (83), 152 (75).
α-glucosidase inhibitory activity
α-glucosidase inhibitory activity was determined using the modified version of the method according to Li et al.Citation46 α-glucosidase (25 µL, 0.2 U/mL), 25 µL of various concentrations of samples, and 175 µL of 50 mM sodium phosphate buffer (pH 7.0) were mixed at room temperature for 10 min. The reaction was started by the addition of 25 µL of 2.5 mM p-nitrophenyl-α-d-glucopyranoside. The activities of glucosidase were detected in 96-well plate, and the absorbance was determined at 405 nm (for p-nitrophenol) in a microplate reader (Corona Electric Co., Ltd). Control sample contained 25 µL DMSO in place of test samples. Percentage of enzyme inhibition was calculated as (B/A) × 100, where A represents absorbance of control without test samples, and B represents absorbance in presence of test samples. All the tests were run in triplicate. Acarbose and 1-deoxynojirimycin were used as the positive control in this study. The fifty percent inhibitory concentration (IC50) values were expressed as mean ± SD (n = 3).
Results and discussion
The synthesis of ester analogues 2–4, 6–9 of bergenin at C-11 position were accomplished by coupling various benzoic acids with bergenin () by employing Mitsunobu protocol in dry THF. For the synthesis of analogues 5, the phenolic hydroxy groups in protocatechualdehyde were selectively allylated. Then, the same procedure as for 2–4, 6–9 was used by starting from 1a (). The pure compound 2, 3, 5, 7, 9 were identified through analysis of spectroscopic data and comparison with previous studiesCitation39,Citation40. The structures of the new bergenin derivatives (4, 6 and 8) have been assigned from 1H and 13C NMR spectra (), IR and EI-MS.
The α-glucosidase inhibitory activities of bergenin derivatives (2–9) were investigated and compared with that of acarbose and 1-deoxynojirimycin, a clinically used α-glucosidase inhibitors, were employed as a reference drugs. These results were shown in . Compounds 2–7 demonstrated a significant inhibition on α-glucosidase in a dose dependent manner. In particular, compound 6, new bergenin derivative, showed excellent inhibitions on yeast α-glucosidase a with extremely high potency, and its 50% inhibition of enzyme activity (IC50) values were calculated as 24.6 µM which were much lower than that of reference drugs, acarbose (907.5 µM) and 1-deoxynojirimycin (278.0 µM). Other active compounds, compound 2–4 and 7 also exhibited a potent inhibition with IC50 values of 118.3 µM, 143.2 µM, 143.8 µM and 186.9 µM, which were much more potent to that of 1-deoxynojirimycin, respectively, on the other hand, compound 5 (IC50: 289.6 µM) showed much the same effect as a potent inhibition of 1-deoxynojirimycin.
Figure 2. Dose-dependent inhibitory effects of bergenin (1) and its derivatives (2–9). Acarbose and 1-deoxynojirimycin were used a positive control. The result represents the means of triplicate experiments.
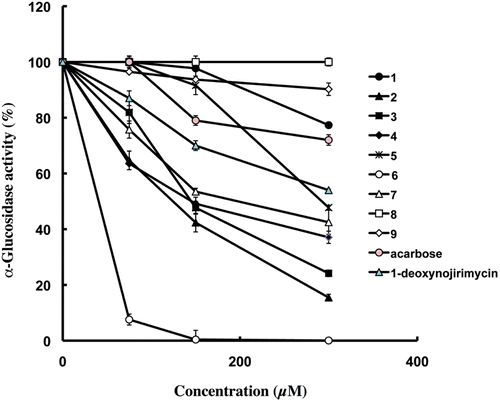
Furthermore, we evaluated the inhibitory activities of benzoic acid derivatives and bergenin (1) to clarify whether inhibitory activities of compound 2–7 are due to benzoic acid moiety or bergenin moiety. Benzoic acid (inhibition: 7.4 ± 0.9% at 300 µM), p-hydroxybenzoic acid (inhibition: 3.1 ± 1.3% at 300 µM), anisic acid (inhibition: 1.6 ± 1.4% at 300 µM), protocatechuic acid (inhibition: 10.7 ± 2.1% at 300 µM), 3,4-dimethoxybenzoic acid (inhibition: 17.7 ± 3.0% at 300 µM), vanilic acid (inhibition: 22.3 ± 1.0% at 300 µM) and bergenin (1) (inhibition: 22.7 ± 0.6% at 300 µM) exhibited low inhibitory activity. Each benzoic acid derivatives exhibited a relatively poor inhibitory activity as compared with these bergenin derivatives (). Therefore, we considered the linkage of benzoic acid and bergenin as well as benzoic acid moiety and bergenin moiety are critical factor of α-glucosidase inhibitory activity. These results accorded with previous reports that α-glucosidase was inhibited to a great extent when the compound contained a fused-aromatic ring and a heteroaromatic componentCitation47.
Table 2. Inhibition effects of bergenin (1) and bergenin derivatives (2–10) against α-glucosidase.
From the structural-activity point of view, the presence of methoxy group at 3′-position on bergenin derivative moiety was potent factor of the α-glucosidase inhibitory activity. In the case of compound 3, 4′ position replacement with OMe group (compound 4) is similar to that of compound 3, however, the introduction of another methoxy group in an ortho-position to a methoxy group (compound 7) led to dramatically increase inhibitory activity relative to compound 4. When the α-glucosidase inhibitory activities of compound 5 and 7 were compared, it was found that the potency increased in the order of 7 > 5. The observation revealed that replacement of the 3′-hydroxy substituted in the compound 5 by OMe group increased α-glucosidase inhibitory activity. In addition to these reports, the observation of the inhibitory activities of compound 5 and 7 indicated that 4′ position OMe group is also potent factor of the α-glucosidase inhibitory activity. The evidence supported is the previous studyCitation48. On the other hand, compound 9 had no effect on α-glucosidase inhibitory activity. The inhibitory effect of each compound on α-glucosidase activity is shown in .
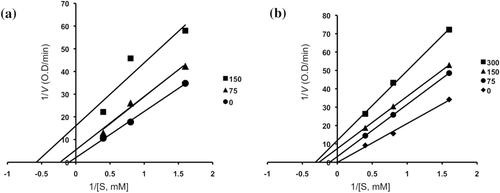
Inhibitory mechanisms of compounds 2, 3, 4, 6, acarbose and 1-deoxynojirimycin on α-glucosidase were analysed by a Lineweaver–Burk plot (–). This analysis ( and ) showed that Vmax decreased without changing Km in the presence of increasing concentration of inhibitors: as can be seen directly from the graph, −1/Km (the x-intercept) was unaffected by inhibitor concentration, whereas 1/Vmax became more positive. This behaviour indicates that compounds 2 and 6 exhibit noncompetitive inhibition characteristics for α-glucosidase. The inhibitory constants (Ki) were determined to be 104.9 µM for compound 2, 19.5 µM for compound 6, by using Dixon plot ( and ). In contrast, as shown representatively in , the kinetic plot shows that acarbose and 1-deoxynojirimycin were noncompetitive inhibitors with Ki value of 943.15 µM and 353.6 µM, respectively ( and ). On the other hand, shows that the plot of 1/V versus 1/[S] give a family of parallel straight lines with the same slopes. Accompanying the increase of the inhibitor concentration the values of both Km and Vm increase, but the ratio of Km/Vm remained unchanged. The slopes were independent of the concentration of compound 3 or 4 which indicated that compounds 3 and 4 were uncompetitive inhibitors of the enzyme. The Ki values of compound 3 and 4 were 217.5 µM and 235.8 µM, respectively.
Figure 3. Lineweaver–Burk plot (panel A) and Dixon plot (panel B) for inhibition of compound 2 (0, 75, 150 and 300 µM) on α-glucosidase activity.
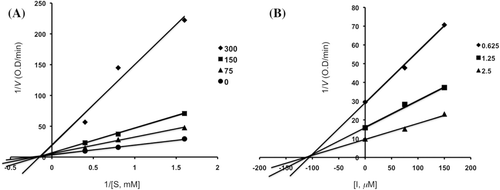
Figure 4. Lineweaver–Burk plot (panel A) and Dixon plot (panel B) for inhibition of compound 6 (0, 7.5, 15 and 30 µM) on α-glucosidase activity.
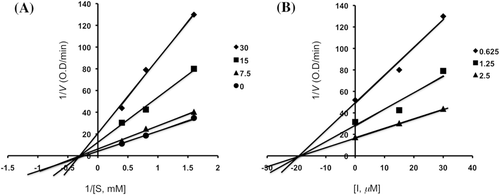
Figure 5. Lineweaver–Burk plot (panels A and C) and Dixon plot (panels B and D) for inhibition of acarbose (0, 0.5, 1.0 and 2.0 mM) and 1-deoxynojirimycin (0, 200, 400 and 800 μM) on α-glucosidase activity.
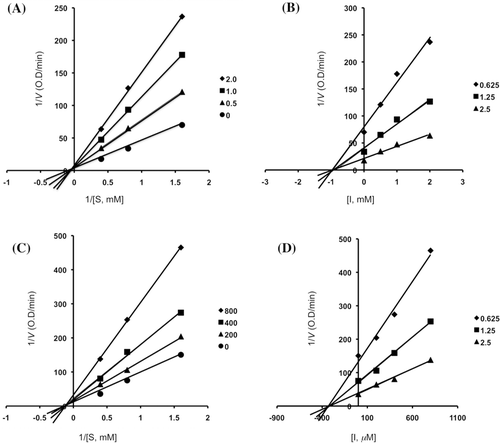
Figure 6. Line weaver–Burk plots for the inhibition of the hydrolysis activity of α-glucosidase by compounds 3 (0, 75 and 150 mM) (a) and 4 (0, 75, 150 and 300 mM) (b).
To date, the microbial α-glucosidase is known to be structurally different to those of mammalial origins. The microbial α-glucosidase inhibitors are not necessarily the mammalial α-glucosidase inhibitors. For example, (+)-catechin, a natural inhibitor of yeast α-glucosidase does not show any inhibitory activity on mammalial α-glucosidaseCitation49. On the other hand, voglibose shows very high inhibitory activity on porcine small intestine α-glucosidase, but both of them show very low inhibitory activity on microbial α-glucosidase, suggesting that ongoing experiments should be focused on the inhibitory activity of these compounds against mammalian intestinal α-glucosidases. Nevertheless, the inhibition of yeast α-glucosidase by bergenin derivatives served as an interesting structural activity relationship of this group of inhibitors.
In conclusion, we have undertaken synthesis of bergenin derivatives which abounds in many plants and a thorough investigation into the α-glucosidase inhibition of these compounds. In this study, we reported the correlation between α-glucosidase inhibition and structures of bergenin derivatives for the first time. Compound 6 (IC50 = 24.6 µM) showed a most potent α-glucosidase inhibition. Furthermore, we have uncovered some of more potent inhibitors of α-glucosidase than positive control. From the structural-activity point of view, linkage of benzoic acid and bergenin as well as benzoic moiety and bergenin moiety are critical factor of α-glucosidase inhibitory activity. We hope that these compounds could be employed as a lead for the design of new potential α-glucosidase inhibitors.
Acknowledgments
I am grateful to Professor I. Horibe for number of valuable suggestions and discussions on this study. The author is grateful to assistant Professor T. Minemastsu (School of Pharmaceutical Sciences, Kinki University) for the NMR spectrometry experiments on this study.
Declaration of interest
The authors report no conflicts of interest.
References
- Porte D Jr. Banting lecture 1990. β-cells in type II diabetes mellitus. Diabetes 1991;40:166–180.
- Taylor SI, Accili D, Imai Y. Insulin resistance or insulin deficiency. Which is the primary cause of NIDDM? Diabetes 1994;43:735–740.
- O’Dea K, Turton J. Optimum effectiveness of intestinal α-glucosidase inhibitors: importance of uniform distribution through a meal. Am J Clin Nutr 1985;41:511–516.
- Samad AH, Ty Willing TS, Alberti KG, Taylor R. Effects of BAYm 1099, new α-glucosidase inhibitor, on acute metabolic responses and metabolic control in NIDDM over 1 mo. Diabetes Care 1988;11:337–344.
- Bischoff H. The mechanism of α-glucosidase inhibition in the management of diabetes. Clin Invest Med 1995;18:303–311.
- Floris AL, Peter LL, Reinier PA, Eloy HL, Guy ER, Chris W. α-glucosidase inhibitors for patients with type 2 diabetes: results from a Cochrane systematic review and meta-analysis. Diabetes Care 2005;28:154–163.
- Fernandes B, Sagman U, Auger M, Demetrio M, Dennis JW. β 1-6 branched oligosaccharides as a marker of tumor progression in human breast and colon neoplasia. Cancer Res 1991;51:718–723.
- Ozawa S, Maruyama A, Odagiri T, Yuasa H, Hashimoto H. Synthesis and biological evaluation of α-l-fucosidase inhibitors: 5a-carba-a-l-fucopyranosylamine and related compounds. Eur J Org Chem 2001;5:967–974.
- Humphries MJ, Matsumoto K, White SL, Olden K. Inhibition of experimental metastasis by castanospermine in mice: blockage of two distinct stages of tumor colonization by oligosaccharide processing inhibitors. Cancer Res 1986;46:5215–5222.
- Gruters RA, Neefjes JJ, Tersmette M, de Goede RE, Tulp A, Huisman HG et al. Interference with HIV-induced syncytium formation and viral infectivity by inhibitors of trimming glucosidase. Nature 1987;330:74–77.
- Tattersall R. α-glucosidase inhibition as an adjunct to the treatment of type 1 diabetes. Diabet Med 1993;10:688–693.
- Sim L, Quezada-Calvillo R, Sterchi EE, Nichols BL, Rose DR. Human intestinal maltase-glucoamylase: crystal structure of the N-terminal catalytic subunit and basis of inhibition and substrate specificity. J Mol Biol 2008;375:782–792.
- Henrissat B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 1991;280 (Pt 2):309–316.
- Henrissat B, Bairoch A. Updating the sequence-based classification of glycosyl hydrolases. Biochem J 1996;316 (Pt 2):695–696.
- Henrissat B, Callebaut I, Fabrega S, Lehn P, Mornon JP, Davies G. Conserved catalytic machinery and the prediction of a common fold for several families of glycosyl hydrolases. Proc Natl Acad Sci USA 1995;92:7090–7094.
- Stam MR, Danchin EG, Rancurel C, Coutinho PM, Henrissat B. Dividing the large glycoside hydrolase family 13 into subfamilies: towards improved functional annotations of α-amylase-related proteins. Protein Eng Des Sel 2006;19:555–562.
- Henrissat B, Davies G. Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol 1997;7:637–644.
- Ly HD, Withers SG. Mutagenesis of glycosidases. Annu Rev Biochem 1999;68:487–522.
- Park H, Hwang KY, Oh KH, Kim YH, Lee JY, Kim K. Discovery of novel α-glucosidase inhibitors based on the virtual screening with the homology-modeled protein structure. Bioorg Med Chem 2008;16:284–292.
- Pluempanupat W, Adisakwattana S, Yibchok-Anun S, Chavasiri W. Synthesis of N-phenylphthalimide derivatives as α-glucosidase inhibitors. Arch Pharm Res 2007;30:1501–1506.
- Zhang R, McCarter JD, Braun C, Yeung W, Brayer GD, Withers SG. Synthesis and testing of 2-deoxy-2,2-dihaloglycosides as mechanism-based inhibitors of α-glycosidases. J Org Chem 2008;73:3070–3077.
- Choi CW, Choi YH, Cha MR, Yoo DS, Kim YS, Yon GH et al. Yeast α-glucosidase inhibition by isoflavones from plants of Leguminosae as an in vitro alternative to acarbose. J Agric Food Chem 2010;58:9988–9993.
- Kim JY, Lee JW, Kim YS, Lee Y, Ryu YB, Kim S et al. A novel competitive class of α-glucosidase inhibitors: (E)-1-phenyl-3-(4-styrylphenyl)urea derivatives. Chembiochem 2010;11:2125–2131.
- Li GL, He JY, Zhang A, Wan Y, Wang B, Chen WH. Toward potent α-glucosidase inhibitors based on xanthones: a closer look into the structure-activity correlations. Eur J Med Chem 2011;46:4050–4055.
- Bahl CP, Murari R, Parthasarathy MR, Seshadri TR. Components of Bergenia strecheyi and B. ligulata. Ind J Chem 1974;12:1038–1039.
- Tucci AP, Delle Monache F, Marini-Bettòlo GB. The occurrence of (+)afzelechin in Saxifraga ligulata Wall. Ann Ist Super Sanita 1969;5:555–556.
- Dixit BS, Srivastava SN. Tannin constituents of Bergenia ligulata roots. Ind J Nat Prod 1989;5:24–25.
- Chandrareddy UD, Chawla AS, Mundkinajeddu D, Maurya R, Handa SS. Paashaanolactone from Bergenia ligulata. Phytochemistry 1998;47:907–909.
- Kashima Y, Yamaki H, Suzuki T, Miyazawa M. Insecticidal effect and chemical composition of the volatile oil from Bergenia ligulata. J Agric Food Chem 2011;59:7114–7119.
- Swarnalakshmi T, Sethuraman MG, Sulochana N, Arivudainambi R. A note on the anti-inflammatory activity of bergenin. Curr Sci 1984;53:917.
- Afran M, Amin H, Karamac M, Kosinska A, Wiczkowski W, Amarowicz R. Antioxidant activity of phenolic fractions of Mallotus philippinensis bark extract. Czech J Food Sci 2009;27:109–117.
- Jahromi MAF, Chansouri JPN, Ray AB. Hypolipidaemic activity in rats of bergenin, the major constituent of Flueggea microcarpa. Phytother Res 1992;6:180–183.
- Takahashi H, Kosaka M, Watanabe Y, Nakade K, Fukuyama Y. Synthesis and neuroprotective activity of bergenin derivatives with antioxidant activity. Bioorg Med Chem 2003;11:1781–1788.
- Piacente S, Pizza C, De Tommasi N, Mahmood N. Constituents of Ardisia japonica and their in vitro anti-HIV activity. J Nat Prod 1996;59:565–569.
- Pu HL, Huang X, Zhao JH, Hong A. Bergenin is the antiarrhythmic principle of Fluggea virosa. Planta Med 2002;68:372–374.
- Prithiviraj B, Singh UP, Manickam M, Srivastava JS, Ray AB. Antifungal activity of bergenin, a constituent of Flueggea microcarpa. Plant Pathol 1997;46:224–228.
- Goel RK, Maiti RN, Manickam M, Ray AB. Antiulcer activity of naturally occurring pyrano-coumarin and isocoumarins and their effect on prostanoid synthesis using human colonic mucosa. Indian J Exp Biol 1997;35:1080–1083.
- Lee YY, Jang DS, Jin JL, Yun-Choi HS. Anti-platelet aggregating and anti-oxidative activities of 11-O-(4′-O-methylgalloyl)-bergenin, a new compound isolated from Crassula cv. ‘Himaturi’. Planta Med 2005;71:776–777.
- Jia Z, Mitsunaga K, Koike K, Ohmoto T. New bergenin derivatives from Ardisia crenata. Nat Med 1995;49:187–189.
- Fuji M, Miyaichi Y, Tomimori T. Studies on nepalese crude drugs. XXII on the phnolic constituents of the rhizome of Bergenia ciliata (Haw.) STERNB. Nat Med 1996;50:404–407.
- Saijo R, Nonaka G, Nishioka I. Gallic acid esters of bergenin and norbergenin from Mallotus japonicus. Phytochemistry 1990;29:267–270.
- Yoshida T, Seno K, Takama Y, Okuda T. Bergenin derivatives from Mallotus japonicus. Phytochemistry 1982;21:1180–1182.
- Girard N, Rousseau C, Martin OR. Intramolecular C-glycosylation of 2-O-benzylated pentenyl mannopyranosides: remarkable 1, 2-trans stereoselectivity. Tetrahedron Lett 2003;44:8971–8974.
- Wang D, Zhu HT, Zhang YJ, Yang CR. A carbon-carbon-coupled dimeric bergenin derivative biotransformed by Pleurotus ostreatus. Bioorg Med Chem Lett 2005;15:4073–4075.
- Pearl IA. Vanilic acid. Org Synth 1963;4:972–977.
- Li W, Wei K, Fu H, Koike K. Structure and absolute configuration of clerodane diterpene glycosides and a rearranged cadinane sesquiterpene glycoside from the stems of Tinospora sinensis. J Nat Prod 2007;70:1971–1976.
- Liang PH, Cheng WC, Lee YL, Yu HP, Wu YT, Lin YL et al. Novel five-membered iminocyclitol derivatives as selective and potent glycosidase inhibitors: new structures for antivirals and osteoarthritis. Chembiochem 2006;7:165–173.
- Adisakwattana S, Sookkongwaree K, Roengsumran S, Petsom A, Ngamrojnavanich N, Chavasiri W et al. Structure-activity relationships of trans-cinnamic acid derivatives on α-glucosidase inhibition. Bioorg Med Chem Lett 2004;14:2893–2896.
- Oki T, Matsui T, Osajima Y. Inhibitory effect of α-glucosidase inhibitors varies according to its origin. J Agric Food Chem 1999;47:550–553.