Abstract
A series of 5,7-dihydroxyflavanone derivatives were synthesized and identified as reversible and competitive protein tyrosine phosphatase (PTP) 1B inhibitors with IC50 values in the micromolar range. Compound 4k had the most potent in vitro inhibition activity against PTP1B (IC50 = 2.37 ± 0.37 μM) and the greatest selectivity (3.7-fold) for PTP1B relative to T-cell protein tyrosine phosphatase. Cell-based studies revealed that 4k was membrane-permeable and enhanced insulin receptor tyrosine phosphorylation in CHO/hIR cells.
| Abbreviations: | ||
| CDC25B, | = | cell division cycle 25 homolog B; |
| CHO/hIR, | = | Chinese hamster ovary cells expressing human insulin receptor; |
| IR, | = | insulin receptor; |
| LAR, | = | leukocyte antigen-related phosphatase; |
| MOPs, | = | 3-[N-morpholino]propane-sulfonic acid; |
| pNPP, | = | p-nitrophenyl phosphate; |
| PTP, | = | protein tyrosine phosphatase; |
| SHP, | = | Src homology phosphatase; |
| TCPTP, | = | T-cell protein tyrosine phosphatase |
Introduction
Protein tyrosine phosphatases (PTPs) and protein tyrosine kinases (PTKs) play essential roles in intracellular signal transduction by regulating the phosphorylation level of tyrosine residue of cellular proteins. Cellular processes controlled by them include growth, differentiation, metabolism, migration, gene transcription, ion channel activity, immune responses, apoptosis and bone developmentCitation1–4. Unregulated activity of PTPs is responsible for several human diseases, including cancer, diabetes, obesity and dysfunction of the immune systemCitation5–7. One member of the PTP family, PTP1B, is an intracellular PTP expressed in insulin responsive tissues and shown to be a negative regulator in the insulin and leptin receptor pathwaysCitation8–11. Compelling evidence for this rationale came from two independent studies on the targeted disruption of PTP1B gene in mice. These PTP1B knock out mice exhibit phenotypes of increased insulin sensitivity, improved glucose tolerance and resistance to diet-induced obesityCitation12,Citation13. Furthermore, treatment of diabetic mice with PTP1B antisense oligonucleotides reduced the expression level of the enzyme and subsequently normalized blood glucose and improved insulin sensitivityCitation14–17. Thus, PTP1B has become an attractive therapeutic target for the treatment of type 2 diabetes mellitus, obesity and cancer, and small molecular inhibitors of PTP1B could be promising candidate drugs for these diseases.
In recent years, following the elucidation of the protein structure of PTP1B, many synthetic PTP1B inhibitors with submicromolar or nanomolar activities have been discovered through high-throughput screening and structure-based designCitation18,Citation19. However, the low selectivity and poor pharmacokinetic properties of these synthetic inhibitors mean that novel PTP1B inhibitors with improved pharmacological properties are still sought afterCitation19. In addition, very few of the proposed inhibitors were derived from natural productsCitation20–23, which represent a very diverse library of candidate drugs.
Flavanones have attracted much attention because they are physiologically active compounds with a wide range of biological activities, including free radical scavenging; stimulation of apoptosis; and inhibition of nitric oxide production, cell proliferation, aromatase activity and trypsin activityCitation24–28. Recently, Cui et al. reported that several flavanones isolated from the stem bark of Erythrina abyssinica (including prenylated flavanones, abyssinoflavanones and flavanones bearing a 2,2-dimethylpyrano ring) exhibited moderate inhibitory effects against PTP1BCitation29–32. Subsequently, Nguyen et al. reported that several deoxyflavonoids isolated from the root bark of Erythrina abyssinica also exhibited moderate inhibitory effects against PTP1BCitation33. These reports suggest that hydroxyflavanones might be promising PTP1B inhibitors.
Although flavanones, which widely exist in plants, exert various biological activities, including PTP1B inhibition, the structure–activity relationships and action mechanism of flavanones against PTP1B inhibition have not fully examined due to insufficient compounds to be synthesized and tested. Therefore, in the present study, we designed and synthesized a series of 5,7-dihydroxyflavanone derivatives in which the hydroxyl molecule was substituted with halogens (F, Cl and Br) or alkoxyl. We then evaluated their inhibitory effects on PTP1B and elucidated the structure–activity relationship. Selected compound were also subjected to kinetics study, selectivity and cell-based studies to determine whether their biological properties made them suitable for further development. Docking simulations of the compounds into the active site of PTP1B were also done to rationalize the results.
Materials and methods
The major chemicals were purchased from Sigma-Aldrich and Fluka. All other chemicals were of the analytical grade. Melting points were determined in open capillary tubes and are uncorrected. Reaction courses were monitored by TLC on silica gel-precoated F254 Merck plates. Developed plates were examined with UV lamps (254 nm). IR spectra were recorded (in KBr) on a FTIR 1730. 1H-NMR spectra were measured on a Bruker AV-300 spectrometer using TMS as the internal standard. Mass spectra were measured on an HP 1100LC (Agilent Technologies, USA). Biological activity was studied at National Center for Drug Screening, Shanghai, China.
Synthesis
We have already reported the synthesis and spectral data of compounds 4a–nCitation34.
Biological activity
Biological assays for PTP1B and related PTPs
The enzymatic assays for PTP1B and related PTPs were performed as described elsewhereCitation35. Briefly, the enzymatic activity of the PTP1B catalytic domain was determined at 30°C by monitoring the hydrolysis of pNPP. Dephosphorylation of pNPP generates the product pNP, which was monitored at an absorbance of 405 nm using an EnVision multilabel plate reader (PerkinElmer Life Sciences, Boston, MA, USA). Assays were performed in a total volume of 100 µL containing 50 mmol/L 3-[N-morpholino]propane-sulfonic acid (MOPs), pH = 6.5, 2 mmol/L pNPP, and 30 nmol/L recombinant PTP1B, as well as the indicated concentrations of the inhibitor. The production of pNP was continuously monitored and the initial rate of the hydrolysis was determined from the early linear region of the enzymatic reaction kinetic curve.
Enzyme kinetic analysis
The enzyme kinetic assays of compound 4k were performed as previouslyCitation35,Citation36. Assays were performed in a total volume of 100 µL containing 50 mmol/L MOPS, pH = 6.5, 30 nmol/L PTP1B, pNPP in two-fold dilutions from 80 mmol/L, and different concentrations of the inhibitor. In the presence of the competitive inhibitor, the Michaelis-Menten equation is described as 1/v = (Km/(Vmax[S]))(1 + [I]/Ki) + 1/Vmax, where v = initial rate, Vmax = maximum rate and [S] = substrate concentration. The Ki value was determined using a linear replot of the apparent Km/Vmax slope from the primary reciprocal plot versus inhibitor concentration [I] according to the equation Km/Vmax = 1 + [I]/Ki.
Insulin receptor phosphorylation cellular assay
The insulin receptor phosphorylation cellular assay of compound 4k were performed as previouslyCitation35,Citation37. CHO cells were transfected with an expression plasmid encoding human IR (CHO/hIR) and were cultured in twelve-well plates. Nearly confluent CHO/hIR cells were starved for 2 h with Ham’s serum-free F-12 medium. Cells were incubated with the indicated compound for 3 h, and then stimulated with 10 nM insulin for 10 min. Cells were scraped and lysed with loading buffer. The samples were resolved by 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, eletrotransferred to nitrocellulose membranes, and probed with anti-pIR (PY20; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-IRβ (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies. The blots were developed using Odyssey Infared Image System (LI-COR Biosciences, GmbH, Germany).
Molecular docking
Molecular docking was done using Discovery Studio software (Accelrys, San Diego, CA, USA). The crystal structure of PTP1B (Protein Data Bank, 1NL9) was defined as the receptor, and the active site of PTP1B was regarded as the binding site. Compound 4k was docked in a flexible manner, and conformations are determined by the stochastic Monte Carlo conformation search method (maximum, 50,000 trials).
Results and discussion
Chemistry
The synthetic pathways are illustrated in . 1-(2,4,6-Trihydroxy-phenyl)-ethanone was treated with chloromethyl methyl ether and potassium carbonate in acetone at room temperature to produce 1-(2-hydroxy-4,6-bis-methoxymeth-oxy-phenyl)-ethanone 1. Intermediates 2a–n were prepared by Claisen-Schmidt condensation of 1 with appropriate aromatic aldehydes, followed by cyclization in refluxing ethanol in the presence of NaOAc, which yielded flavanones 3a–n. Demethoxymethylation of 3a–n was carried out in 5 M HCl in refluxing methanol to yield 4a-nCitation38,Citation39. The spectral data obtained from infrared spectroscopy, 1H-NMR and mass spectrometry were consistent with the structures proposed.
Scheme 1. Synthesis of compounds 4a–n. Reagents and conditions: (a) MOMCl, K2CO3, acetone 0°C, rt; (b) corresponding benzaldehyde, 10% KOH, ethanol/H2O, rt; (c) 3 M HCl, MeOH/THF, reflux; (d) NaOAc, EtOH, reflux.
Biological evaluation
The inhibitory activities of the synthesized compounds against PTP1B were measured using p-nitrophenyl phosphate (pNPP) as a substrate. The results are summarized in . Ursolic acid, a known PTP1B inhibitor (IC50 = 3.20 ± 0.31 μM), was used as a positive controlCitation37.
Table 1. Inhibitory activity of compounds 4a–n against PTP1B.
We first examined the inhibitory effects of the synthesized compounds on PTP1B. All of the tested compounds showed potent PTP1B inhibitory activity (73.66–99.13%) at a concentration of 20 μg/mL. Furthermore, the compounds dose-dependently inhibited PTP1B with IC50 values ranging from 2.37 to 36.73 μM. The IC50 values of compounds 4c, 4f, 4g, 4j and 4k (2.66 ± 0.23, 3.60 ± 0.41, 2.40 ± 0.22, 3.48 ± 0.19, 2.37 ± 0.37 μM, respectively) were more potent or similar to that of ursolic acid. Of these, 4k showed excellent inhibition (99.13%) at 20 μg/mL and a reasonable IC50 value (2.37 ± 0.37 μM).
We next analyzed the structure–activity relationships of compounds 4a–n. The substituted compounds 4a–k and 4m, n (IC50 = 2.37–14.09 μM), which contained electron-donating or electron-withdrawing groups, showed more potent PTP1B inhibitory activity than compound 4l (IC50 = 36.73 ± 7.16 μM). These results suggest that the B ring of flavanones might be important in the inhibition of PTP1B. Compounds with electron-withdrawing groups on the B ring (i.e., 4a–k, IC50 = 2.37–10.23 μM) showed more potent activity than the compounds containing electron-donating groups (i.e., 4m, n, IC50 = 13.12–14.09 μM). These results indicated that electron-withdrawing groups facilitated PTP1B inhibition. We also found that the position of the substituent on the B ring significantly influenced PTP1B inhibition, with an activity order of p-F > m-F > o-F for fluorinated compounds, p-Br > o-Br > m-Br for brominated compounds, and 2,4-(Cl)2 > p-Cl > m-Cl > o-Cl for chlorinated compounds. The 2,4-dichlorated derivative 4g (IC50 = 2.40 ± 0.22 μM) was approximately two–four-fold more potent than the 2-chlorinated compound 4d (IC50 = 10.23 ± 2.45 μM) and the 3-chlorinated compound 4e (IC50 = 4.18 ± 0.94 μM). Similarly, the 3-trifluorinated derivative 4k (IC50 = 2.37 ± 0.37 μM) was six-fold more potent than the 4-methylated compound 4m (IC50 = 14.09 ± 1.38 μM). These results suggest that increasing the lipophilicity of the B ring improved inhibitory potency against PTP1B. This is consistent with earlier studies in which lipophilic moieties stabilized the enzyme-compound complex via hydrophobic interactions with the active site and surrounding subpockets, a common feature of PTP1B-inhibitor complexesCitation40,Citation41. This concept is also supported by our docking simulations described below. From these results, we focused on compound 4k in subsequent studies.
We next investigated the selectivity of compound 4k for other PTPs. As shown in , the inhibitory activities of 4k against T-cell protein tyrosine phosphatase (TCPTP) were simultaneously investigated using the same methodCitation37. Compound 4k showed a 3.7-fold greater selectivity for PTP1B than for TCPTP. Besides TCPTP, several critical PTPs were reported to regulate insulin dephosphorylation negatively, including leukocyte antigen-related phosphatase (LAR), Src homology phosphatase (SHP)-1 and SHP-2. CDC25B, a dual-specific phosphatase, is an attractive antitumor target for small molecule intervention because of its central role in controlling malignant cell proliferation by regulating cyclin dependent kinases, and because it is highly expressed in many human tumorsCitation42. Therefore, we also tested the inhibitory activities of 4k against the purified recombinant human enzymes LAR, SHP-1, SHP-2 and CDC25B, as previously describedCitation18,Citation35. As shown in , 4k did not show any inhibitory activity against LAR, SHP-1 or SHP-2, but 3.6-fold more selective for PTP1B than for CDC25B.
Table 2. Inhibitory activity of compounds 4k against related PTPs.
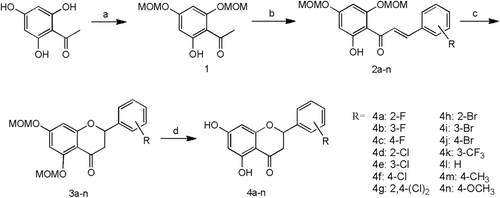
A kinetic study was performed to shed light on the inhibitory mechanism of compound 4kCitation36. As shown in , we determined the inhibition modality of 4k on PTP1B that exhibited characteristics typical of a competitive inhibitor, including increased Km values and unchanged Vmax values following increased inhibitor concentration. The Lineweaver-Burk plot also indicated that 4k is a competitive inhibitor of PTP1B because a nest of lines for increasing inhibitor concentrations intersect on the y-axis (). The results indicate that 4k binds to the catalytic pocket of PTP1B and behaves as a competitor to the physiological substrate. The Ki value calculated from was 3.4 μM.
Figure 1. Characterization of 4k to PTP1B. (A) At various fixed concentrations of 4k the initial velocity was determined with various concentrations of pNPP. (B) Typical competitive inhibition of 4k shown by Lineweaver-Burk plot. (C) Ki determination.
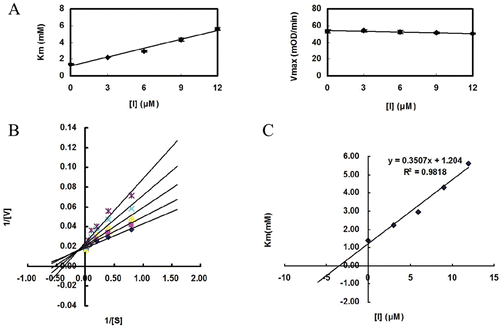
To further examine the cellular effects of compound 4k, we tested its effects on the phosphorylation level of IR in Chinese hamster ovary cells transfected with an expression plasmid encoding human IR (CHO/hIR). Cells were treated with the indicated compound and sodium vanadate (0.5 mM) as a positive control, and then stimulated with 10 nM insulin. As shown in , compound 4k significantly increased the IR phosphorylation in a dose-dependent manner, indicating the compound is cell-permeable and inhibiting PTP1B in cells.
Figure 2. Effect of compound 4k on tyrosine phosphorylation of IRβ in CHO/hIR cells. Sodium vanadate (V = 0.5 mM) and DMSO were used as postitive and negative controls. The levels of IRβ and β-actin were used to normalization.
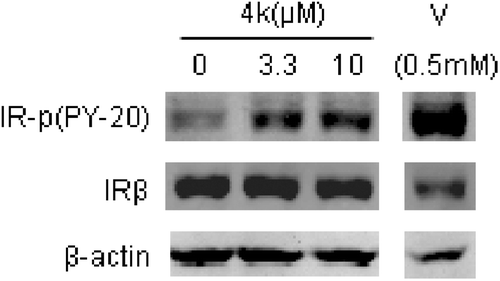
In order to understand the inhibitory activity of 4k against PTP1B, we performed molecular docking analysis of 4k using CDOCKER in concert with Discovery Studio 2.1 (http://www.accelrys.com). The X-ray crystal structure of PTP1B with a resolution of 2.40 Å (Protein Data Bank, 1NL9) was used for the docking studies. The preferred coordination modes of 4k are shown in . Fragment A of 4k binds to the active site via H-bond interactions involving 7-OH and 3-carbonyl. The 7-OH of the A ring forms an H-bond with Glu115, while the carbonyl of the C ring forms an H-bond with Gly200. The analysis also identified hydrophobic interactions between the B ring of the ligands (e.g. the lipophilic 3-(trifluoromethyl)phenyl group) and surrounding lipophilic amino acid residues (e.g. Tyr46).
Figure 3. Docking result of compound 4k in the PTP1B catalytic pocket. (A) Binding pose of 4k in PTP1B active site; (B) Key residues in binding site surrounding 4k.
Conclusion
In conclusion, a series of 5,7-dihydroxyflavanone derivatives were synthesized and identified as reversible and competitive PTP1B inhibitors with IC50 values in the micromolar range. Systemic structure–activity relationship studies of R substituents of the B ring showed that five compounds inhibited PTP1B with stronger or similar activities to that of ursolic acid. We also predicted the mechanisms involved in the interaction between the inhibitors and PTP1B using docking analysis. Of the compound tested, 4k demonstrated excellent selectivity for PTP1B relative to LAR, SHP-1 and SHP-2, and moderate selectivity relative to CDC25B. More importantly, 4k exhibited 3.7-fold greater selectivity for PTP1B than for TCPTP. Cell-based studies revealed that 4k could cross the membrane and enhanced tyrosine phosphorylation of IR in CHO/hIR cells. The cellular results provided evidence that this group of inhibitors is potent and cell-permeable. The results also indicate that the compounds tested here represent a foundation for developing novel PTP1B inhibitors that show good cell permeability, bioavailability and improved pharmacological properties.
Declaration of interest
This work was supported by the National Natural Science Foundation of China (Grants 20962021 and 81125023). and the National Program on Key Basic Research Project of China (973 Program, 2012CB524906).
References
- Neel BG, Tonks NK. Protein tyrosine phosphatases in signal transduction. Curr Opin Cell Biol 1997;9:193–204.
- Tonks NK, Neel BG. Combinatorial control of the specificity of protein tyrosine phosphatases. Curr Opin Cell Biol 2001;13:182–195.
- Hunter T. Signaling–2000 and beyond. Cell 2000;100:113–127.
- Lee S, Wang Q. Recent development of small molecular specific inhibitor of protein tyrosine phosphatase 1B. Med Res Rev 2007;27:553–573.
- Cook WS, Unger RH. Protein tyrosine phosphatase 1B: a potential leptin resistance factor of obesity. Dev Cell 2002;2:385–387.
- Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature 2001;411:355–365.
- Montalibet J, Kennedy BP. Therapeutic strategies for targeting PTP1B in diabetes. Drug Discov Today: Therap Strateg 2005;2:129–135.
- Johnson TO, Ermolieff J, Jirousek MR. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat Rev Drug Discov 2002;1:696–709.
- Liu G, Trevillyan JM. Protein tyrosine phosphatase 1B as a target for the treatment of impaired glucose tolerance and type II diabetes. Curr Opin Investig Drugs 2002;3:1608–1616.
- Tobin JF, Tam S. Recent advances in the development of small molecule inhibitors of PTP1B for the treatment of insulin resistance and type 2 diabetes. Curr Opin Drug Discov Devel 2002;5:500–512.
- Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ et al. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell 2002;2:497–503.
- Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999;283:1544–1548.
- Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol 2000;20:5479–5489.
- Rondinone CM, Trevillyan JM, Clampit J, Gum RJ, Berg C, Kroeger P et al. Protein tyrosine phosphatase 1B reduction regulates adiposity and expression of genes involved in lipogenesis. Diabetes 2002;51:2405–2411.
- Zinker BA, Rondinone CM, Trevillyan JM, Gum RJ, Clampit JE, Waring JF et al. PTP1B antisense oligonucleotide lowers PTP1B protein, normalizes blood glucose, and improves insulin sensitivity in diabetic mice. Proc Natl Acad Sci USA 2002;99:11357–11362.
- Ramachandran C, Kennedy BP. Protein tyrosine phosphatase 1B: a novel target for type 2 diabetes and obesity. Curr Top Med Chem 2003;3:749–757.
- Gum RJ, Gaede LL, Koterski SL, Heindel M, Clampit JE, Zinker BA et al. Reduction of protein tyrosine phosphatase 1B increases insulin-dependent signaling in ob/ob mice. Diabetes 2003;52:21–28.
- Zhang YN, Zhang W, Hong D, Shi L, Shen Q, Li JY et al. Oleanolic acid and its derivatives: new inhibitor of protein tyrosine phosphatase 1B with cellular activities. Bioorg Med Chem 2008;16:8697–8705.
- Combs AP. Recent advances in the discovery of competitive protein tyrosine phosphatase 1B inhibitors for the treatment of diabetes, obesity, and cancer. J Med Chem 2010;53:2333–2344.
- Na M, Jang J, Njamen D, Mbafor JT, Fomum ZT, Kim BY et al. Protein tyrosine phosphatase-1B inhibitory activity of isoprenylated flavonoids isolated from Erythrina mildbraedii. J Nat Prod 2006;69:1572–1576.
- Wang Y, Shang XY, Wang SJ, Mo SY, Li S, Yang YC et al. Structures, biogenesis, and biological activities of pyrano[4,3-c]isochromen-4-one derivatives from the Fungus Phellinus igniarius. J Nat Prod 2007;70:296–299.
- Combs AP, Zhu W, Crawley ML, Glass B, Polam P, Sparks RB et al. Potent benzimidazole sulfonamide protein tyrosine phosphatase 1B inhibitors containing the heterocyclic (S)-isothiazolidinone phosphotyrosine mimetic. J Med Chem 2006;49:3774–3789.
- Wilson DP, Wan ZK, Xu WX, Kirincich SJ, Follows BC, Joseph-McCarthy D et al. Structure-based optimization of protein tyrosine phosphatase 1B inhibitors: from the active site to the second phosphotyrosine binding site. J Med Chem 2007;50:4681–4698.
- Matsuda H, Wang T, Managi H, Yoshikawa M. Structural requirements of flavonoids for inhibition of protein glycation and radical scavenging activities. Bioorg Med Chem 2003;11:5317–5323.
- Matsuda H, Morikawa T, Ando S, Toguchida I, Yoshikawa M. Structural requirements of flavonoids for nitric oxide production inhibitory activity and mechanism of action. Bioorg Med Chem 2003;11:1995–2000.
- Pouget C, Lauthier F, Simon A, Fagnere C, Basly JP, Delage C et al. Flavonoids: structural requirements for antiproliferative activity on breast cancer cells. Bioorg Med Chem Lett 2001;11:3095–3097.
- Pouget C, Fagnere C, Basly JP, Habrioux G, Chulia AJ. New aromatase inhibitors. Synthesis and inhibitory activity of pyridinyl-substituted flavanone derivatives. Bioorg Med Chem Lett 2002;12:1059–1061.
- Maliar T, Jedinák A, Kadrabová J, Sturdík E. Structural aspects of flavonoids as trypsin inhibitors. Eur J Med Chem 2004;39:241–248.
- Cui L, Na M, Oh H, Bae EY, Jeong DG, Ryu SE et al. Protein tyrosine phosphatase 1B inhibitors from Morus root bark. Bioorg Med Chem Lett 2006;16:1426–1429.
- Cui L, Ndinteh DT, Na M, Thuong PT, Silike-Muruumu J, Njamen D et al. Isoprenylated flavonoids from the stem bark of Erythrina abyssinica. J Nat Prod 2007;70:1039–1042.
- Cui L, Thuong PT, Lee HS, Ndinteh DT, Mbafor JT, Fomum ZT et al. Flavanones from the stem bark of Erythrina abyssinica. Bioorg Med Chem 2008;16:10356–10362.
- Cui L, Lee HS, Ndinteh DT, Mbafor JT, Kim YH, Le TV et al. New prenylated flavanones from Erythrina abyssinica with protein tyrosine phosphatase 1B (PTP1B) inhibitory activity. Planta Med 2010;76:713–718.
- Nguyen PH, Dao TT, Kim J, Phong do T, Ndinteh DT, Mbafor JT et al. New 5-deoxyflavonoids and their inhibitory effects on protein tyrosine phosphatase 1B (PTP1B) activity. Bioorg Med Chem 2011;19:3378–3383.
- Zhao DH, Sui X, Qu YL, Yang LY, Wang X, Guan LP. Synthesis and studies on antidepressant effect of 5,7-dihydroxyflavanone derivatives. Asian J Chem 2011;23:1129–1132.
- Shi L, Yu HP, Zhou YY, Du JQ, Shen Q, Li JY et al. Discovery of a novel competitive inhibitor of PTP1B by high-throughput screening. Acta Pharmacol Sin 2008;29:278–284.
- McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J Med Chem 2003;46:4265–4272.
- Zhang W, Hong D, Zhou Y, Zhang Y, Shen Q, Li JY et al. Ursolic acid and its derivative inhibit protein tyrosine phosphatase 1B, enhancing insulin receptor phosphorylation and stimulating glucose uptake. Biochim Biophys Acta 2006;1760:1505–1512.
- Zhao LM, Jin HS, Sun LP, Piao HR, Quan ZS. Synthesis and evaluation of antiplatelet activity of trihydroxychalcone derivatives. Bioorg Med Chem Lett 2005;15:5027–5029.
- Dong X, Liu Y, Yan J, Jiang C, Chen J, Liu T et al. Identification of SVM-based classification model, synthesis and evaluation of prenylated flavonoids as vasorelaxant agents. Bioorg Med Chem 2008;16:8151–8160.
- Zhao H, Liu G, Xin Z, Serby MD, Pei Z, Szczepankiewicz BG et al. Isoxazole carboxylic acids as protein tyrosine phosphatase 1B (PTP1B) inhibitors. Bioorg Med Chem Lett 2004;14:5543–5546.
- Liu G, Xin Z, Pei Z, Hajduk PJ, Abad-Zapatero C, Hutchins CW et al. Fragment screening and assembly: a highly efficient approach to a selective and cell active protein tyrosine phosphatase 1B inhibitor. J Med Chem 2003;46:4232–4235.
- Ducruet AP, Vogt A, Wipf P, Lazo JS. Dual specificity protein phosphatases: therapeutic targets for cancer and Alzheimer’s disease. Annu Rev Pharmacol Toxicol 2005;45:725–750.