Abstract
A convenient protocol for the multicomponent reaction (MCRs) between malononitrile with an orthoester and hydrazine derivatives, under acid catalyst is described. A series of aminocyanopyrazoles 4 was prepared, isolated and characterized. These pyrazoles reacted with sodium nitrite followed by secondary amine reagent and with formic acid to lead pyrazolotriazines 6 and pyrazolopyrimidinones 7. Some of the aminopyrazoles were converted to the corresponding sulfamides by reaction with sulfamoyl chloride. The aminopyrazoles incorporating phenyl and tosyl moieties were tested as inhibitors of four carbonic anhydrase (CA, EC 4.2.1.1) isoforms, the human (h) hCA I, II, IX and XII. Many of them showed low micromolar or submicromolar inhibition of these enzymes. The corresponding sulfamides were low nanomolar CA inhibitors.
Introduction
Due to the convenience and high degree of atom economy, multicomponent reactions (MCRs) have become one of the most efficient tools for rapid scaffold construction and introduction of molecular diversityCitation1,Citation2. MCRs are important synthetic tools that can be useful in the preparation of biologically active compoundsCitation3. In our search for operationally simple, resource and cost-effective processes, we have been investigating pyrazole MCRs. Their classical syntheses in two steps were well described in the literatureCitation4–6. In fact, pyrazoles constitute an important family of compoundsCitation7,Citation8 due to their applications as antiviralCitation9, as xanthine oxidase inhibitorsCitation10, as dual MAO-B inhibitors and anti-inflammatory analgesicsCitation11 and have considerable chemical and pharmacological importanceCitation12–15. In this work, our objective is to achieve valuable MCR aminocyanopyrazoles and to study their application by diazotation in hydrochloric acid and by action of formic acid.
Results and discussion
Synthesis of 5-amino-4-cyano-1-substituted pyrazoles 4
The first step of our strategy was the formation of MCR 5-amino-4-cyano-1- substituted pyrazoles 4. Several works mention the synthesis of the 5-amino-4-cyano pyrazolesCitation4–6 prepared via a standard addition of hydrazine derivatives to ketene ethoxymethylene compounds. However, we were delighted to observe the sole formation of pyrazoles 4 when introducing equivalent amounts of malononitrile 1 with orthoester 2 and hydrazine derivatives 3 in the presence of few drops of acetic acid in the ethanol heated under reflux overnight. This procedure gave satisfying results: phenyl hydrazines afforded the pyrazole in good yields ().
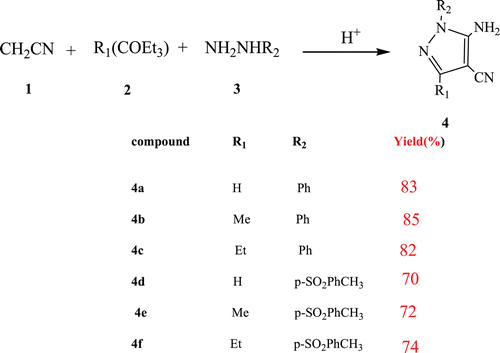
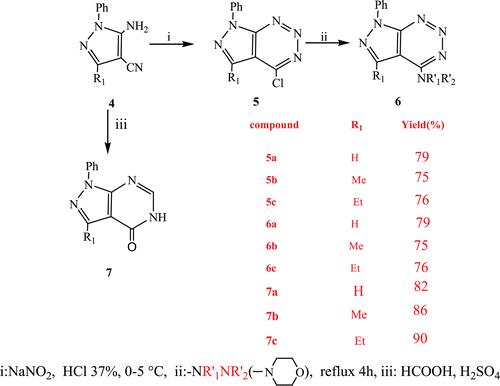
Synthesis of pyrazolotriazines 6 and pyrazolopyrimidinones 7
Taking advantage of the presence of the two functional groups in 5-amino-4-cyano-1-substituted pyrazoles 4, we prepared the 4-chlorotriazine by diazotation in hydrochloric acidCitation16–20. In the literature amino cyano pyrazoles undergo diazotization reaction with NaNO2 in a mixture of HCl/AcOH and leads to the pyrazolotriazin-4-onesCitation21. To the best of our knowledge 5-substituted chloropyrazolotriazines 5a–c are not described in the literature. The chlorine atom was easily substituted by a secondary amine to afford compounds 6a–c. We note that the mass spectra of triazines 5 and 6 showed a molecular mass decrease by the mass of two nitrogen atoms. This is has been mentioned in literatureCitation22.
The reaction of formic acid in the presence of H2SO4 on anthranilonitrile and analogs is well described in the literatureCitation23–27. We used classical conditions to synthesize compound 7. 5-amino-4-cyano-1-substituted pyrazoles 4 was refluxed with formic acid in the presence of H2SO4 under stirring providing pyrazolopyrimidinones 7 from good to very good yields (). This protocol allows to generalize the synthesis of pyrazolo pyrimidinones and increase in some cases the yield of the reaction. The structures of compounds 7 were determined by IR, 1H, 13C NMR spectra, mass spectroscopy and elemental analysis. These data spectrum demonstrate in particularly the disparities of CN group in the aminocyanopyrazoles.
Some of the amino-containing pyrazoles investigated here were converted to the corresponding sulfamides by reaction with sulfamoyl chloride. It is in fact well know that the sulfamide function is a good zinc-binding group for generating potent CA inhibitorsCitation28,Citation29. Several such compounds, of type 8a-8c were prepared in this way.
Carbonic anhydrase inhibitory action
Compounds 4a–f and 8a–c have been tested as inhibitors of four carbonic anhydrase (CA, EC 4.2.1.1) isoforms, the human (h) hCA I, II, IX and XII (). The inhibition/activation of CAs are well understood processes, with most classes of inhibitors binding to the metal centerCitation28–34, whereas activators bind at the entrance of the active site cavity and participate in proton shuttling processes between the metal ion – bound water molecule and the environmentCitation35. It should be mentioned that recently other inhibition mechanisms than the binding to the metal center were reported for α-CAs, which do not directly involve the metal ion from the enzyme active site. For example polyamines bind to the enzyme by anchoring to the zinc-coordinated water/hydroxide ionCitation36, whereas coumarins act as prodrugs and bind at the entrance of the active site cavity, rather far away from the metal ionCitation37–39. The compounds 4 reported here contain an amino moiety attached to the pyrazole ring which may bind to the enzyme similar to the polyamine spermine, which is anchored by means of one of its primary amino moieties to the zinc-coordinated water moleculeCitation36. Furthermore, some secondary/tertiary sulfonamides as those present in derivatives 4d–f, were recently shown to be inhibitor, probably binding at the entrance of the cavity, in the coumarin-binding siteCitation40.
Table 1. hCA I, II, IX and XII I nhibition data with compounds 1a–1c and 4.
Data of show that compounds 4 act as medium potency, micromolar or submicromolar inhibitors of the four investigated CA isoforms. Against the cytosolic hCA I they showed activity with inhibition constants in the range of 3.1–12.6 µMCitation41–45. Sulfonamides 4d–f were more active than the corresponding phenyl derivatives 4a–c. The second cytosolic isoform, hCA II, the physiologically dominant one, was better inhibited by compounds 4 compared to hCA I, with KIs in the range of 0.79–7.14 µM. The structure activity relationship (SAR) was also similar for the two isoforms.
The tumor-associated, transmembrane isoforms hCA IX and XII were better inhibited by compounds 4 compared to the cytosolic ones mentioned above. Thus, against hCA IX, compounds 4 showed inhibition in the range of 0.27–1.24 µM, with the phenyl derivatives more active than the sulfonamides in this case. For hCA XII the inhibition constants were in the range of 0.15–3.12 µM and the SAR more complicated as the best inhibitor was the sulfonamide 4f, but the phenyl derivatives 4a–c also showed quite significant inhibition ().
As these compounds may not bind to the Zn(II) ion from the CA active site, we hypothesize that they inhibit the enzyme either binding similar to spermine (anchoring to the zinc-coordinated water molecule) or as the coumarins, at the entrance of the active site cavity. This hypothesis should be verified by solving the X-ray crystal structure of adducts of these compounds with some CA isoformsCitation46.
The sulfamides 8 were much more potent inhibitors of all CA isoforms compared to the previously mentioned compounds, being low nanomolar inhibitors of all four CA isoforms investigated here (). This is probably due to their direct binding to the metal ion within the enzyme active siteCitation28.
In conclusion, we report a novel multicomponent reaction of malononitrile with orthoesters and hydrazine derivatives that leads to the aminocyanopyrazoles 4 with good yields. These pyrazoles are open for further transformation due to an already existing amino and cyano groups in their structures. These pyrazoles react with sodium nitrite followed by secondary amine reagent and with formic acid to lead respectively to pyrazolotriazines 6 and pyrazolopyrimidinones 7. This new protocol was employed for the rapid synthesis of compounds 5 and 6 and for the amelioration of yield of compounds 7. Many of therse compounds showed interesting CA inhibitory activity.
Materials and methods
Anhydrous solvents and all reagents were purchased from Sigma-Aldrich, Alfa Aesar and TCI. All reactions involving air- or moisture-sensitive compounds were performed under a nitrogen atmosphere using dried glassware and syringes techniques to transfer solutions. IR spectra were determined in KBr on a JASCO FT-IR-420 spectrometer which precision is of 2 cm−1 covering field 400–4000 cm−1. The spectra of NMR 1H and NMR 13C were recorded on an AC Bruker 250 MHz or Bruker Advance III 400 MHz spectrometers in CDCl3 or DMSO-d6 and the chemical shifts are expressed in ppm. The multiplicities of the signals are indicated by the following abbreviations: s: singlet, d: doublet, t: triplet, q: quadruplet, m: multiplet, brs: broad singlet and the coupling constants are expressed in Hz. The assignment of exchangeable protons (OH and NH) was confirmed by the addition of D2O. The melting points were determined in Electrothermal 9100 apparatus and are uncorrected. The reactions were monitored by thin layer chromatography (TLC) using aluminium sheets with silica gel 60 F254 (230–400 mesh ASTM) as the stationary phase and ethylacetate/n-hexane or MeOH/DCM were used as eluants. The mass spectrometer was operated in EI mode at 70 eV and MS spectra were recorded from m/z 50 to 650.
General procedure for synthesis of aminocyanopyrazoles (4)
A solution of malononitrile 1 (33 mmol), orthoester 2 (34 mmol), hydrazine (33 mmol) 3 and few drops of acetic acid in ethanol (30 mL) was heated under reflux overnight. The product, which precipitates, was filtered and recrystallized from ethanol.
5-Amino-1-phenyl-1H-pyrazolo-4-carbonitrile (4a)
Yield 83%, white solid; mp = 166°C (methanol); IR (cm−1): 1594, 1640, 2217 (CN), 2917, 3432, 3221, 1H NMR (δ ppm, DMSO): 6.82 (2H, s, NH2), 7.40–7.55 (6H, m), 13C NMR (δ ppm, DMSO): 144.60 (C-3), 73.48 (C-4), 152.74 (C-5), 114.17 (CN), 117.08–137.52 (C-arom).
5-Amino-3-methyl-1-phenyl-1H-pyrazolo-4-carbonitrile (4b)
Yield 85%, white solid; mp = 133°C (methanol); IR (cm−1) 1598, 1645, 2215 (CN), 3331, 3227, 1H NMR (δ ppm, DMSO) 2.15 (3H, s, CH3), 6.66 (2H, s, NH2), 7.35–7.52 (5H, m);13C NMR (δ ppm, DMSO) 11.28 (CH3), 74.23 (C-4), 115.57 (CN), 124.40–138.03 (C-arom), 150.56 (C-5), 151.96 (C-3); MS m = e/z 199 [M + 1]+, 100%.
5-Amino-3-ethyl-1-phenyl-1H-pyrazolo-4-carbonitrile (4c)
Yield 82%, white solid; mp = 137°C (methanol); IR (cm−1) 1597, 1647, 2209 (CN), 3433, 3296; 1H NMR (δ ppm, DMSO) 1.18 (3H, t, J = 7.1 Hz, CH3), 2.53 (2H, q, J = 7.1 Hz, CH2), 6.61 (2H, s, NH2), 7.35–7.53 (5H, m); 13C NMR (δ ppm, DMSO) 12.88 (CH3), 21.13 (CH2), 73.22 (C-4), 115.47 (CN), 124.43–138.11 (C-arom), 152.13 (C-3), 155.58 (C-5); MS: m = e/z 213 [M + 1]+, 100%, 158 [M-54]+, 5%.
5-Amino-1-tosyl-1H-pyrazolo-4-carbonitrile (4d)
Yield 70%, white solid; mp = 192°C (methanol); IR (cm−1) 1527, 1616, 2211 (CN), 3347, 3225; 1H NMR (δ ppm, DMSO) 2.27 (3H, s, CH3), 7.11 (2H, s, NH2), 7.13–7.51 (5H, m); 13C NMR (δ ppm, DMSO) 21.28 (CH3), 75.51 (C-4), 113.81 (CN), 138.58 (C-3), 145.55 (C-5), 124.66–129.07 (C-arom).
5-Amino-3-methyl-1-tosyl-1H-pyrazolo-4-carbonitrile (4e)
Yield 72%, white solid; mp = 185°C (methanol); IR (cm−1) 1534, 1628, 2216 (CN), 3381, 3227; 1H NMR (δ ppm, DMSO) 2.04 (3H, t, CH3), 2.39 (3H, s CH3), 7.63 (2H, s, NH2), 7.47–7.87 (4H, m); 13C NMR (δ ppm, DMSO) 13.06 (CH3), 21.53 (CH3), 73.97 (C-4), 113.71 (CN), 127.89–133.44 (C-arom), 146.80 (C-3), 154.87 (C-5).
5-Amino-3-ethyl-1-tosyl-1H-pyrazolo-4-carbonitrile (4f)
Yield 74%, white solid; mp = 178°C (methanol); IR (cm−1) 1526, 1643, 2215 (CN), 3346, 3222; 1H NMR (δ ppm, DMSO) 0.87 (3H, t, J = 7.0 Hz, CH3), 2.16 (2H, q, J = 7.0 Hz, CH2), 2.38 (3H, s, CH3), 7.38 (2H, s, NH2) 7.38–7.68 (5H, m); 13C NMR (δ ppm, DMSO) 10.81 (CH3), 21.52 (CH2), 23.30 (CH3), 73.50 (C-4), 114.32 (CN), 128.20–136.42 (C-arom), 143.49 (C-3), 144.26 (C-5).
General procedure for synthesis of chloropyrazolotriazines (5)
A solution of sodium nitrite (11.4 mmol) in water (7 mL) was added over 15 min to a suspension of the foregoing 5-amino-4-cyano-1-substituted pyrazole (8.1 mmol) at 0-5°C in concentrated hydrochloric acid (16 mL). The resulting mixture was stirred at 0°C for a further 40 min and then allowed to stand at room temperature overnight. The reaction mixture was quenched in water (100 mL). The precipitate was washed twice with 15 mL of water, and dried under room temperature, recrystallized in methanol.
4-Chloro-7-phenyl-7H-pyrazolo[3,4-d][1,2,3]triazine(5a)
Yield 79%; mp = 170°C [methanol]; IR (cm−1) 1590 (C=N); NMR 1H (δ ppm, DMSO) 7.75 (1H, s, H5), 7.23 ppm (5H, s); 13C NMR (δ ppm, DMSO) 93.01 (C-4a), 131.15 (C-7a), 136.63 (C-5), 159.87 (C-4), 124.54–129.33 (C-arom); GC-MS: m/z (%) 204 (100), 154 (24), 141 (17); C10H6ClN5 Calculated (%): C 51.85, H 2.61, N 30.23, Found (%) C 51.79, H 2.60, N 30.25.
4-Chloro-5-methyl-7-phenyl-7H-pyrazolo[3,4-d][1,2,3]triazine(5b)
Yield 75% mp = 159°C [methanol]; IR (cm−1) 1586 (C=N); 1H NMR (δ ppm, DMSO) 2.38 (3H, s, CH3), 7.26–7.64 ppm (5H, m); 13C NMR (δ ppm, DMSO) 13.52 (CH3), 94.54 (C-4a), 112.26 (C-7a), 137.38 (C-5), 153.08 (C-4), 120.01-133.11 (C-arom); GC-MS: m/z (%)218 (100), 168 (36), 155 (14); C11H8ClN5 Calculated (%): C 53.78, H 3.28, N 28.51, Found (%) C 53.79, H 3.30, N 28.53.
4-Chloro-5-ethyl-7-phenyl-7H-pyrazolo[3,4-d][1,2,3]triazine(5c)
Yield 76%; mp = 179°C [methanol]; IR (cm−1) 1587 (C=N); NMR 1H (δ ppm, DMSO) 1.35 (3H, t, J = 7.1 Hz, CH3), 2.80 (2H, q, J = 7.1 Hz, CH2), 7.51 ppm (5H, s); 13C NMR (δ ppm, DMSO) 12.74 (CH3), 21.52 (CH2), 93.13 (C-4a), 112.01 (C-7a), 137.03 (C-5) 157.91 (C-4), 125.04-132.89 (C-arom), GC-MS: m/z (%) = 232 (100), 204 (43), 182 (51), 169 (15), 142 (30), C12H10ClN5 Calculated (%): C 55.50, H 3.88, N 26.97, Found (%) C 55.48, H 3.89, N 26.96.
General procedure for synthesis of aminopyrazolotriazines (6)
The chloropyrazolotriazine 5a–c (1 mmol) and corresponding amine (10 mL) were refluxed for 6 h. The mixture was cooled at room temperature. When a precipitate was formed, it was filtered, washed twice with 15 mL of water and twice with 8 mL of diethyl ether, and dried at room temperature overnight. The products were recrystallised in methanol to give products 6.
4-Morpholino-7-phenyl-7H-pyrazolo[3,4-d][1,2,3]triazine (6a)
Yield 79% mp = 170°C [methanol]; IR (cm−1) 1556 (C=N), 1453 (C=N), 1422 (N=N); NMR 1H (δ ppm, DMSO) 3.15 (4H, t, J = 7.1 Hz, CH2), 3.64 (4H, t, J = 7.1 Hz, CH2), 7.34-7.68 ppm (5H, m), 7.75 (1H, s, H5); 13C NMR (δ ppm, DMSO) 51.10 (CH2), 68.12 (CH2), 115.15 (C-7a), 93.01 (C-4a), 156.63 (C-5), 159.87 (C-4), 124.54 (C-arom); GC-MS m/z (%) 255 (100), 197 (23), 119 (66), C14H14N6O Calculated (%): C 59.50, H 5.00, N 29.77, O 5.67 Found (%) C 59.52, H 5.03, N 29.78, O 5.65.
5-Methyl-4-morpholino-7-phenyl-7H-pyrazolo[3,4-d][1,2,3]triazine (6b)
Yield: 75%. mp = 159°C [methanol]. IR (cm−1): 1560 (C=N), 1467 (C=N), 1431 (N=N); NMR 1H (δ ppm, DMSO): 2.28 (3H, s, CH3), 3.17 (4H, t, J = 7.1 Hz, CH2), 3.66 (4H, t, J = 7.1 Hz, CH2); 7.35-7.62 ppm (5H, m); 13C NMR (δ ppm, DMSO) 13.31 (CH3), 50.53 (CH2), 66.63 (CH2), 82.18 (C-4a), 115.39 (C-7a), 152.41 (C-5), 153.80 (C-4), 124.17–139.23 (C-arom); GC-MS m/z (%) 269 (100), 211 (53), 133 (22), C15H16N6O: Calculated(%): C 60.80, H 5.44, N 28.36, O 5.40 Found (%): C 60.82, H 5.45, N 28.34, O 5.42.
5-Ethyl-4-morpholino-7-phenyl-7H-pyrazolo[3,4-d][1,2,3]triazine (6c)
Yield 76%; mp = 170°C [methanol]; IR (cm−1) 1590 (C=N), 1526 (C=N), 1460 (N=N); NMR 1H (δ ppm, DMSO) 1.30 (3H, t, J = 7.1 Hz, CH3), 2.72 (2H, q, J = 7.3 Hz, CH2), 3.18 (4H, t, J = 7.3 Hz, CH2), 3.66 (4H, t, J = 7.3 Hz, CH2), 7.33-7.63 ppm (5H, m); 13C NMR (δ ppm, DMSO) 12.86 (CH3), 21.28 (CH2), 50.18 (CH2), 66.32 (CH2), 80.71 (C-4a), 115.12 (C-7a), 153.52 (C-5), 157.26 (C-4), 123.85–138.89 (C-arom); GC-MS: m/z(%) 283 (100), 213 (25), 147 (36). C16H18N6O: Calculated (%): C 61.92, H 5.85, N 27.08, O 5.16 Found (%): C 61.90, H 5.83, N 27.10, O 5.17.
General procedure for synthesis of pyrazolopyrimidinones (7)
5-amino-4-cyano-1-substituted pyrazole 4 (10 mmol) was added portionwise over 1 h to a mildly refluxing formic acid solution (20 mL) containing concentrated sulphuric acid (1.2 mL). After an additional 30 min, the solution was cooled to 0°C and poured on crushed ice. The resulting precipitate was collected by filtration, washed with water, and dried to give pyrazolopyrimidinones 7.
1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (7a)
Yield 82% mp = 164°C [methanol]; IR (cm−1) 1663 (C = O), 1583 (C=N), 1545 (C=N), 1500 (C=C); NMR 1H (δ ppm, DMSO) 7.34-7.65 (5H, m);7.72 (1H, s, H3); 7.75 (1H, s, H6); 8.01 ppm (1H, s, NH); 13C NMR (δ ppm, DMSO): 93.01 (C-3a), 131.15 (C-7a), 136.63 (C-3), 142.00 (C-6), 159.87 (C-4), 124.54–129.33 (C-arom); GC-MS: m/z (%) 213 (100), 168 (28), 102 (58); C11H8N4O: Calculated (%): C 62.26, H 3.80, N 26.40, O 7.54 Found (%): C 62.28, H 3.81, N 26.42, O 7.55.
3-Methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (7b)
Yield: 86%. mp = 159°C [methanol]. IR (cm−1): 1665 (C=O); 1580 (C=N); 1541 (C=N); 1506 (C=C); NMR 1H (δ ppm, DMSO): 2.25 (3H, s, CH3), 7.34-7.68 (5H, m), 7.72 (1H, s, H6); 8.00 ppm (1H, s, NH); 13C NMR (δ ppm, DMSO) 13.76 (CH3), 93.00 (C-3a), 135.55 (C-3), 135.55 (C-7a), 139.76 (C-6), 160.30 (C-4), 124.54–129.33 (C-arom); GC-MS: m/z (%) 227 (100), 200 (36), 182 (12), 168 (50), 131 (33), 116 (44), 104 (20).
3-Ethyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (7c)
Yield 90%; mp = 168°C [methanol]; IR (cm−1) 1665 (C = O), 1580 (C = N), 1541 (C = N), 1506 (C=C); NMR 1H (δ ppm, DMSO) 3.15 (3H, t, J = 7.1 Hz, CH3); 3.64 (2H, q, J = 7.1 Hz, CH2); 7.34–7.60 (5H, m, CH = C); 7.78 (1H, s, H6); 8.05 ppm (1H, s, NH); 13C NMR (δ ppm, DMSO): C2 93.01 (C-3a); C3131.15 (C-7a); C1 136.63 (C-3); C5 145.76 (C-6); C4 159.87 (C-4); 124.54–129.33 (C-arom); GC-MS m/z (%) = 241 (100), 186 (60), 141 (45), C13H12N4O: Calculated (%): C 64.99, H 5.03, N 23.32, O 6.66 Found (%): C 64.97; H 5.00; N 23.35; O 6.65.
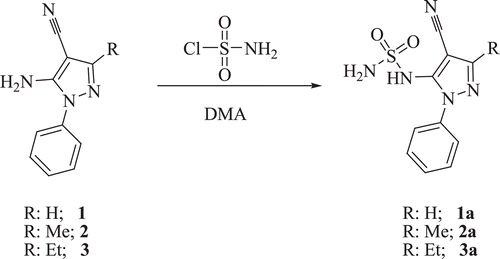
General procedure for the synthesis of compounds (8a–c)Citation47
shows the preparation of sulfamides 8a–c. Freshly prepared sulfamoyl chloride (2.0 eq) was added to a 2.0 M solution of 5-amino-1-phenyl-1H-pyrazole-4-carbonitrile 4a–c in dry DMA under a nitrogen atmosphere and the solution was stirred at r.t. until starting material was consumed (TLC monitoring). Then the solution was quenched with slush and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with H2O (4 × 20 mL), brine (3 × 20 mL) dried over Na2SO4, filtered and concentrated in vacuo to give a sticky residue that was purified to afford the desired sulfamides 8a–c as white solids.
Scheme 3. Preparation of sulfamides 8a–c.
Synthesis of 1-phenyl-5-sulphamido-1H-pyrazole-4-carbonitrile (8a)
5-Amino-1-phenyl-1H-pyrazole-4-carbonitrile 4a (0.05 g, 1.0 eq) was treated according to the general procedure previously described. The obtained residue was purified by silica gel column chromatography eluting with 50% ethyl acetate/n-hexane to afford 8a as a white solid ().
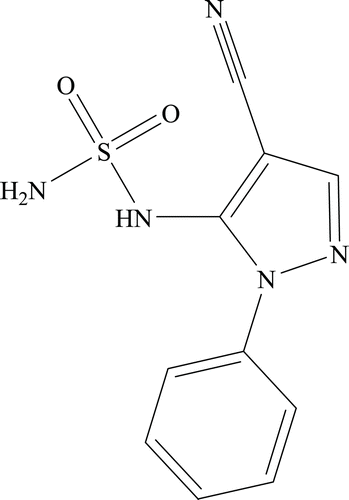
1-Phenyl-5-sulphamido-1H-pyrazole-4-carbonitrile 8a: 54% yield; silica gel TLC Rf 0.09 (50% ethyl acetate/n-hexane); δH (400 MHz, DMSO-d6) 9.60 (1H, brs, exchange with D2O, NH), 7.60 (8H, m, Ar-H, 3-H, SO2NH2), 7.42 (2H, s, exchange with D2O, SO2NH2); δC (100 MHz, DMSO-d6) 155.5, 154.1, 141.3, 130.2, 125.0, 120.0, 117.0, 79.8.
Synthesis of 3-methyl-1-phenyl-5-sulphamido-1H-pyrazole-4-carbonitrile 8b
5-Amino-3-methyl-1-phenyl-1H-pyrazole-4-carbonitrile 4b (0.05 g, 1.0 eq) was treated according to the general procedure previously described. The obtained residue was purified by silica gel column chromatography eluting with 70% ethyl acetate/n-hexane to afford 8b as a white solid ().

3-Methyl-1-phenyl-5-sulphamido-1H-pyrazole-4-carbonitrile 8b: 48% yield; silica gel TLC Rf 0.11 (50% ethyl acetate/n-hexane); δH (400 MHz, DMSO-d6) 9.99 (1H, brs, exchange with D2O, NH), 7.53 (5H, m, Ar-H), 7.42 (2H, s, exchange with D2O, SO2NH2), 2.18 (3H, s CH3); δC (100 MHz, DMSO-d6) 156.0, 155.1, 141.0, 130.1, 125.2, 119.0, 117.2, 80.0, 14.9.
Synthesis of 3-ethyl-1-phenyl-5-sulphamido-1H-pyrazole-4-carbonitrile (8c)
5-Amino-3-ethyl-1-phenyl-1H-pyrazole-4-carbonitrile 4c (0.05 g, 1.0 eq) was treated according to the general procedure previously described. The obtained residue was purified by silica gel column chromatography eluting with 70% ethyl acetate/n-hexane to afford 8c as a white solid ().
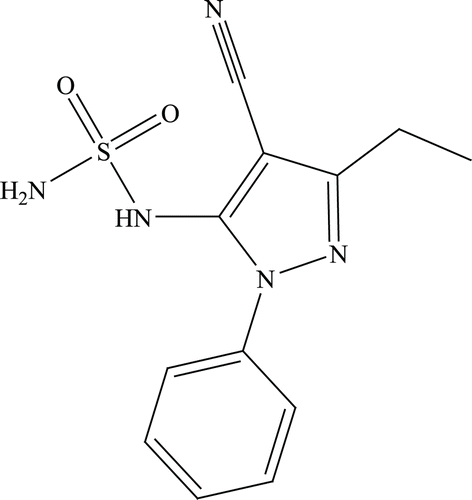
3-Ethyl-1-phenyl-5-sulphamido-1H-pyrazole-4-carbonitrile 8c: 52% yield; silica gel TLC Rf 0.13 (50% ethyl acetate/n-hexane); δH (400 MHz, DMSO-d6) 9.98 (1H, brs, exchange with D2O, NH), 7.66 (2H, d, J 9.6, Ar-H), 7.54 (3H, m, Ar-H), 7.41 (2H, s, exchange with D2O, SO2NH2), 4.10 (2H, q, J 7.2, CH2), 1.21 (3H, t, J = 7.2 Hz, CH3); δC (100 MHz, DMSO-d6) 157.2, 155.0, 141.0, 130.2, 125.1, 119.0, 117.4, 79.8, 23.9, 14.2.
CA inhibition
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activityCitation41. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes/TRIS (pH 7.5 for α-CAs, and 8.4, for β-CAs) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 sCitation43. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionized water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3, as reported earlierCitation42, and represent the mean from at least three different determinations. All CA isofoms were recombinant ones obtained in house as reported earlierCitation43–45.
Declaration of interest
The authors declare no conflict of interest.
References
- Zhu J, Bienayme H. Multicomponent Reactions. Wiley-VCH, Verlag GmbH & Co.KgaA: Weinheim, 2005: 311–341.
- Hulme C, Gore V. “Multi-component reactions: emerging chemistry in drug discovery” ‘from xylocain to crixivan’. Curr Med Chem 2003;10:51–80.
- Yamada T, Ichino T, Hanyu M, Ninomiya D, Yanagihara R, Miyazawa T et al. A novel intramolecular hydrogen bonding between a side-chain pyridine ring and an amide hydrogen of the peptide backbone in tripeptides containing the new amino acid, alpha,alpha-di(2-pyridyl)glycine. Org Biomol Chem 2004;2:2335–2339.
- Anderson JD, Cottam HB, Larson SB, Nord LD, Revankar GR, Robins RK. Synthesis of certain pyrazolo[3,4-d] pyrimidin-3-one nucleoside. J Heterocyclic Chem 1990; 27: 439–453.
- Petrie CR, Cottam HB, Mekernam PA, Robins RK, Revankar GR. Synthesis and biological activity of 6-azacadeguomycin and certain 3,4,6-trisubstituted pyrazolo[3, 4-d] pyridine ribonucleosides. J Med Chem 1985; 28: 1010–1016
- Allouche F, Chabchoub F, Salem M, Kirsch G. Synthesis of New Pyrazolopyrimidinedithiones and Pyrazolopyrimidinephosphines from Aminocyanopyrazoles. Synthetic Commun 2011; 41: 1500–1507.
- Gupta S, Rodrigues LM, Esteves AP, Oliveira-Campos AM, Nascimento MS, Nazareth N et al. Synthesis of N-aryl-5-amino-4-cyanopyrazole derivatives as potent xanthine oxidase inhibitors. Eur J Med Chem 2008;43:771–780.
- Ranjana A, Vinod K, Rajiv K, Shiv PS. Approaches towards the synthesis of 5-aminopyrazoles. J Org Chem 2011; 7: 179–197.
- Aymn ER, Mohamed IH, Randa EAM, Jehan AM, Farouk MEAM. Synthesis and antiviral evaluation of some new pyrazole and fused pyrazolopyrimidine derivatives. Bioorg Med Chem 2008; 16:7102–7106.
- Kunal N, Gurinderdeep S, Anil T, Amit A, Sameer S, Raj K, Uttam CB, Prabhakar KV, Naresh KS, Manish KG, Om PS, Dhar KL. A rational approach for the design and synthesis of 1-acetyl-3,5-diaryl-4,5-dihydro(1H)pyrazoles as a new class of potential non-purine xanthine oxidase inhibitors. Bioorg Med Chem 2011; 19: 1950–1958.
- Nesrin GK, Samiye Y, Esra K, Umut S, Ozen O, Gulberk U, Erdem Y, Engin K, Akgul Y, Altan B. A new therapeutic approach in Alzheimer disease: Some novel pyrazoles derivatives as dual MAO-B inhibitors and antiinflammatory analgesics. Bioorg Med Chem 2007; 15: 5775–5786.
- Smyth LA, Matthews TP, Horton PN, Hursthouse MB, Collins I. Divergent cyclisations of 2-(5-amino-4-carbamoyl-1H-pyrazol-3-yl)acetic acids with formyl and acetyl electrophiles. Tetrahedron 2007; 63: 9627–9634.
- Schenone S, Brullo C, Bruno O, Bondavalli F, Mosti L, Maga G et al. Synthesis, biological evaluation and docking studies of 4-amino substituted 1H-pyrazolo[3,4-d]pyrimidines. Eur J Med Chem 2008;43:2665–2676.
- Daniels RN, Kim K, Lebois EP, Muchalski H, Hughes M, Lindsley CW. Microwave-assisted protocols for the expedited synthesis of pyrazolo[1,5-a] and [3,4-d]pyrimidines. Tetrahedron Lett 2008; 49: 305–310.
- Rashad AE, Hegab MI, Abdel-Megeid RE, Micky JA, Abdel-Megeid FME. Synthesis and antiviral evaluation of some new pyrazole and fused pyrazolopyrimidine derivatives. Bioorg Med Chem 2008; 16: 7102–7106.
- Guerrera F, Sarvà MC, Siracusa MA, Caruso A, Leone MG, Cutuli V et al. Synthesis and pharmacological properties of 3-alkylthio derivatives of isothiazolothieno-1,2,3-triazine. Farmaco 1991;46:33–44.
- El-Saghier AMM. Asimple synthesis of some new thienopyridine and Thieno- pyrimidine derivatives. Molecules 2002; 7: 756–766.
- Blanco G, Quintela JM, Peinador C. Synthesis of new heteroaromatic nitrogen ligands : Pyrimido[4’’,5’’: 4’,5’]thieno[3, 2-d]1,2,3–triazines. J Heterocyclic Chem 2006; 43: 1051–1056.
- Thomae D, Perspicace E, Hesse S, Kirsch G, Seck P. Synthesis of substituted[1,3] thiazolo [4, 5-b] pyridines and thiazolo [1,3][4, 5-b] triazines. Tetrahedron 2008; 64: 9309–9314.
- Diana P, Barraja P, Lauria A, Montalbano A, Almerico AM, Dattolo G et al. Pyrrolo[2,1-d][1,2,3,5]tetrazine-4(3H)-ones, a new class of azolotetrazines with potent antitumor activity. Bioorg Med Chem 2003;11:2371–2380.
- Colomer JP, Moyano EL. New application of heterocyclic diazonium salts. Synthesis of pyrazolo[3,4-d][1,2,3]triazin-4-ones and imidazo[4,5-d][1,2,3]triazin-4-ones. Tetrahedron Lett 2011; 52: 1561–1565.
- Thomae D, Perspicace E, Hesse S, Kirsch G, Seck P. Synthesis of substituted[1,3] thiazolo [4, 5-b] pyridines and thiazolo [1,3][4, 5-b] triazines. Tetrahedron 2008; 64: 9309–9314.
- Domarkas J, Dudouit F, Williams C, Qiyu Q, Banerjee R, Brahimi F et al. The combi-targeting concept: synthesis of stable nitrosoureas designed to inhibit the epidermal growth factor receptor (EGFR). J Med Chem 2006;49:3544–3552.
- Hess S, Perspicace E, Kirsch G. Microwave-assisted synthesis of 2-aminothiophene-3-carboxylic acid derivatives, 3H-thieno[2,3-d] pyrimidin-4-one and 4-chlorothieno[2,3-d] pyrimidine. Tetrahedron Lett 2007; 48: 5261–5264.
- Davoodnia A, Rahimizadeh M, Rivadeh Sh, Bakavoli M, Roshani M. Synthesis of new substituted pyrazolo [3,4-d] pyrimidin-4-ones under microwave irradiation. Indian J Heterocylic Chem 2006; 16: 151–154.
- Cheng CC, Rolande KR. Potential purine antagonists. VI. Synthesis of-1-alkyl and-1-aryl-4-substituted pyrazolo[3, 4-d]pyrimidines 1956; 21: 1240–1256.
- Press JB, Eudy NH, Morthon GO. Aryl coupling reactions of pyrazolo[3, 4-d] pyrimidin-4-yl radicals. J Org Chem 1983; 48: 4605–4611.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–181.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–777.
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–3474.
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229.
- Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011;3:1165–1180.
- Supuran CT. Bacterial carbonic anhydrases as drug targets: toward novel antibiotics? Front Pharmacol 2011;2:34.
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–189.
- Temperini C, Scozzafava A, Vullo D, Supuran CT. Carbonic anhydrase activators. Activation of isozymes I, II, IV, VA, VII, and XIV with l- and d-histidine and crystallographic analysis of their adducts with isoform II: engineering proton-transfer processes within the active site of an enzyme. Chemistry 2006;12:7057–7066.
- Carta F, Temperini C, Innocenti A, Scozzafava A, Kaila K, Supuran CT. Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J Med Chem 2010;53:5511–5522.
- Maresca A, Temperini C, Vu H, Pham NB, Poulsen SA, Scozzafava A et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–3062.
- Maresca A, Temperini C, Pochet L, Masereel B, Scozzafava A, Supuran CT. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–344.
- Touisni N, Maresca A, McDonald PC, Lou Y, Scozzafava A, Dedhar S et al. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J Med Chem 2011;54:8271–8277.
- Alp C, Maresca A, Alp NA, Gültekin MS, Ekinci D, Scozzafava A et al. Secondary/tertiary benzenesulfonamides with inhibitory action against the cytosolic human carbonic anhydrase isoforms I and II. J Enzyme Inhib Med Chem 2012.
- Khalifah RJ. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971, 246:2561–2573.
- Fabrizi F, Mincione F, Somma T, Scozzafava G, Galassi F, Masini E et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–147.
- Baranauskiene L, Hilvo M, Matuliene J, Golovenko D, Manakova E, Dudutiene V et al. Inhibition and binding studies of carbonic anhydrase isozymes I, II and IX with benzimidazo[1,2-c][1,2,3]thiadiazole-7-sulphonamides. J Enzyme Inhib Med Chem 2010;25:863–870.
- Maresca A, Vullo D, Scozzafava A, Manole G, Supuran CT. Inhibition of the beta-class carbonic anhydrases from Mycobacterium tuberculosis with carboxylic acids. J Enzyme Inhib Med Chem 2012.
- Maresca A, Vullo D, Scozzafava A, Supuran CT. Inhibition of the alpha- and beta- carbonic anhydrases from the gastric pathogen Helicobacter pylori with anions. J Enzyme Inhib Med Chem 2012.
- Alterio V, Di Fiore A, D’Ambrosio K, Supuran CT, De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–4468.
- Okada M, Iwashita S, Koizumi N. Efficient general method for sulfamoylation of a hydroxyl group. Tetrahedron Lett 2000, 41:7047–7050.