Abstract
Plant cytochrome P450 is a key enzyme responsible for the herbicide resistance but the molecular basis of the mechanism is unclear. To understand this, four typical plant P450s and a widely resistant herbicide chlortoluron were analysed by carrying out homology modelling, molecular docking, molecular dynamics simulations and binding free energy analysis. Our results demonstrate that: (i) the putative hydrophobic residues located in the F-helix and polar residues in I-helix are critical in the herbicide resistance; (ii) the binding mode analysis and binding free energy calculation indicate that the distance between catalytic site of chlortoluron and heme of P450, as well as the binding affinity are key elements affecting the resistance for plants. In conclusion, this work provides a new insight into the interactions of plant P450s with herbicide from a molecular level, offering valuable information for the future design of novel effective herbicides which also escape from the P450 metabolism.
Introduction
Herbicide application has been a prominent weed control measure in recent decades for the purpose of crop cultivation. However, the occurrence of herbicide-resistant weeds worldwide has become a severe problem, which could lead to higher food prices and more pollutionCitation1. For plant, herbicide resistance refers to the ability of a plant biotype to survive and reproduce under a normally lethal dose of herbicide, normally involving two primary mechanisms: (i) a mutation associated with target site of the herbicide (target-site resistance) and (ii) non-target-site resistance which normally involves the biochemical modification of the herbicide and/or the compartmentation of the herbicideCitation2. The non-target herbicide resistance comes out of the plant detoxification process which usually comprises a four-phase schema: conversion by enzymes; conjugation of a hydrophilic molecular to activated metabolites; transporting into the vacuole or extracellular; degradationCitation3.
It is known that cytochrome P450 monooxygenases can metabolize (either oxidize or reduce) a large number of structurally different endogenous and exogenous compoundsCitation4. Meanwhile in the conversion process of herbicide molecules through the oxidation and peroxidation reactions P450 play the major rolesCitation5–7. In plants, many subtypes of P450s have been cloned and most of them exhibited limited activity and specificity toward exogenous compoundsCitation8,Citation9. Recently, several lines of evidence have suggested that the development of herbicide resistance in plant is frequently associated with elevated levels of P450 activityCitation5. In phenylurea herbicide resistant populations of Phalaris minor in Asia, prosulfuron, diclofop and chlortoluron were converted by P450s into several metabolitesCitation10.
Recent advances made in the isolation and genetic manipulations of P450 enzymes have created new opportunities for their application in engineering herbicide tolerance. CYP76B1, one of the P450 genes, was identified for chlortoluron resistance, which was cloned from Jerusalem artichoke (Helianthus tuberosus)Citation11. It catalyzes double N-dealkylation reactions and can be strongly induced by many phenylurea herbicides, such as chlortoluronCitation12. In the transgenic soybean and tobacco, CYP71A10 has also been demonstrated with the enhanced tolerance to chlortoluron for these plantsCitation13. And the overexpression of CYP71B1 in yeast also showed resistance to chlortoluron metabolismCitation14. In wheat, the CYP71C6V1 was able to metabolize sulfonylurea herbicides such as triasulfuron, chlorsulfuronCitation15. However, the tolerance to the chlortoluron has not been reported to date. As compared among the four P450s, the metabolite of CYP76B1-dependent metabolism of chlortoluron is processed by N-demethylation, whereas ring-methyl group oxidation is performed in the other three P450s.
Presently, the molecular mechanism of herbicide resistance mediated by P450 in plants is still unclear. Thus a theoretical study integrating homology modelling, molecular docking, molecular dynamics, and binding free energy analysis was performed. Up to date, to our knowledge, the crystal structures for most plant P450s are unavailableCitation11,Citation16. The homology modelling could provide a suitable model for the unknown proteins, accelerating the understanding the structural basis of P450 recognition of substrates. The typical molecule chlortoluron was applied as a probe to investigate the features of P450 catalytic sites in plants. Molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method was adopted to calculate the ligand binding affinity. With the energy decomposition analysis, the key residues which are responsible for substrate specificity are uncovered. The results obtained in this study will be helpful for the further understanding of the substrate specificity and regioselectivity, as well as herbicide resistance-mediated by P450 enzymes in plants.
Materials and methods
Herbicide
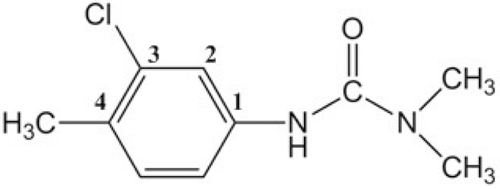
Chlortoluron (3-(3-chloro-4-methylphenyl)-1-1-dimethylurea) show in , as a member of the phenylureas family derivatives, is widely used for selective weed control in cereals cropsCitation14. In the present work, this herbicide was selected for the reason that the tolerance to the chlortoluron in plant is due to enhanced rates of metabolism catalyzed by P450s.
Figure 1. Chemical structure of the chlortoluron.
Homology modelling
For homology modelling, the modelling results based on the tertiary structures would be reasonable if the protein sequences are similar or have high homology in conservation regions between the target protein and the homology protein. The primary sequences of the four P450s (CYP71A10, CYP71B1, CYP71C7V1, CYP76B1) were obtained from the NCBI Web site (http://www.ncbi.nlm.nih.gov). The template proteins 2PG5 (resolution: 1.95 Å), 1PQ2 (resolution: 2.60 Å), 2F9Q (resolution: 3.00 Å), 3PM0 (resolution: 2.70 Å) identified by Blast program (http://blast.ncbi.nlm.nih.gov/Blast.cgi) were employed as the templates for CYP76B1, CYP71B1, CYP71A10 and CYP71C6V1, respectively. The multiple sequence alignments between the template and the four P450s were produced using Clustalx 1.83 programCitation17.
In present work, based on the alignment result, MODELLER 9V7Citation18 was used to build the initial structures of the four P450s. In construction of these backbone structures, the coordinates for heme were obtained from the template and inserted in the models. Then the covalent bond between the heme’s iron atom and the sulphur atom of conserved axial cysteine residue was createdCitation19. As plant P450s are membrane-bound proteinsCitation20, the N-terminal amino acids containing this membrane anchor were left out for the reason that no appropriate template for modelling the spanning segments was available and the region in the four P450s is not included in the active site.
Molecular docking
In order to drive a starting configuration for the molecular dynamics (MD) studies, the identification and selection of an initial receptor-ligand complex is necessary. Molecular docking was carried out by the AutoDock module to understand the detailed binding modes for the compound. AutoDock Tools (version 1.4.5) were used for protein and ligand preparation. Generally, polar hydrogen atoms were added and Kollman charges were assigned to all atoms of the proteins. The docking site was defined using AutoGrid 4, for ligand atoms, a 126 × 126 × 126 Å 3D grids centred on the binding site with 0.375 Å spacing were calculated. For the ligand, Gasteiger chargesCitation21 were added to the ligand, and nonpolar hydrogens were mergedCitation22. For docking simulations, the Lamarckian genetic algorithm (LGA) was selected for ligand conformational and all bond rotations for ligands were ignored. Finally, the docking results were further analysed as the initial structures in the MD simulations.
Molecular dynamics simulations
Molecular dynamics simulations were performed to examine the quality of the model structures by checking their stability over long simulations at room temperature. All the MD simulations were carried out using the AMBER 10 packageCitation23. Each initial structure for the four models with ligand was modelled using Xleap module. For MD simulations, four models were solvated with the TIP3P water moleculesCitation24 in a truncated octahedron periodic box with a margin of 10 Å along each dimension. Periodic boundary conditions were employed and Particle Mesh EwaldCitation25 was applied to calculate the long-range electrostatic interactions, then the cut-off distance was set to 10 Å to compute the nonbonded interactions. The SHAKE algorithmCitation26 was used to constrain all covalent bonds to hydrogen atoms.
Prior to the production phase, each system was gradually energy-minimized using 5000 steps of minimization procedure (2500 steepest descent steps and 2500 conjugate-gradient steps) to relax any steric conflicts generated. The force field energy of each starting structure was minimized in the following equilibration protocols. First, the solvent was relaxed by energy minimization while restraining the protein atomic positions with a harmonic potential. After the system was heated to 300 K from the initial temperature of 0 K using Langevin dynamics parametersCitation27, a 50 ps pressure-constant period to raise the density while still keeping the complex atoms constrained was followed. Finally, a 5 ns MD simulation with a 2 fs time step was conducted at 1 atm and 300 K with the NPT ensemble under the periodic boundary conditions for each system. During the simulations, the ff99SB all atom Amber force fieldCitation28 was used for proteins, and the force field parameters for the heme group were adopted from previously workCitation29,Citation30. The parameters of atom charges of the ligand were performed by general AMBER force field (GAFF). The optimized structures were subjected to quality assessment and used as the model for structure and binding mode analysis.
MM-PBSA calculation
In order to obtain a detailed view of the interaction between the protein residues and the substrates and identify certain key residues responsible for the substrate binding, the binding free energy was calculated using the MM-PBSA approachCitation31. We extracted the 500 snapshots from the last 1 ns of each system to estimate the binding free energies. The binding free energy was calculated by equation as follows:
Where ΔGcomplex, ΔGprotein and ΔGligand are the free energies of the complex, the protein and the ligand, respectively. Each of them is calculated by summing the total molecular mechanical energy ΔGMM, the solvation free energy (ΔGsol), and the entropy (TΔSsolute).
Since the calculation of the entropic contribution (−TΔSsolute) requires several approximations and provides only a rough estimate, especially in the case of a simulation in which only a small portion of the protein moves, the entropic contribution was not considered. The solvation free energy (ΔGsol) is divided into two contributions: the polar (ΔGsolpolar) and nonpolar (ΔGsolpolar) components. The ΔEMM is the molecular mechanics interaction energy between the protein and the ligand, the term ΔEMM contains the internal stain energy (Eint), van der Waals energy (vdWtot), and electrostatic energy (ELEtot). Eint is further divided into Ebond, Eangle and Etorsion to take into account energies associated with bonds, angles, and torsions. The formula is as follows:
The electrostatic component, Eele is calculated from Coulomb’s expression using a dielectric constant of 1Citation32 and van der Waal’s energy, EvdW is calculated from the Lennard-Jones 6-12 potential.
The ΔGsolv contains the non-polar solvation free energy and the electrostatic solvation free energy is computed with
The nonpolar contribution was determined based on solvent-accessible surface area determined with the LCPO methodCitation33. Where SASA is the solvent-probe radius of 1.4 Å, γ and β are empirical constants set to 0.0072 kcal·mol−1·Å−2 and 0.00 kcal·mol−1, respectively.
The electrostatic solvation free energy (ΔGpol) is the cost of charging the discharged solute in the cavity. Here, the polar contribution (ΔGpol) was calculated by solving the Poisson-Boltzmann (PB) equation using the program Delphi αCitation34. In Delphi calculations, the grid spacing was set to 0.5 Å, and the radii of atoms were taken from the PARSE parameter setCitation35. The values of the interior dielectric constant and the exterior dielectric constant were set to 1–80, respectively.
Results and discussion
Sequence alignment and homology modelling
To guide and examine the molecular basis of the resistance conferred by P450, the 3D structural homology models were constructed on the basis of structural alignment of amino acid residues. In homology modelling, selection of an appropriate template is an important step for constructing the target model. The template selection was performed on the basis of sequence similarly, residues completeness, and crystal resolution. For this reason, using more than one template therefore generates a set of models that hopefully provide a better representation of the target protein than if a single template was used. In the present work, all four sequences were individually aligned against with known three-dimensional structures CYP2A6 (PDB code (2PG5), CYP2C8 (1PQ2), CYP2D6 (2F9Q) and CYP1B1 (3PM0)). In each case, the amino acid sequence identity of CYP76B1 was 28% to 2A13; CYP71B1 was 28% to 2C8; CYP71A10 was 26% to 2A6 and 25% to 2D6 in CYP71C6V1, respectively. () The sequence alignment between the template and models were displayed in Figure S1Citation36 in the Supporting Information.
Table 1. The amino acid sequence identity.
In the case of generating plant P450 models, it is important to note that there is a ≤40% identity between a P450 in one family and that in another familyCitation37. Although the sequence identity is very low, their structures surrounding the buried catalytic site and contributing to the overall fold are highly conservedCitation38–40, indicating a common mechanism of oxygen activation. The conserved core is formed of a four-helix (D, E, I and L) bundle, helices J and K, two sets of β sheets (). These regions comprise: the heme-binding loop; the absolutely conserved motif in helix K and central part of I helixCitation41.
Figure 2. Ribbon schematic representations of the refined homology models of the four P450s. Heme is shown as red stick. Major helices are labelled. Image was generated using PyMOL (http://www.pymol.org).
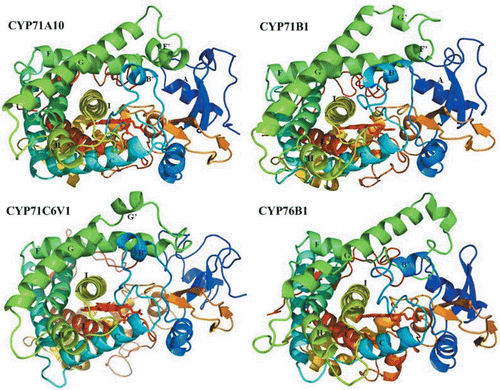
The heme coordinates which are considered important for the structural integrity, were copied from the template crystal structure with a covalent bond created between the heme’s iron atom and the sulphur of the conserved cysteine axial ligandCitation42. Then, the initial 3D structural models were energy-minimized to release the bad atomic contact and tune unreasonable local structural conformations. After that, the final models were assessed by ProcheckCitation43. Concerning the Procheck assessment, the percentage of the residues in core Ramachandran region are 89.5%, 85.0%,85.7% and 87.3% for CYP71A10, CYP71B1, CYP71C6V1 and CYP76B1, respectively and the template was also evaluated for comparison ().
Table 2. Sequence identity scores and Ramachandran plot statistics for the four P450 models presented. Scores indicate identity with amino acid sequence of the template.
The results of the Procheck indicate that approximately 87% of residues are located in the favoured or additional allowed regions of Ramachandran plot, only 1.4%, 1.7%, 1.2% and 1.0% of residues in each structure are in the disallowed regions. Residues located in the unfavourable regions are far from the substrate-binding domain, indicating that these residues did not affect the ligand-protein binding simulations. The final step of testing was the packing quality of each residue as evaluated by the Verify-3D method, which represents the profile achieved with respect to the residues. In summary, the results of quality assessment suggest that the model of the four plant P450s are of reasonable quality compared to the crystal structure of the templates.
Molecular docking analysis
As an efficient technique to predict the potential ligand binding site on the whole protein target, molecular docking is a valuable tool for studying substrate orientation within the P450 active siteCitation44. In order to select the suitable binding mode of chlortoluron in the four complexes, the structures were judged on the basis of their structural and chemical soundness, as well as their consistency between proteins. The lowest energy docking models were selected from the fine grid docking using several scoring functions for configuration analysis; for each receptor, the distance between the heme’s iron atom and 4′ carbon in chlortoluron moiety was measured. The selected results in each system were analysed on the basis of the conditions that the ligand has the closest distance between active site and the heme’s iron atom, and the favourable hydrophobic interactions in the catalytic site between the ligand and the side chain’s residues.
The docking results with free binding energy of the four modes were −6.4, −7.0, −6.9 and −6.5 kcal/mol for CYP71A10, CYP71B1, CYP71C6V1 and CYP76B1, respectively. The low distance corresponds to the highest electrophilic character of this positions was found in this study. Since docking studies do not consider protein backbone movement during the substrate-binding process, MD simulations are employed to determine the different conformation of enzymes acquired during their catalysis process. Binding modes with the lowest total energies and the methyl group of chlortoluron closet to the heme were chosen for second-round energy minimizations during which all protein side chains were allowed to move freely while the heme coordinates were fixed to avoid distortions of the heme plane.
Molecular dynamic simulation
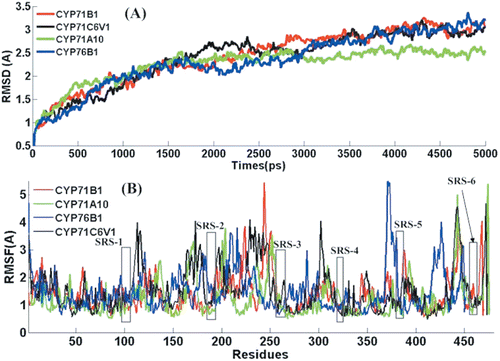
Molecular dynamic simulations have been widely used to obtain the ‘real’ bioactive conformation when the crystal structure of protein-ligand complex is unavailable. In the present work, this method was performed to further examine the quality of the model structures by checking their stability over long simulations at room temperature (300 K). The root-mean-square deviations (RMSDs) between the Cα atoms of the structures obtained during the trajectories and the initial structures are shown in for the four systems. The dynamic behaviour and the structural changes of the complexes in the course of the simulation period were analysed as follows.
Figure 3. RMSD (A) and RMSF (B) of Ca backbone as a function of time for CYP71A10-CT (green), the CYP71B1-CT (red), CYP71C6V1-CT (black) and CYP76B1-CT (blue).
Stability of the simulations
To evaluate the deviation of the trajectory from the initial structure, the RMSD was monitored along the trajectory. shows RMSD of Cα backbone with respect to the initial structure. The RMSD plot indicates that the RMSD of the backbone atoms in the four complexes increases sharply within 4000 ps and then remains stable to the end of simulation. The averaged RMSD values during the MD simulations are 2.42, 2.39, 2.25 and 2.33 for CYP71B1, CYP71C6V1, CYP71A10 and CYP76B1 respectively, which demonstrated that the generated MD trajectories of the four complexes are stable. In the four cases, the complex structures have reached a stable state after 4 ns equilibration, where the RMSD value converged to a value around 3.0 Å (CYP71B1 and CYP76B1, CYP71C6V1). For CYP71A10, the protein atoms do not undergo significant structural changes and the RMSD values of this system converge to about 2.5 Å, a relatively small deviation from the minimized structure. It is an obvious reflection of relatively weaker binding affinity that the three complexes show large conformation changes of protein than CYP71A10. From the structural and energetic analysis, we can see that the whole complexes did not significantly deviate from their starting structures after equilibrium from both the RMSD and binding energies during the molecular dynamics simulation. Therefore, our subsequent energy analyses in each case were based on the MD trajectory truncated between 4 and 5 ns.
Root mean square fluctuation (RMSF) of Ca atoms by residues
In order to locate the flexible regions, the RMSF was examined for the Cα atoms of each of the residue representing the average displacement of these atoms. We focused our analysis on those structure elements that are involved in the formation of the substrate binding cavity, because conformational changes in this region of the protein have been identified to change the shape of the active site containing cavity.
In a typical RMSF pattern, a low RMSF value indicates the well-structured regions while the high values indicate the loosely structured loop regions or domains terminalCitation45. As shown in , the major backbone fluctuation was seen to occur in the loop regions, whereas regions with low RMSF correspond exclusively to the rigid beta-alpha-beta fold. Our RMSFavg values for CYP71A10, CYP71B1, CYP71C6V1 and CYP76B1 are 1.24, 1.37, 1.54 and 1.49 Å, respectively. Detailed analysis of RMSF shows that large peaks are observed in the loop portions, especially those located on the protein surface. The relatively larger RMSF per residue of the four complexes may be explained by the relatively weaker binding of chlortoluron.
In contrast to the loops, the substrate binding residues located in the core part of the structure barely fluctuate in the whole simulations. The conserved regions remained stable over the entire simulation which seems to be more energetically preferred. During the dynamics simulation, very few fluctuations in the SRS regions go beyond 1.5 Å, indicating that the regions in the four complexes show a similar fluctuation during the MD simulation. The regions SRS1, SRS4, SRS5 and SRS6 indicated in their orientations relative to the heme catalytic centre shown the low fluctuation than the other SRS regions. In summary, the RMSF for the simulation point to that the surface loops are flexible and the core parts are rigid, the SRS structure itself seems to remain fairly unchanged, though the size and shape of the active site cavity changes in adapting to substrates.
Binding mode analysis
Chlortoluron is metabolized in plant via two major pathways of hydroxylation and N-dealkylation. The N-dealkylation of chlortoluron in 1-position is mediated by CYP76B1 in Jerusalem artichokeCitation46, whereas the hydroxylation is performed in the 4-position predominantly by CYP71A10, CYP71B1 and CYP71C6V1. The binding conformation of the four P450s with chlortoluron is showed in . In order to gain insights into mechanisms of the compound and identify key residues responsible for the binding of P450, the average structures were generated from the last 1 ns on the MD trajectory.
Figure 4. A close view of the binding mode of the four P450s with (A) CYP71A10, (B) CYP71B1, (C) CYP71C6V1 and (D) CYP76B1. Heme is represented by a red stick. Key residues are represented by stick, with red, gray, blue, yellow, and light gray representing oxygen, nitrogen, sulphur, and carbon, respectively. The hydrogen bonds are shown in green dotted lines and ligand with green stick.
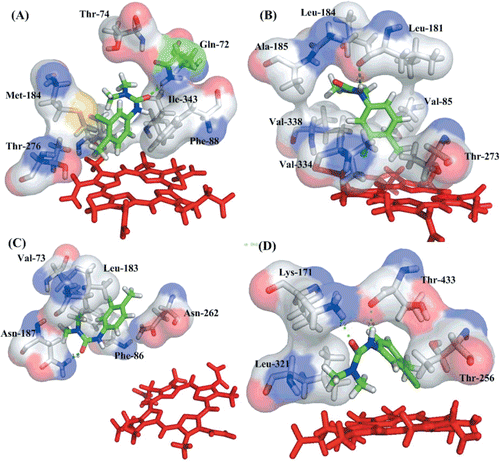
In general, chlortoluron is stabilized by hydrophobic interactions in the active site, which is constructed by hydrophobic and few polar residues. All the residues are close to each other in space and form the hydrophobic binding site as suggested by Gotoh based on a comparison between family 2 enzymesCitation47. The residues responsible for binding are located in six SRSs regions, especially the regions SRS-1 (B-C loop), SRS-2 (F-helix), SRS-4 (I-helix) and SRS-5 (the loop connects β3). As displayed, Leu187 in CYP71A10, Leu181 in CYP71B1 and leu183 in CYP71C6V1 located in SRS-2 show a highly conservation. Polar residues Thr276 in CYP71A10, Thr273 in CYP71B1, Glu262 in CYP71C6V1 and Thr256 in CYP76B1 in the SRS-4 region play a key role in orienting the ligand.
In addition, there are some differences in binding modes among the four complexes. The residues formed hydrogen bonds with the chlortoluron in the binding site are Gln72 in CYP71A10, Leu181 in CYP71B1, Asn187 in CYP71C6V1 and Lys171, Thr433 in CYP76B1. In CYP71A10, the benzene ring of chlortoluron interacts by π–π stacking interactions with aromatic amino acids Phe88. Two polar residues Gln72 and Thr276 in the binding pocket may dictate the position of the ligand. The distance between the methyl carbon atoms from the heme is considered as a key condition in the binding of chlortoluron to P450s. While the substrate reaches a stable state the distance are 5.3, 4.19, 12.70 and 5.22 Å for CYP71A10, CYP71B1, CYP71C6V1 and CYP76B1, respectively. The average binding free energy for these four complexes are −23.30, −21.40, −13.69 and −23.64 kcal/mol, respectively. It indicates that the binding mode of CYP76B1 is stronger than the others, and the CYP71C6V1 shows a weaker binding ability with the ligand. To CYP71B1, the distance is 4.19 Å, which demonstrate that the action could easily occur. For CYP71C6V1, the distance may be too far to allow the action to occur as proposed by previous studiesCitation48. For CYP76B1, there are two residues Lys171 and Thr433 forming hydrogen bond with chlortoluron in the binding site. The distance is 4.62 Å, indicated that the action is easy to take place in this position. The simulation based result is in a good agreement with the experiment data that chlortoluron is metabolized in N-dealkylation way in CYP76B1 in Jerusalem artichoke.
Binding free energy analysis
The binding free energies calculated using MM-PBSA method for the four complexes is summarized in . As seen from , the total binding energies for CYP71A10, CYP71B1, CYP71C6V1 and CYP76B1 are −23.30, −21.40, −13.69 and −23.64, respectively. These results suggested that the CYP76B1 was more stable than the other three complexes and experimental observations show that the resistance caused by CYP76B1 in plant is obvious than the othersCitation11. Lower value of ΔGbind for the CYP76B1 complex is in correspondence with an increase in the binding affinity.
Table 3. Interaction energies of four complexes with substrate (kcal/mol).
Further analysis suggests that, both the intermolecular van der Waals and electrostatics interactions are major contributions to the binding, whereas polar solvation polar interaction has a relatively small influence especially for binding. Nonpolar solvation terms, which correspond to the burial of SASA upon binding, contribute slightly favourably. The van der Waals interactions energies about the four models are −32.06, −33.86, −29.56 and −31.95 kcal/mol for CYP71A10, CYP71B1, CYP71C6V1 and CYP76B1, respectively. As compared with the four proteins, with the lowest total energy among the four complexes, the value of van der Waals contribution to CYP71C6V1 in binding affinity is higher than the others. And the distance between the iron atom and the methyl carbon atom in chlortoluron is the longest. All these results demonstrate that the resistance caused by CYP71C6V1 might be the lowest in the four complexes. The calculated electrostatics contributions to the binding free energies for the four enzymes are list in . As can be seen from the comparison, the electrostatic interaction in CYP76B1 was quite strong, which correspond with the binding mode analysis that polar residues in the catalytic site play a critical role in the substrate-receptor interaction.
Free energy decomposition
In order to detect certain key residues responsible for substrate binding, MM-GBSA was used to calculate the interaction energies between the substrate and the four complexes. The residues in the binding site identified by the total interaction energy between the ligand and individual are considered as the key residue. In general, the residues having interaction energy less than −1 kcal/mol with chlortoluron are considered to be important in substrate binding. lists the interaction energies contributions of certain structurally important residues for the protein-ligand interactions in the four cases.
Table 4. Energy contribution of key residues to the binding energies.
shows that the side chains of Gln72, Thr74, Phe88, Met184, Leu187, Gly272, Thr276, Leu339 and Ile343 residues have the largest contributions to the binding energy, suggesting that these residues play an important role in the binding interaction. By analysing the detailed interaction between these residues and substrate, we can see that the charged Gln72 can form stronger interactions with ligand. This indicates that polar interaction plays a crucial role during the binding of substrate. For this enzyme, five residues (Gln72, Phe88, Met184, Leu187 and Ile343) in CYP71A10, which interactions with chlortoluron (−3.132, −2.935, −1.589, −1.412 and −1.271) were determined to be important in distinguishing the binding modes. The residues considered to play a key role in the binding site in the three proteins are listed in the . Analysis of interactions shows that the total, vdW, and electrostatic energies are included. According to the individual energy components of the binding free energies, the primary favourable contributor to the ligand binding is the van der Waals interaction. The result is in line with the fact that the binding pocket is mainly composed of hydrophobic residues. Comparison of the four binding pocket show that most of these residues are located in the six SRSs regions, and the highly conserved regions include the important residues are SRS2, SRS4 and SRS5. The residues Met184, Leu187 in CYP71A10, Leu181, Leu184, and Ala185 in CYP71B1, Leu183 in CYP71C6V1, and Lys171 in CYP76B1 are located in SRS2 (F-helix). Residues Gly273 and Thr276 in CYP71A10, Ala269 and Thr273 in CYP71B1, Asn262 and Glu265 in CYP71C6V1, Thr256 in CYP76B1 are in the SRS4 (I-helix). Ile343 in CYP71A10, Val334 and Val338 in CYP71B1, Leu338 in CYP71C6V1, Leu321 in CYP76B1 are placed in SRS5 (the loop connect β3).
Comparison of different binding patterns and free energy decomposition shows that, several residues identified by binding mode have been shown to play a key role in the interaction of substrate by P450s. Special attention has been paid to those residues with their special role to binding free energies, such as Leu187 in CYP71A10, Leu184 in CYP71B1 and Leu183 in CYP71C6V1. In the opposite site of the ligand in the pocket, residues Thr276 in CYP71A10, Thr273 in CYP71B1, and Glu265 in CYP71C6V1 and Thr256 in CYP76B1 in SRS-4 play an important role in orienting the ligand at the active site by electrostatic interactions. The conserved hydrophobic residues located in SRS-2 and polar residues in SRS-4 provide van der Waals and electrostatic interactions in catalyzing the interaction between protein and the ligand. According to the results of the free energy decomposition analysis, it is implied that van der Waals interactions and hydrophobic effects provide a reasonable basis for understanding binding affinities. Seen from , the primary favourable contributor to the ligand binding is the van der Waals interaction. For CYP71A10, residue Phe88 shows a π–π stacking interaction with chlortoluron in the binding site, where the van der Waals interaction (−1.816 kcal/mol) is shown a major contribution. In CYP71B1, Leu181 and Leu184 are here found to have either highly favourable electrostatic or van der Waals interactions with the ligand, indicating their importance for tight binding and inhibition. To CYP71C6V1, there are only four residues (Phe86, Leu183, Val73, Asn62) in the binding site contribute energy lower than −1 kcal/mol, which is in accordance with the hypothesis that the action is difficult to occur. In CYP76B1, the ligand formed two hydrogen bonds with residues Lys171 and Thr433, which indicated that the ligand is anchored in the binding pocket via hydrogen bonds. For the polar residue Lys171, the electrostatic calculations provide perhaps the main contributions to the binding process. We put this hypothesis that the different binding pocket construction is the main reason for chlortoluron metabolized by CYP76B1 in N-dealkylation pathwayCitation49. According to the individual energy components of the binding free energies, we can see that the primary favourable contributor to the ligand binding is the van der Waals interaction. The hydrophobic interactions were formed between the residues and the ligand in the active site and specific residues form important hydrogen bonds. This indicates that polar interaction plays an important role during the binding of ligand.
In summary, the current results indicated that the hydrophobic residues located in the SRS2 and polar residues in SRS4 have been shown to affect substrate binding or specificity in P450s. SRS-2 located in F-helix is located on the opposite wall of the active site from the heme ring. SRS-4 (I-helix) protrudes through the whole enzyme and includes residues that are highly conserved among the CYP450 enzymes. Key amino acid residues in the P450 active sites are important contact points for the binding of substrates such that metabolism occurs in specific positions, being governed by the enzyme-mediated orientation of the substrate molecules relatives to the heme moiety. Taken together, the current results indicate the regions are important for ligand binding, and thus could further affect the metabolic activity of the four P450s.
Conclusion
In the present study, to elucidate the molecular mechanism of herbicide resistance between the plant P450s and chlortoluron, a series of models was conducted. For the first time, we provide four validated 3D models of CYP71A10, CYP71B1, CYP71C6V1 and CYP76B1, with the emphasis on the regions conserved in the ligand binding site that is crucial for the herbicide resistance.
On the basis of the calculated binding free energies, we are able to assess several key residues (Gln72 and Leu187 in CYP71A10, Leu184 in CYP71B1, Asn187 and Leu183 in CYP71C6V1, Lys171 and Thr433 in CYP76B1) responsible for the substrate specificity. Hydrophobic residues located in SRS-2 and polar residues in SRS-4 were identified to be important in the substrate binding. In addition, the ligand site and pose is considered to be orientated by these two regions. The analysis of individual energy terms demonstrates that both the van der Waals and electrostatic contribution are essential for the chlortoluron binding. It is observed that the van der Waals contribution is more crucial for distinguishing the binding affinities among these four complexes. The identification of the molecular mechanism for herbicides resistance provides valuable information to understand the herbicide resistance to plant P450s and thus aid in the design of new potential herbicides.
Declaration of interest
This work is financially supported by State Key Laboratory of the Discovery and Development of Novel Pesticide (No. 201101). The research is also supported by high-performance computing platform of Northwest A & F University.
References
- Dill GM. Glyphosate-resistant crops: history, status and future. Pest Manag Sci 2005;61:219–224.
- Yuan JS, Tranel PJ, Stewart CN Jr. Non-target-site herbicide resistance: a family business. Trends Plant Sci 2007;12:6–13.
- Heinrich S Jr.. Molecular ecotoxicology of plants. Trends Plant Sci 2004;9:406–413.
- de Groot MJ. Designing better drugs: predicting cytochrome P450 metabolism. Drug Discov Today 2006;11:601–606.
- Werck-Reichhart D, Hehn A, Didierjean L. Cytochromes P450 for engineering herbicide tolerance. Trends Plant Sci 2000;5:116–123.
- Schuler MA, Werck-Reichhart D. Functional genomics of P450s. Annu Rev Plant Biol 2003;54:629–667.
- Yun MS, Yogo Y, Miura R, Yamasue Y, Fischer AJ. Cytochrome P-450 monooxygenase activity in herbicide-resistant and -susceptible late watergrass (Echinochloa phyllopogon). Pestic Biochem Phys 2005;83:107–114.
- Holt JS, Powles SB, Holtum JAM. Mechanisms and agronomic aspects of herbicide resistance. Plant Mol Biol 1993;44:203–229.
- De Prado RA, Franco AR. Cross-resistance and herbicide metabolism in grass weeds in Europe: biochemical and physiological aspects. Weed Sci 2004;52:441–447.
- Kemp Malcolm S, Moss Stephen R, Thomas Tudor H. Herbicide resistance in Alopecurus myosuroides. In: Managing Resistance to Agrochemicals. American Chemical Society 1990;421:376–393.
- Robineau T, Batard Y, Nedelkina S, Cabello-Hurtado F, LeRet M, Sorokine O et al. The chemically inducible plant cytochrome P450 CYP76B1 actively mebalolizes phenylureas and other xenobiontics. Plant Physiol 1998;118:1049–1056.
- Didierjean L, Gondet L, Perkins R, Lau SM, Schaller H, O’Keefe DP et al. Engineering herbicide metabolism in tobacco and Arabidopsis with CYP76B1, a cytochrome P450 enzyme from Jerusalem artichoke. Plant Physiol 2002;130:179–189.
- Siminszky B, Corbin FT, Ward ER, Fleischmann TJ, Dewey RE. Expression of a soybean cytochrome P450 monooxygenase cDNA in yeast and tobacco enhances the metabolism of phenylurea herbicides. Proc Natl Acad Sci USA 1999;96:1750–1755.
- Lamb DC, Kelly DE, Hanley SZ, Mehmood Z, Kelly SL. Glyphosate is an inhibitor of plant cytochrome P450: functional expression of Thlaspi arvensae cytochrome P45071B1/reductase fusion protein in Escherichia coli. Biochem Biophys Res Commun 1998;244:110–114.
- Xiang W, Wang X, Ren T. Expression of a wheat cytochrome P450 monooxygenase cDNA in yeast catalyzes the metabolism of sulfonylurea herbicides. Pestic Biochem Phys 2006;85:1–6.
- Lamb SB, Lamb DC, Kelly SL, Stuckey DC. Cytochrome P450 immobilisation as a route to bioremediation/biocatalysis. FEBS Lett 1998;431:343–346.
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 1997;25:4876–4882.
- Andrej A. Comparative protein modeling by satisfaction of spatial restraints. Mol Med Today 1995;1:270–277.
- Meng XY, Zheng QC, Zhang HX. A comparative analysis of binding sites between mouse CYP2C38 and CYP2C39 based on homology modeling, molecular dynamics simulation and docking studies. Biochim Biophys Acta 2009;1794:1066–1072.
- Bolwell GP, Bozak K, Zimmerlin A. Plant cytochrome P450. Phytochemistry 1994;37:1491–1506.
- Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 1980;36:3219–3228.
- Medina-Franco JL, López-Vallejo F, Kuck D, Lyko F. Natural products as DNA methyltransferase inhibitors: a computer-aided discovery approach. Mol Divers 2011;15:293–304.
- Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, et al. AMBER 10 University of California, San Francisco 2008.
- Jorgensen WL. Revised TIPS for simulations of liquid water and aqueous solutions. J Chem Phys 1982;77:4156–4163.
- Darden T, York D, Pedersen L. Particle mesh Ewald: An N [center-dot] log(N) method for Ewald sums in large systems. J Chem Phys 1993;98:10089–10092.
- Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 1977;23:327–341.
- Brünger A, Brooks Iii CL, Karplus M. Stochastic boundary conditions for molecular dynamics simulations of ST2 water. Chem Phys Lett 1984;105:495–500.
- Lee MC, Duan Y. Distinguish protein decoys by using a scoring function based on a new AMBER force field, short molecular dynamics simulations, and the generalized born solvent model. Proteins 2004;55:620–634.
- Harris DL, Park JY, Gruenke L, Waskell L. Theoretical study of the ligand-CYP2B4 complexes: effect of structure on binding free energies and heme spin state. Proteins 2004;55:895–914.
- Li W, Ode H, Hoshino T, Liu H, Tang Y, Jiang H. Reduced catalytic activity of P450 2A6 mutants with coumarin: a computational investigation. J Chem Theory Comput 2009;5:1411–1420.
- Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 2000;33:889–897.
- Hou T, Wang J, Li Y, Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 2011;51:69–82.
- Weiser J, Shenkin PS, Still WC. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J Comput Chem 1999;20:217–230.
- Gilson MK, Sharp KA, Honig BH. Calculating the electrostatic potential of molecules in solution: Method and error assessment. J Comput Chem 1988;9:327–335.
- Sitkoff D, Sharp KA, Honig B. Accurate calculation of hydration free energies using macroscopic solvent models. J Phys Chem 1994;98:1978–1988.
- Yano JK, Hsu MH, Griffin KJ, Stout CD, Johnson EF. Structures of human microsomal cytochrome P450 2A6 complexed with coumarin and methoxsalen. Nat Struct Mol Biol 2005;12:822–823.
- Shimazu S, Inui H, Ohkawa H. Phytomonitoring and phytoremediation of agrochemicals and related compounds based on recombinant cytochrome P450s and aryl hydrocarbon receptors (AhRs). J Agric Food Chem 2011;59:2870–2875.
- Graham SE, Peterson JA. How similar are P450s and what can their differences teach us? Arch Biochem Biophys 1999;369:24–29.
- Poulos T, Johnson E. (2005). Cytochrome P450: Structure, Mechanism, and Biochemistry. In: Ortiz de Montellano PR, ed. New York: Springer US, 87–114.
- CDS. Cytochrome P450 Conformational Diversity. Structure 2004;12:1921–1922.
- Werck-Reichhart D, Feyereisen R. Cytochromes P450: a success story. Genome Biol 2000;1:REVIEWS3003.
- Jensen K, Osmani SA, Hamann T, Naur P, Møller BL. Homology modeling of the three membrane proteins of the dhurrin metabolon: catalytic sites, membrane surface association and protein-protein interactions. Phytochemistry 2011;72:2113–2123.
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 1993;26:283–291.
- Schoch GA, Attias R, Le Ret M, Werck-Reichhart D. Key substrate recognition residues in the active site of a plant cytochrome P450, CYP73A1. Homology guided site-directed mutagenesis. Eur J Biochem 2003;270:3684–3695.
- Saladino AC, Xu Y, Tang P. Homology modeling and molecular dynamics simulations of transmembrane domain structure of human neuronal nicotinic acetylcholine receptor. Biophys J 2005;88:1009–1017.
- Robineau T, Batard Y, Nedelkina S, Cabello-Hurtado F, LeRet M, Sorokine Oet al. The chemically inducible plant cytochrome P450 CYP76B1 actively metabolizes phenylureas and other xenobiotics. American Society of Plant Physiologists.
- Gotoh O. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J Biol Chem 1992;267:83–90.
- Wang H, Cheng JD, Montgomery D, Cheng KC. Evaluation of the binding orientations of testosterone in the active site of homology models for CYP2C11 and CYP2C13. Biochem Pharmacol 2009;78:406–413.
- Werck-Reichhart D, Hehn A, Didierjean L. Cytochromes P450 for engineering herbicide tolerance. Trends Plant Sci 2000;5:1360–1385.