Abstract
Carbonic anhydrase IX (CA IX) is a hypoxia-regulated enzyme, overexpressed in many types of human cancer. CA IX is involved in pH homeostasis, contributing to extracellular acidification and tumourigenesis. Acidification of the extracellular milieu can impact upon cellular uptake of chemotherapeutic drugs by favouring weak acids (e.g. melphalan), but limiting access of weak bases (e.g. doxorubicin). We investigated whether alterations of CA IX activity affected anti-cancer drug uptake and toxicity. CA inhibitor acetazolamide (AZM) enhanced doxorubicin toxicity but reduced melphalan toxicity in cell lines that highly expressed CA IX under anoxic conditions (HT29 and MDA435 CA9/18). The toxicity changes reflected modification of passive drug uptake. AZM did not alter toxicity or uptake in cells with low CA IX activity (HCT116 and MDA435 EV1). AZM lowered intracellular pH in HT29 and MDA435 CA9/18 cells under anoxic conditions. CA IX activity has chemomodulatory properties and is an attractive target for anti-cancer therapy.
Introduction
Areas of low oxygen tension (termed hypoxia) are a common feature of solid tumours as the growth rate of tumours often exceeds the rate of angiogenesis, resulting in an abnormal vasculatureCitation1. Chronic hypoxia results from cells being too distant from a blood vessel for oxygen to diffuse. Temporary obstructions in blood flow in the disorganized vasculature can lead to areas of transient hypoxiaCitation2,Citation3. The presence of hypoxia has been found to contribute to tumour progression, for example by selecting for cells with reduced apoptotic potentialCitation4. The central mediator of the cellular response to hypoxia is the hypoxia inducible factor (HIF-1) transcription factor. HIF-1 regulates a wide range of genes that are involved in adaptation to hypoxic stress, including erythropoietin, glycolytic enzymes, VEGF (vascular endothelial growth factor) and carbonic anhydrase IX (CA IX)Citation5,Citation6. Evidence is also emerging that hypoxic tumour cells in primary tumours contribute to metastatic growth in distant organs through the upregulation of HIF-associated genes like lysyl oxidase (LOX)Citation7. In patients, tumour hypoxia reduces the survival rate by increasing the resistance to radio- and chemotherapyCitation8,Citation9 affecting the loco-regional control of tumour growth as well as the development of lymph node and distant metastasesCitation10–13.
CA IX is a member of the α carbonic anhydrases (CA) that reversibly catalyze the hydration of carbon dioxide (H2O + CO2 ↔ H+ + HCO3−) and serve a diverse range of physiological functionsCitation14. CA IX is one of four transmembrane CA (the others being IV, XII, XIV) with an extracellular catalytic unit. CA IX converts the CO2 diffusing out of a cell into carbonic acid, thus producing protons in the extracellular spaceCitation15. CA IX is known to be strongly associated with tumours, being rarely expressed in normal tissues but overexpressed in corresponding neoplastic tissuesCitation16. Wycoff et alCitation17 demonstrated a hypoxia-dependent upregulation of CA IX in several tumour cell lines in vitro and correlated this to the presence of a hypoxia response element (HRE), to which HIFs bind, within the CA IX promoter. Increased CA IX activity enhanced extracellular acidification in cultures of various tumour cell linesCitation18. CA IX is currently under investigation as an endogenous marker of hypoxia in various tumour types, in hope of avoiding problems associated with invasive techniques or administration of chemical markers prior to biopsyCitation17,Citation19–23.
Microelectrode and non-invasive measurements of tumour pH have revealed that human tumours have acidic extracellular pH (pHe), but the intracellular pH (pHi) remains neutral to alkaliCitation24,Citation25. CA IX has been shown to be a key contributor to the acidification process in tumours with the use of membrane ghost preparationsCitation26 and tumour cell lines in which CA IX activity was either silenced or overexpressedCitation27. Tumour acidification is positively correlated with enhance proliferation, invasiveness and metastasisCitation27,Citation28. Many chemotherapeutic agents are weak electrolytes and enter cells by passive diffusion. Low pHe has implications for their uptake. The plasma membrane is less permeable to ionized species and any pH gradient between intra- and extracellular compartments can result in ion trapping and altered drug distributionCitation29. The ion trapping model predicts that weakly basic chemotherapeutic drugs such as doxorubicin, mitoxantrone and daunorubicin concentrate in the more acidic extracellular compartment, hindering their uptake and toxicity. However, weak acids chemotherapeutic drugs (e.g. melphalan, chlorambucil) should be easily taken up into the relatively neutral intracellular spaceCitation30.
The fact that CA IX has strong tumour association and is regulated by a common physiological feature of solid tumours (i.e. hypoxia) has in the last decade accelerated the research into the use of CA IX as a diagnostic and therapeutic target in patients with cancerCitation31,Citation32. This includes the development of more selective CA inhibitors which target the tumour-associated CA isoformsCitation33,Citation34. In some instances the use of CA IX-based therapy has already progressed into clinical trials, as in the case of monoclonal antibodies targeting CA IX in renal cell carcinoma (RCC)Citation35. The use of CA inhibitors to reduce tumour acidity and possibly modulate uptake and toxicity of anti-cancer drugs in vitro has not previously been demonstrated. This study utilizes acetazolamide (AZM), a broad spectrum CA inhibitor in the low nanomolar range, to inhibit CA IX function under aerobic and anoxic conditions in several cell lines in vitro and observe if changes in CA IX activity reflect the uptake and toxicity of two anti-cancer drugs, the weak acid melphalan (MEL) and the weak basic doxorubicin (DOX). DOX has a pKa of 8.34 and MEL is known to be more toxic under acidic pHeCitation36, despite being actively transported into cells.
Materials and methods
Materials
Unless otherwise stated all chemicals were purchased from BDH Chemicals Ltd (Poole, UK). Cell culture materials were obtained from Invitrogen Ltd (Paisley, UK), with the exception of G418 (PAA Laboratories, Colbe, Germany). Sigma-Aldrich (Gillingham, UK) provided the following materials: acetazolamide (AZM), melphalan (MEL), doxorubicin (DOX), protease inhibitors, Tween-20, reduced glutathione (GSH), trichloroacetic acid (TCA) and 5′5′-Dithio-bis(2-nitrobenzoic acid) (DTNB). Triethanolamine-HCl (TEA) was purchased from Fluka (Gillingham, UK). Nitrocellulose membranes and ECL kits were purchased from Amersham (Buckinghamshire, UK). Seminaphtharhodafluor (SNARF-1) and nigericin were obtained from Molecular Probes (Eugene, OR,USA). The chemical structures of AZM, MEL and DOX are shown in .
Figure 1. Chemical structures of doxorubicin, melphalan and acetazolamide.
Cell culture
HCT116 and HT29 colon carcinoma cells were maintained in RPMI 1640 media with the addition of 10% foetal calf serum (FCS) and 1% glutamine. MDA435 human melanoma cells, stably transfected to express empty vector (EV1) or CA9 (CA9/18) as described in Robertson et alCitation37, were routinely cultured in RPMI 1640 media with the addition of 10% FCS, 1% glutamine and 3 mg/ml G418. Cells were exposed to normoxia (21% O2) in a 5% CO2 humidified 37°C incubator. Anoxic (< 10 ppm O2) exposures took place in a 5% CO2, 37°C anaerobic chamber. AZM stock solution was dissolved in dimethyl sulfoxide (DMSO) and added to the culture medium. MEL was dissolved in acidified ethanol and added to culture medium. DOX was dissolved in deionized water before addition to culture medium. Bicarbonate free, HEPES buffered RPMI was used for AZM, MEL and DOX exposures to reduce buffering of pH changes.
Preparation of cell membrane extracts
Membrane extracts of each cell line following treatment with AZM for 18 hrs in air and anoxia were prepared as follows. Cells were lysed in a hypotonic buffer (10 mM HEPES, 10 mM NaCl, 1 mM KH2PO4, 5 mM NaHCO3, 1 mM CaCl2, 0.5 mM MgCl2) with protease inhibitors added just prior to use (1 mM PMSF, 10 µg/ml aprotinin, 10 µg/ml leupeptin, 1 µg/ml pepstatin) and kept on ice for 5 min. After sonication, cells were centrifuged at 7500 rpm for 5 min at 4°C and the supernatant collected. The supernatant was centrifuged at 13000 rpm at 4°C for 1 hr and the cytosolic supernatant discarded. The crude membrane pellet was resuspended in PBS and stored at −80°C. Protein content was determined by Bradford assay.
Carbonic anhydrase activity assay
The CA activity of the membrane extracts was assayed by an electrometric assay adapted from Wilbur & AndersonCitation38, in which the time required for a saturated CO2 solution to lower the pH of 0.02M Tris-HCl buffer from 8.3 to 6.3 at 0°C is determined. Briefly, the pH of 6 ml chilled 0.02M Tris-HCl was recorded and 4 ml chilled saturated water quickly added. The time taken for the pH to drop to 6.3 was recorded with (T) or without (T0) the addition of 100 µl sample. The units of activity were calculated using the equation 2 x (T0-T)/ T x mg protein.
Western blotting
Following exposure to 18 hrs of air or anoxia, whole cells extracts were prepared and assayed for protein content by Bradford assay. Samples (25 µg/lane) were resolved by 10% SDS-PAGE and transferred to a nitrocellulose membrane and blocked overnight in blocking buffer (10% milk in 0.1% tris buffered saline-Tween 20, TBS-T). Membranes were washed in TBS-T and incubated with anti-CA IX (M75) antibody for 1 hr, washed several times and incubated for 30 min with an anti-mouse peroxidase conjugated secondary antibody. Bands were detected by ECL.
Clonogenic survival assay
Single cell suspensions of each cell lines were seeded at 500 cells per well of a 6-well plate in triplicate and allowed to recover for at least 24 hrs. The cells were pre-exposed to AZM for 18 hrs in air or anoxia before 3 hrs exposure to drug in air/anoxia. The media was replaced and cells incubated for 10–14 days until visible colonies were observed. Methylene blue was used to visualize the colonies for counting and the colony efficiency calculated.
Glutathione cytotoxicity assay
The cellular levels of GSH were measured in whole cell extracts of cells treated with AZM and MEL using an adapted Ellman assayCitation39. 1 × 106 cells were lysed in air and anoxia following 18 hrs AZM exposure and additional 3 hrs MEL treatment. 1 mM GSH stock was serially diluted to produce a series of standard concentrations in 20 mM triethanolamine-HCl (TEA). In Eppendorfs, 100 µl sample/standard was added to 100 µl 10% trichloroacetic acid (TCA) solution and spun down at 1500 rpm for 15 min at room temperature. In a 96-well plate, 200 µl Tris-HCl pH 8.9, 20 µl 150 µM 5′5′-Dithio-bis(2-nitrobenzoic acid) (DTNB) and 50 µl supernatant from TCA precipitation were added to each well and absorbance measured at 414 nm. The GSH concentrations of samples were calculated from the standard curve and corrected for the cell number per sample.
Doxorubicin uptake measurement
The intracellular accumulation of DOX was assessed by flow cytometry. DOX autofluorescence (590 nm) was detected in the FL-2 channel. 1 × 105 cells were treated with DOX for 3 hrs with or without 18 hrs AZM pre-incubation in air and anoxia. The cells were harvested by trypsinisation, pelleted, washed twice with PBS and resuspended in 0.5 ml PBS. A total of 10000 events were analyzed per sample and the amount of drug uptake was recorded as mean fluorescence collected in the FL-2 channel (arbitrary fluorescence units).
Intracellular pH measurement
The pHi of cells treated with AZM for 18 hrs in air or anoxia was analyzed using seminaphtharhodafluor (SNARF-1). A total of 5 µM SNARF-1 was incubated with the sample and control cells for 30 min at 37°C in bicarbonate free media prior to centrifugation at 1500 rpm for 10 min. The cell pellets were stored at 22°C in the dark until analysis. Analysis was performed using a flow cytometer, collecting mean fluorescence at 670 nm and 575 nm. For calibration of SNARF-1 fluorescence ratios to pH, buffers A (135 mM KH2PO4, 20 mM NaCl, 1 mM MgCl, 1 mM CaCL2, 10 mM glucose) and buffer B (135 mM K2HPO3, 20 mM NaCl, 1 mM MgCl, 1 mM CaCl2, 10 mM glucose) were mixed in various ratios to produce a series of solutions with pH between 6 and 8. 2 µg/ml of the ionophore, inhibitor nigericin was added to each buffer and 5 × 105 cells/ml suspended in the calibration buffers and analyzed by flow cytometry. For each experiment and cell line, a calibration curve was determined to relate fluorescence as function of pHi.
Results and discussion
Different cell lines express different levels of CA IX activity
CA IX enzyme activity cannot be measured directly; however, membrane extracts from tumour cells isolated the four transmembrane CA enzymes, of which only CA IX (and XII) are known to be overexpressedCitation40. In , range of cell lines were tested for activity levels under aerobic and anoxic conditions and a subset with varying levels and inductions selected for further investigation. Two colon cell lines were shown to have different activity patterns, HCT116 cells revealing low activity in both conditions with no anoxic induction (10.4 units/mg protein and 10.45 units/mg protein under aerobic and anoxic conditions respectively, p = 0.5). In contrast, HT29 cells had slightly higher normoxic activity (17.4 units/mg protein) but substantially elevated levels in response to anoxic conditions (108.3 units/mg protein, p = 0.019). MDA435 cells had not been found to express CA IX or CA XII (data not shown) and were selected for transfection to overexpress CA IX (CA9/18) and empty vector control (EV1)Citation37. CA9/18 cells showed increased activity under aerobic and anoxic conditions compared to empty vector control (EV1) cells. A similar pattern was observed in CA IX protein expression in . Incubation with 100 µM of the CA inhibitor AZM lowered CA IX activity under aerobic and anoxic conditions and abrogated the anoxic induction in most cell lines. An apparent exception was EV1 cells, however the basal levels were low in comparison with other cell lines and difference was not statistically significant (p = 0.13) (). shows that AZM inhibited CA activity at concentrations between 10 and 100 µM, in all four of the cell lines (data not shown for HCT116 and HT29), but toxicity was observed beyond this range (data not shown).
Table 1. Inhibition of CA IX activity by AZM.
Figure 2. CA activity of membrane extracts in air and anoxia. (A) Wide range of aerobic (black bars) and anoxic (white bars) levels of CA activity observed, for example HCT116 and EV1 cells exhibited lower activity levels with minimal anoxic induction. CA9/18 cells had high aerobic activity and moderate induction under anoxic conditions. Activity in HT29 cells was greatly enhanced under anoxic conditions. (B) CA IX protein expression in the four cell lines specified reflected the activity profiles. (C) 10–100 µM AZM reduced activity in EV1 (black line) and CA9/18 (grey line) cells in a dose dependent manner under aerobic conditions. *p < 0.05. Error bars indicate SEM of n = 3 samples. AZM, acetazolamide; CA IX, carbonic anhydrase IX.
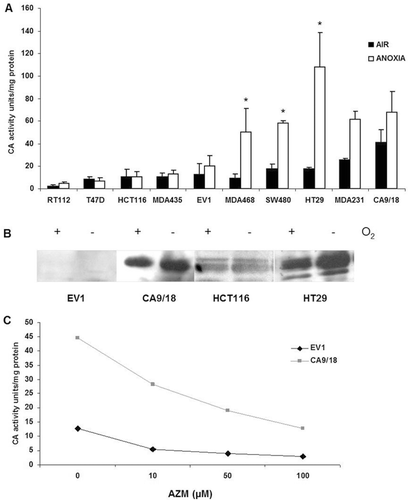
Variation was observed in the aerobic levels in terms of protein and activity in the panel of cell lines. The overexpression noted in cell lines of different tissues of origin correlates with the overexpression found in many human tumoursCitation41–43. However, not all the cell lines substantially induced CA IX activity in response to anoxia despite the identification of a HRE within the CA IX promoterCitation17,Citation44. Direct measurement of CA IX activity is not currently possible. This study isolated membrane extracts, which contain the four transmembrane CA isozymes, of which CA IX is by far the most activeCitation45. While it is possible that the activity of CA IX is not accurately measured, the activity correlated well with the protein levels, suggesting that CA IX is the predominant detected activity.
Four cell lines were selected from the panel with differential CA IX expression and activity for further investigation. Two colon adenocarcinoma cell lines had different patterns of activity, HCT116 with relatively low aerobic activity and minimal anoxic induction of CA IX. In contrast, slightly higher aerobic activity with substantial anoxic induction of CA IX activity was identified in HT29 cells. A similar pattern was noted in MDA435 cells stably transfected with a control vector (EV1) and to overexpress CA IX (CA9/18), but with lower anoxic induction compared to the colon lines (). The CA inhibitor (AZM) reduced activity under normoxic and anoxic conditions in the four cell lines following an 18 hrs exposure. Inhibition was observed from 10 to 100 µM range, but studies revealed that the cell lines were sensitive to CA-independent toxic effects above that dose (data not shown), so 50 µM AZM was used in combination with anti-cancer drugs MEL or DOX. AZM strongly inhibits CA IX but also affects other isoforms including CA IICitation45–47. AZM is slightly more membrane impermeable than other general inhibitorsCitation14, so will effectively inhibit CA IX as it is the most active transmembrane form. AZM has been shown to inhibit invasion of other cell lines in vitro and has itself been investigated as an anti-cancer drugCitation48,Citation49. Based on the structure of AZM, other derivates have been chemically synthesized with many showing a high in vitro CA IX inhibition activity with Ki at low nanomolar levelsCitation47. Sulphonamide of which AZM is a derivate shows great potential as a chemical group as CA IX-specific inhibitors in both in vitro as well as in vivo assaysCitation50,Citation51.
Acetazolamide reduces the toxicity of melphalan
AZM was utilized in combination with the weakly acidic drug MEL to elucidate if changes in CA IX activity reflected in altered drug toxicity, as assessed by clonogenic assay. In , 50 µM AZM increased survival from MEL exposure in HT29 cells under aerobic and anoxic conditions. Interestingly, 50 µM AZM was more effective against 50 µM MEL than 10 µM MEL under anoxic conditions, but nevertheless indicates that CA IX inhibition reduced toxicity to MEL. Changes in survival of cells exposed to 10 µM and 50 µM MEL plus 50 µM AZM compared to MEL alone for each of the cell lines under aerobic and anoxic conditions are given in . Pre-treatment with 50 µM AZM for 18 hrs did not significantly affect toxicity of 50 µM MEL in any of the lines under aerobic conditions (, left panel). No effect of 50 µM AZM on MEL toxicity was observed in HCT116 cells (which exhibited minimal CA IX induction) under anoxic conditions, however EV1 cells appeared to show potentiation of 10 µM MEL toxicity, p = 0.006 (, right panel). In contrast, HT29 cells which induced CA IX activity under anoxic conditions, exhibited a reduction in MEL toxicity in combination with 50 µM AZM (2.3 fold at 10 µM MEL, p = 0.0009 and 6.3 fold at 50 µM MEL, p = 0.0008). AZM alone was shown to alter survival in the majority of the cell lines so any influence of AZM on survival had been corrected for in the analysis, indicating the effect was due to the combination, and not due to AZM toxicity.
Figure 3. AZM reduces toxicity of melphalan. AZM alone had a minor impact on cell survival, but the results were corrected for any effects of AZM. (A) 50 µM AZM reduced MEL toxicity in aerobic (left panel) and anoxic (right panel) HT29 cells. (B) No reduction in MEL toxicity following 50 µM AZM exposure under aerobic conditions was noted in HCT116 (black bars) and EV1 (white bars) cells (left panel). 50 µM AZM significantly reduced 50 µM MEL toxicity in HT29 cells (6.3 fold) under anoxic conditions (horizontal lined bars) (right panel). *p < 0.05. Error bars indicate SEM of n = 3 samples. AZM, acetazolamide; MEL, melphalan.
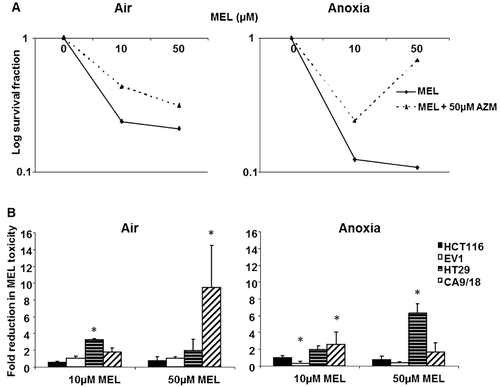
Pre-exposure to AZM increased the clonogenic survival from MEL exposure in highly inducible cell lines, indicating reduced toxicity of the weakly acidic drug. No effect was seen in HCT116 cells with minimal CA IX induction, suggesting that inhibition of CA IX was primarily responsible for the modification of MEL toxicity. However, it was noted that EV1 cells (which also showed low CA IX activity) actually revealed minor enhancement of MEL toxicity in combination with AZM under anoxic conditions. It is possible that others mechanisms of action of AZM may be responsible for this phenomenon. The toxicity results are largely consistent the possibility of a role of CA IX in pH regulation, as inhibition of CA IX would be thought to increase the extracellular pH (pHe) and relatively decrease the pHi. Intracellular acidification would limit the uptake of weak bases (e.g. MEL) and conversely increase the uptake of weak acids (e.g. DOX). Previous studies using media with altered pH have shown that activity of MEL increased in vitro at low pHeCitation36,Citation52.
Acetazolamide enhances the toxicity of doxorubicin
AZM altered the toxicity of a weakly acidic anti-cancer agent MEL and hence clonogenic experiments were repeated with DOX to investigate if the opposite occurred for a weakly basic agent.
shows that in the cell lines that exhibited minimal CA IX activity and anoxic induction (HCT116 and EV1), DOX toxicity was unaffected by 50 µM AZM in either condition. However, the highly inducible HT29 cells showed potentiation by 50 µM AZM under anoxic conditions, producing a 5.1 fold increase in the toxicity of 1 µM DOX (p = 0.026) and 4.3 fold increase of 5 µM DOX toxicity (p = 0.042) (). CA IX overexpressing CA9/18 cells showed a 5 fold increase in the toxicity of 5 µM DOX in combination with 50 µM AZM under aerobic conditions (p = 0.05) (). A similar increase was observed with 50 µM AZM and 1 µM DOX toxicity (p = 0.001) but a 12.3 fold increase of 5 µM DOX toxicity under anoxic conditions in CA9/18 cells (p = 0.007). It was noted that in all the cell lines tested, DOX toxicity was slightly reduced under anoxic versus aerobic conditions (data not shown).
Figure 4. AZM enhances toxicity of doxorubicin. The minimal effect of AZM alone on toxicity has been corrected. (A) The addition of 50 µM AZM increased DOX toxicity in HT29 cells under aerobic and anoxic conditions. (B) 50 µM AZM had a minimal effect on doxorubicin toxicity in HCT116 (black bars) and EV1 (white bars) cells under aerobic conditions (left panel), however CA9/18 (diagonal lined bars) cells showed a 5.2 fold increase in toxicity of 5 µM DOX. 50 µM AZM did not alter DOX toxicity in HCT116 and EV1 cells under anoxic conditions (right panel), but HT29 (horizontal lined bars) cells exhibited an approximately 5 fold increase in toxicity at both doses of DOX. Anoxic CA9/18 cells showed a 5.2 fold increase in toxicity of 1 µM DOX and 12.13 fold increase of 5 µM DOX toxicity (right panel). *p < 0.05. Error bars indicate SEM of n = 3 samples. AZM, acetazolamide; DOX, doxorubicin.
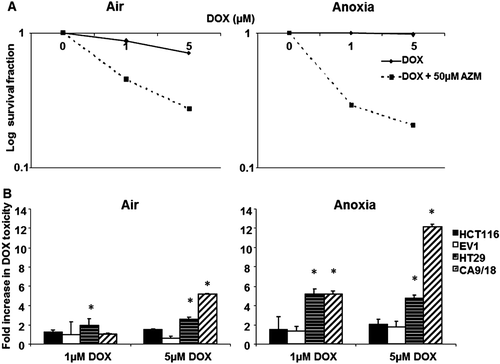
AZM enhanced the toxicity of the weak base DOX in highly inducible cell lines (HT29, CA9/18) under anoxic conditions, but had no effect in minimally inducible cells. Again, this is consistent with the theory that CA IX inhibition would result in increased uptake and toxicity of weakly basic drugs. The reduced toxicity of DOX under anoxic conditions seen in all the lines tested has been noted in other studiesCitation53, but others have found increased activity in anoxia depending on the cell lineCitation54,Citation55. However, hypoxic resistance is consistent with the ion trapping model, as low pHe would restrict uptake of weak basesCitation56. This theory does however not take into account that part of the efficiency of DOX uptake/toxicity are pH-insensitive as result of the binding of DOX to slowly-releasing nuclear binding sitesCitation57.
AZM antagonizes the uptake of melphalan
illustrated reduced toxicity of MEL following treatment with AZM. In order to determine whether the change in toxicity was due to altered passive uptake of the drug, cellular reduced glutathione levels (GSH) were assayedCitation58. MEL conjugates with GSH resulting in intracellular depletion of GSH, providing a surrogate marker for the amount of melphalan entering cells.
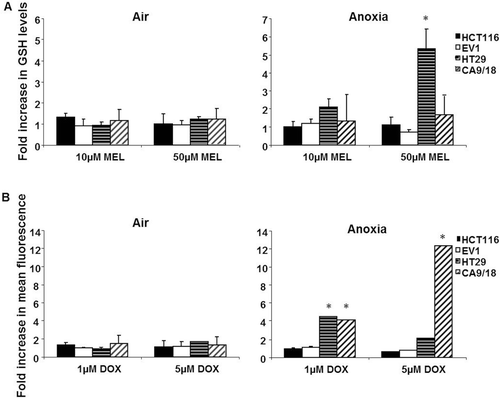
GSH levels in untreated cells ranged from 50 pmol/cell in EV1 cells to 450 pmol/cell in HT29 cells (data not shown). A total of 50 µM MEL depleted GSH levels in all the cell lines under aerobic and anoxic conditions (data not shown). The data were corrected for any affect of AZM alone on GSH levels. (left panel) revealed that none of the cell lines altered GSH depletion when AZM was combined with MEL under aerobic conditions. Under anoxic conditions however, HT29 cells exhibited increased GSH levels (5.3 fold, p = 0.007) with the combination of 50 µM AZM and 50 µM MEL, compared to 50 µM MEL alone (, right panel). Although not a direct measurement of MEL uptake, these results support the contention that MEL uptake was possibly abrogated by CA IX inhibition. This would be consistent with the theory that AZM affects uptake via pH regulation as weak acids would be trapped in the extracellular compartment.
Figure 5. AZM limited GSH depletion by MEL but enhanced uptake of doxorubicin. (A) Under aerobic conditions no cell line revealed significant increased GSH levels upon the addition of 50 µM AZM to MEL (left panel). Under anoxic conditions, HT29 cells (horizontal lined bars) showed a 5.33 fold increase in GSH levels at 50 µM MEL in combination with 50 µM AZM, but other cell lines were unaffected (right panel). (B) AZM alone had no effect on background fluorescence in the cell line tested in air or anoxia. DOX increased the mean fluorescence value relative to concentration (data not shown), but was observed to be lower in anoxia than normoxia. Fluorescence was unaffected in all the cell lines by the addition of 50 µM AZM under aerobic conditions (left panel). Under anoxic conditions, 50 µM AZM significantly enhanced the uptake of 1 µM DOX in HT29 cells (horizontal lined bars) and induced a 12 fold increase in the toxicity of 5 µM DOX in CA9/18 cells (diagonal lined bars) (right panel). *p < 0.05 Error bars indicate SEM of n = 3 samples. AZM, acetazolamide; DOX, doxorubicin; GSH, glutathione; MEL, melphalan.
AZM enhances doxorubicin uptake
The uptake of DOX was assessed in order to determine if the enhanced toxicity in combination with AZM was reflected in increased uptake. DOX has auto-fluorescent properties so its intracellular accumulation could be measured.
AZM alone did not modify background fluorescence in any of the cell lines or conditions tested, but mean fluorescence increased with increasing concentrations of DOX in all the lines tested (data not shown). It was observed that the uptake of DOX increased under normoxic conditions, however not to a statistically significant level (data not shown). In the lines and conditions where toxicity was unaffected by 50 µM AZM (e.g. HCT116, EV1), intracellular mean fluorescence was unchanged (). A total of 50 µM AZM potentiated the uptake of 1 µM DOX in anoxic HT29 cells by 4.4 fold (p = 0.002). CA9/18 cells exhibited a 12 fold increase in fluorescence of 5 µM DOX in combination with 50 µM DOX under anoxic conditions (p = 0.001). These results indicate that AZM alters passive uptake of DOX.
Uptake of DOX was enhanced in HT29 and CA9/18 cells under anoxic conditions, but uptake was unaffected in the low-activity cell lines. This suggests that potentiation of toxicity by CA IX inhibition was a result of increased uptake. The increased uptake did not correlate precisely with the enhanced toxicity profiles, for example 50 µM AZM enhanced toxicity in air, but did not affect uptake, but was largely consistent with CA IX activity levels and toxicity. The intracellular accumulation of DOX alone in the cell lines, regardless of CA IX status, was lowered in anoxia, correlating with the slight anoxic resistance observed in the toxicity experiments.
AZM causes intracellular acidification
One explanation for altered uptake/toxicity is a change in the pH gradient across cell membranes. The pHi was assessed following 18 hrs incubation with AZM under aerobic and anoxic conditions.
shows the pHi measurements for each cell line in air and anoxia following incubation with the concentrations of AZM used in the combination studies. HCT116 and EV1 cells (neither of which revealed any alteration in toxicity or uptake) had relatively stable pHi measurements upon treatment with AZM under aerobic and anoxic conditions; however pHi slightly increased in anoxic conditions. In contrast, HT29 and CA9/18 cells exhibited decreased pHi with AZM treatment in anoxia. HT29 cells had the largest reduction (1 pH unit) and were more sensitive than CA9/18 cells. AZM acidified the intracellular environment in the cells with highest CA IX levels, correlating with altered toxicity and uptake.
Figure 6. AZM reduced intracellular pH (pHi). The pHi of all the cell lines tested was around neutral and was not substantially affected by AZM under aerobic conditions (left panel). Under anoxic conditions the control pHi (0 µM AZM) in all lines slightly increased in comparison with air. HCT116 and EV1 pHi remained unaltered by AZM treatment under anoxic conditions (solid lines). CA9/18 cells (grey dashed line) exhibited decreased pHi at 50 µM AZM in anoxia. The pHi of anoxic HT29 cells significantly reduced with both concentrations of AZM (right panel). *p < 0.05. Error bars indicate SEM of n = 3 samples. AZM, acetazolamide.
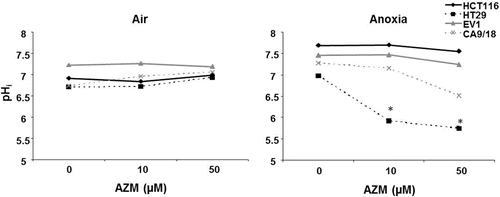
CA IX inhibition resulted in decreased pHi under anoxic conditions in the lines that exhibited alterations in uptake/toxicity. No significant changes in pHi were noted in any of the cell lines in air, in spite of the alterations in toxicity/uptake seen in aerobic CA9/18 cells. pHi was assessed using the pH sensitive fluorescent probe SNARF-1, but it is possible that very subtle changes were not discernable. In addition, other mechanisms could be involved in the aerobic effects of AZM. The pHi of anoxic HT29 cells was altered by a lower AZM concentration than CA9/18, but this is consistent with the changes in uptake/toxicity at 10 µM and 50 µM AZM observed in HT29 cells, but only at 50 µM AZM in CA9/18 cells (10 µM AZM data not shown). Such a shift in the pH balance would feasibly affect the uptake of weak electrolytes, resulting in the enhanced uptake of weak bases such as DOX. The toxicity of anti-cancer agents has been modified by altering the extracellular pHe in vitro, for example by changing the pH of media and/or using ionophore inhibitors to equilibrate the intracellular pHCitation36. Teicher et alCitation59 utilized AZM in combination with several anti-cancer agents in vivo and observed additive growth delays. It was also noted that tumour growth was significantly delayed by administration of AZM alone; however, the doses were relatively high. Intracellular acidification is thought to promote growth inhibition and apoptosis, perhaps contributing to some of the effects of AZMCitation60. In addition, there are other mechanisms that regulate intracellular pH, including the Na+/H+ antiport, Na+ dependent HCO3−/HCl− exchange, which also may have roles in maintaining tumour pH gradientsCitation34,Citation61.
The current results demonstrate that inhibition of a tumour-associated hypoxia-regulated enzyme, CA IX, differentially modified uptake/toxicity of MEL and DOX, by acidifying the intracellular environment. However, AZM is not an isoform specific CA inhibitor and as other isoforms of CA are involved in crucial physiological processes there is a possibility of unwanted side effects with systemic application of this drug. CA IX-specific inhibitors would provide greater selectivity. In this regard, we recently reported about a new generation of CA inhibitors (CAI) specifically targeting the CA IX/XII isoformsCitation62. These compounds belonging to the underexposed sulfamate class of CAI, proved very effective as anti-cancer drugs by themselves in in vitro assays. The lead compound S4 also showed a clear anti-metastatic potential in vivoCitation62. Whether CAI S4 potentiates the effectiveness of DOX is under investigation.
Declaration of interest
Work supported by Program grants to IJS from Medical Research Council (MRC), to ALH from Cancer Research UK (CRUK) and to KJW from European Commission (EC FP7 METOXIA). The authors report no conflict of interest.
References
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011;473:298–307.
- Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res 1989;49:6449–6465.
- Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res 1998;58:1408–1416.
- Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996;379:88–91.
- Semenza GL. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell 2001;107:1–3.
- Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol 2002;64:993–998.
- Erler JT, Bennewith KL, Nicolau M, Dornhöfer N, Kong C, Le QT et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006;440:1222–1226.
- Brown JM. The hypoxic cell: a target for selective cancer therapy–eighteenth Bruce F. Cain Memorial Award lecture. Cancer Res 1999;59:5863–5870.
- Goonewardene TI, Sowter HM, Harris AL. Hypoxia-induced pathways in breast cancer. Microsc Res Tech 2002;59:41–48.
- Hockel M, Schlenger K, Aral B, Mitze M, Schaffer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res 1996;56:4509–4515.
- Höckel M, Schlenger K, Höckel S, Vaupel P. Hypoxic cervical cancers with low apoptotic index are highly aggressive. Cancer Res 1999;59:4525–4528.
- Kim CY, Tsai MH, Osmanian C, Graeber TG, Lee JE, Giffard RG et al. Selection of human cervical epithelial cells that possess reduced apoptotic potential to low-oxygen conditions. Cancer Res 1997;57:4200–4204.
- Brizel DM, Scully SP, Harrelson JM, Layfield LJ, Bean JM, Prosnitz LR et al. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res 1996;56:941–943.
- Chegwidden WR, Dodgson SJ, Spencer IM. The roles of carbonic anhydrase in metabolism, cell growth and cancer in animals. EXS 2000:343–363.
- Ivanov S, Liao SY, Ivanova A, Danilkovitch-Miagkova A, Tarasova N, Weirich G et al. Expression of hypoxia-inducible cell-surface transmembrane carbonic anhydrases in human cancer. Am J Pathol 2001;158:905–919.
- Potter CP, Harris AL. Diagnostic, prognostic and therapeutic implications of carbonic anhydrases in cancer. Br J Cancer 2003;89:2–7.
- Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res 2000;60:7075–7083.
- Svastová E, Hulíková A, Rafajová M, Zat’ovicová M, Gibadulinová A, Casini A et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett 2004;577:439–445.
- Beasley NJ, Wykoff CC, Watson PH, Leek R, Turley H, Gatter K et al. Carbonic anhydrase IX, an endogenous hypoxia marker, expression in head and neck squamous cell carcinoma and its relationship to hypoxia, necrosis, and microvessel density. Cancer Res 2001;61:5262–5267.
- Loncaster JA, Harris AL, Davidson SE, Logue JP, Hunter RD, Wycoff CC et al. Carbonic anhydrase (CA IX) expression, a potential new intrinsic marker of hypoxia: correlations with tumor oxygen measurements and prognosis in locally advanced carcinoma of the cervix. Cancer Res 2001;61:6394–6399.
- Olive PL, Aquino-Parsons C, MacPhail SH, Liao SY, Raleigh JA, Lerman MI et al. Carbonic anhydrase 9 as an endogenous marker for hypoxic cells in cervical cancer. Cancer Res 2001;61:8924–8929.
- Airley RE, Loncaster J, Raleigh JA, Harris AL, Davidson SE, Hunter RD et al. GLUT-1 and CAIX as intrinsic markers of hypoxia in carcinoma of the cervix: relationship to pimonidazole binding. Int J Cancer 2003;104:85–91.
- Hoskin PJ, Sibtain A, Daley FM, Wilson GD. GLUT1 and CAIX as intrinsic markers of hypoxia in bladder cancer: relationship with vascularity and proliferation as predictors of outcome of ARCON. Br J Cancer 2003;89:1290–1297.
- Wike-Hooley JL, van den Berg AP, van der Zee J, Reinhold HS. Human tumour pH and its variation. Eur J Cancer Clin Oncol 1985;21:785–791.
- Stubbs M, Rodrigues L, Howe FA, Wang J, Jeong KS, Veech RL et al. Metabolic consequences of a reversed pH gradient in rat tumors. Cancer Res 1994;54:4011–4016.
- Li Y, Tu C, Wang H, Silverman DN, Frost SC. Catalysis and pH control by membrane-associated carbonic anhydrase IX in MDA-MB-231 breast cancer cells. J Biol Chem 2011;286:15789–15796.
- Chiche J, Ilc K, Laferrière J, Trottier E, Dayan F, Mazure NM et al. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res 2009;69:358–368.
- Parks SK, Chiche J, Pouyssegur J. pH control mechanisms of tumor survival and growth. J Cell Physiol 2011;226:299–308.
- Ojugo AS, McSheehy PM, Stubbs M, Alder G, Bashford CL, Maxwell RJ et al. Influence of pH on the uptake of 5-fluorouracil into isolated tumour cells. Br J Cancer 1998;77:873–879.
- Mahoney BP, Raghunand N, Baggett B, Gillies RJ. Tumour acidity, ion trapping and chemotherapeutics I. Acid pH affects the distribution of chemotherapeutic agents in vitro. Biochem Pharmacol 2003;66:1207–1218.
- Supuran CT. Inhibition of carbonic anhydrase IX as a novel anticancer mechanism. World J Clin Oncol 2012;3:98–103.
- McDonald PC, Winum JY, Supuran CT, Dedhar S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget 2012;3:84–97.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–181.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–777.
- Stillebroer AB, Mulders PF, Boerman OC, Oyen WJ, Oosterwijk E. Carbonic anhydrase IX in renal cell carcinoma: implications for prognosis, diagnosis, and therapy. Eur Urol 2010;58:75–83.
- Wood PJ, Sansom JM, Newell K, Tannock IF, Stratford IJ. Reduction of tumour intracellular pH and enhancement of melphalan cytotoxicity by the ionophore Nigericin. Int J Cancer 1995;60:264–268.
- Robertson N, Potter C, Harris AL. Role of carbonic anhydrase IX in human tumor cell growth, survival, and invasion. Cancer Res 2004;64:6160–6165.
- Wilbur KM, Anderson NG. Electrometric and colorimetric determination of carbonic anhydrase. J Biol Chem 1948;176:147–154.
- Sedlak J, Lindsay RH. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal Biochem 1968;25:192–205.
- Truppo E, Supuran CT, Sandomenico A, Vullo D, Innocenti A, Di Fiore A et al. Carbonic anhydrase VII is S-glutathionylated without loss of catalytic activity and affinity for sulfonamide inhibitors. Bioorg Med Chem Lett 2012;22:1560–1564.
- Giatromanolaki A, Koukourakis MI, Sivridis E, Pastorek J, Wykoff CC, Gatter KC et al. Expression of hypoxia-inducible carbonic anhydrase-9 relates to angiogenic pathways and independently to poor outcome in non-small cell lung cancer. Cancer Res 2001;61:7992–7998.
- Kivela AJ, Saarnio J, Karttunen TJ, Kivelä J, Parkkila AK, Pastorekova S et al. Differential expression of cytoplasmic carbonic anhydrases, CA I and II, and membrane-associated isozymes, CA IX and XII, in normal mucosa of large intestine and in colorectal tumors. Dig Dis Sci 2001;46:2179–2186.
- Bartosová M, Parkkila S, Pohlodek K, Karttunen TJ, Galbavý S, Mucha V et al. Expression of carbonic anhydrase IX in breast is associated with malignant tissues and is related to overexpression of c-erbB2. J Pathol 2002;197:314–321.
- Kaluz S, Kaluzová M, Stanbridge EJ. Expression of the hypoxia marker carbonic anhydrase IX is critically dependent on SP1 activity. Identification of a novel type of hypoxia-responsive enhancer. Cancer Res 2003;63:917–922.
- Wingo T, Tu C, Laipis PJ, Silverman DN. The catalytic properties of human carbonic anhydrase IX. Biochem Biophys Res Commun 2001;288:666–669.
- Supuran CT, Scozzafava A. Carbonic anhydrase inhibitors–Part 94. 1,3,4-thiadiazole-2-sulfonamidederivatives as antitumor agents? Eur J Med Chem 2000;35:867–874.
- Turkmen H, Durgun M, Yilmaztekin S, Emul M, Innocenti A, Vullo D et al. Carbonic anhydrase inhibitors. Novel sulfanilamide/acetazolamide derivatives obtained by the tail approach and their interaction with the cytosolic isozymes I and II, and the tumor-associated isozyme IX. Bioorg Med Chem Lett 2005;15:367–372.
- Parkkila S, Rajaniemi H, Parkkila AK, Kivela J, Waheed A, Pastorekova S et al. Carbonic anhydrase inhibitor suppresses invasion of renal cancer cells in vitro. Proc Natl Acad Sci USA 2000;97:2220–2224.
- Whittington DA, Waheed A, Ulmasov B, Shah GN, Grubb JH, Sly WS et al. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc Natl Acad Sci USA 2001;98:9545–9550.
- Pacchiano F, Carta F, McDonald PC, Lou Y, Vullo D, Scozzafava A et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–1902.
- Lou Y, McDonald PC, Oloumi A, Chia S, Ostlund C, Ahmadi A et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res 2011;71:3364–3376.
- Jähde E, Glüsenkamp KH, Rajewsky MF. Increased drug cytotoxicity at reduced pH counteracts cyclophosphamide resistance in cultured rat mammary carcinoma cells. Int J Cancer 1989;44:1082–1087.
- Matthews NE, Adams MA, Maxwell LR, Gofton TE, Graham CH. Nitric oxide-mediated regulation of chemosensitivity in cancer cells. J Natl Cancer Inst 2001;93:1879–1885.
- Kennedy KA, Siegfried JM, Sartorelli AC, Tritton TR. Effects of anthracyclines on oxygenated and hypoxic tumor cells. Cancer Res 1983;43:54–59.
- Potmesil M, Kirschenbaum S, Israel M, Levin M, Khetarpal VK, Silber R. Relationship of adriamycin concentrations to the DNA lesions induced in hypoxic and euoxic L1210 cells. Cancer Res 1983;43:3528–3533.
- Atema A, Buurman KJ, Noteboom E, Smets LA. Potentiation of DNA-adduct formation and cytotoxicity of platinum-containing drugs by low pH. Int J Cancer 1993;54:166–172.
- Swietach P, Hulikova A, Patiar S, Vaughan-Jones RD, Harris AL. Importance of intracellular pH in determining the uptake and efficacy of the weakly basic chemotherapeutic drug, doxorubicin. PLoS ONE 2012;7:e35949.
- Chen G, Waxman DJ. Role of cellular glutathione and glutathione S-transferase in the expression of alkylating agent cytotoxicity in human breast cancer cells. Biochem Pharmacol 1994;47:1079–1087.
- Teicher BA, Liu SD, Liu JT, Holden SA, Herman TS. A carbonic anhydrase inhibitor as a potential modulator of cancer therapies. Anticancer Res 1993;13:1549–1556.
- Goossens JF, Hénichart JP, Dassonneville L, Facompré M, Bailly C. Relation between intracellular acidification and camptothecin-induced apoptosis in leukemia cells. Eur J Pharm Sci 2000;10:125–131.
- Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res 1989;49:4373–4384.
- Gieling RG, Babur M, Mamnani L, Burrows N, Telfer BA, Carta F et al. Antimetastatic effect of sulfamate carbonic anhydrase IX inhibitors in breast carcinoma xenografts. J Med Chem 2012;55:5591–5600.