
Abstract
In this study, in vitro inhibitory effects of some saccharin derivatives on purified carbonic anhydrase I and II were investigated using CO2 as a substrate. The results showed that all compounds inhibited the hCA I and hCA II enzyme activities. Among the compounds, 6-(p-tolylthiourenyl) saccharin (6m) was found to be the most active one for hCA I activity (IC50 = 13.67 μM) and 6-(m-methoxyphenylurenyl) saccharin (6b) was found to be the most active one for hCA II activity (IC50 = 6.54 μM). Structure–activity relationships (SARs) study showed that, generally, thiourea derivatives (6l--v) inhibited more hCA I and hCA II than urea derivatives (6a–k). All compounds (excluding 6c and 6r) have higher inhibitory activity on hCA II than on hCA I.
Introduction
Carbonic anhydrase (CA, EC 4.2.1.1) is a ubiquitous zinc enzyme. Basically, there are several cytosolic forms (CA-I, CA-II, CA-III and CA-VII), four membrane-bound forms (CA-IV, CA-IX, CA-XII and CA-XIV), one mitochondrial form (CA-V), as well as a secreted CA form (CA-VI)Citation1,Citation2. They all catalyze a very simple physiological reaction, the interconversion between carbon dioxide and the bicarbonate ion, and are thus involved in crucial physiological processes connected with respiration and transport of CO2/bicarbonate between metabolizing tissues and the lungs, pH and CO2 homeostasis, electrolyte secretion in a variety of tissues/organs, biosynthetic reactions (such as the gluconeogenesis, lipogenesis and ureagenesis), bone resorption, calcification, tumorigenicity and many other physiologic or pathologic processesCitation1–3. CA inhibitors have now been a mainstay of human clinical intervention for several decades, with at least 25 clinically used drugs that are CA inhibitorsCitation4. Although there are many studies on this enzyme, the CA enzyme family continues to capture the attention of drug discovery scientists and clinicians as the knowledge regarding the therapeutic implications associated with this enzyme class continues to growCitation4,Citation5.
Saccharin, 1,2-benzisothiazole-3-one-1,1-dioxide, is a well-known heterocyclic compound and has been used as a sweetener in the form of its sodium salt since 1885. Yet it is also a heterocycle of pharmaceutical importance, being a key structural element of certain CNS-active drugsCitation6. Chemically, saccharin consists of a sulfimide with a lactam and cyclic sulfonamide moiety. The latter functionality is responsible for the acidic character of the molecule and suggests its potential to interact with the zinc ion at the floor of the binding pocket of carbonic anhydrases. Köhler et al. presented saccharin as zinc-binding portion based on the X-ray structureCitation7.
Sulfonamides are the best known inhibitors of CA enzymes and are used for the treatment of glaucoma in medicinal chemistryCitation8. Acetazolamide (AAZ), dorzolamide (DZA) and brinzolamide (BRZ) are sulfonamide derivatives and are used in the treatment of glaucoma. However, these drugs have several side effects such as numbness and tingling in the fingers and toes, blurred vision, kidney stones, an increase in urination, upset stomach, dry eye and headache or dizzinessCitation2,Citation9.
There are reportsCitation10,Citation11 of many such aromatic sulfonamides or bis-sulfonamide moieties incorporating urea or thiourea groups as very potent inhibitors against three isozymes, human CA I, human CA II and bovine isozyme CA. A small series of five ureido-substituted benzenesulfonamide derivatives were recently investigated as inhibitors of the cytosolic isoform hCA II by one of these groups. It has been observed that their potency varied between 3.3 and 226 nM, and by means of X-ray crystallography a highly variable orientation of the R-ureido moieties was evidenced when the inhibitor was bound within the enzyme active siteCitation11.
In this study, we evaluated 6-(phenylurenyl/thiourenyl) saccharin derivatives (6a--v), synthesized in the previous workCitation12, effects on hCA I and hCA II purified from human erythrocytes. Additionally, we presented SAR analyses.
Materials and methods
General
Sepharose 4B, l-tyrosine, sulfonamide, synthetic starting material, reagents and solvents were purchased from Merck (Darmstadt, Germany), Alfa Easer (Ward Hill, MA), Sigma-Aldrich (Taufkirchen, Germany) and Fluka (Taufkirchen, Germany).
General procedure for 6-(phenylurenyl/thiourenyl) saccharin derivatives
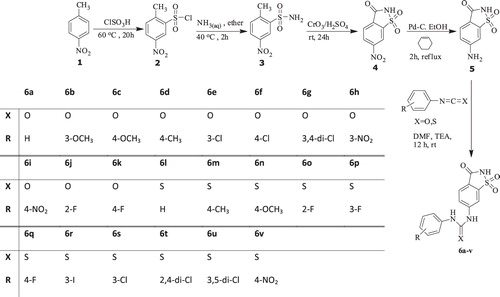
Phenylisocyanate or phenylisothiocyanate derivatives (1 mmol) were added to a solution of 6-aminosaccharin (1 mmol) and triethyl amine (1 mL) in dry DMF. The mixture was stirred at room temperature for 12 h and then poured into cold 1 M HCl. The precipitate was filtered and washed with cold water. The crude products were recrystallized from ethanol over 99% purity. The synthetic procedures are depicted in .
Scheme 1. Synthesis of 6-(phenylurenyl/thiourenyl) saccharin (6a--v) derivatives.
Preparation of hemolysate and purification from blood red cells
Preparation of hemolysate and purification from blood red cells made by the literatureCitation13 was presented in supporting information.
CA enzyme assay
CA activity measured by the Maren methodCitation14 was presented in supporting information.
In vitro inhibition studies
For the inhibition studies of saccharin, different concentrations of these compounds were added to the enzyme. Activity percentage values of CA for different concentrations of each saccharin were determined by regression analysis using Microsoft Office 2000 Excel. CA enzyme activity without a saccharin solution was accepted as 100% activity.
Results and discussion
Chemistry
The synthetic procedures are depicted in . 6-(Phenylurenyl/thiourenyl) saccharin compounds (6a–v) were synthesized from 4-nitrotoluene (1) in five steps by known proceduresCitation12.
Biological evaluation of saccharin derivatives for hCA I and hCA II inhibitory activities
For evaluating the hCA I and hCA II inhibitory effects, all compounds were subjected to hCA I and hCA II inhibition assay with CO2 as a substrate. The result showed that all compounds (6a--v) inhibited the hCA I and hCA II enzyme activity.
The IC50 values and inhibition constants of 6a--v analogues against hCA I and hCA II are summarized in and the IC50 graphs are given in . The IC50 figures are presented as supporting information.
Figure 1. IC50 graphics of saccharin derivatives (5, 6a--v) on hCA I and hCA II.
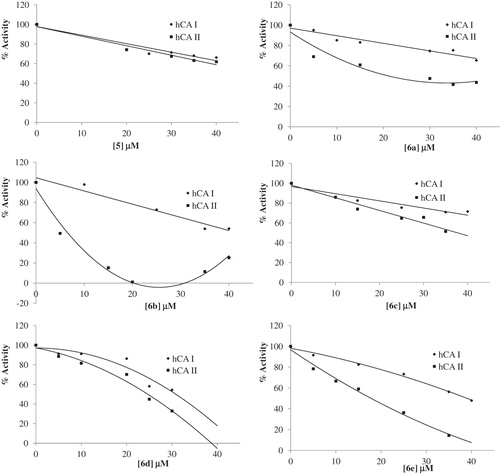
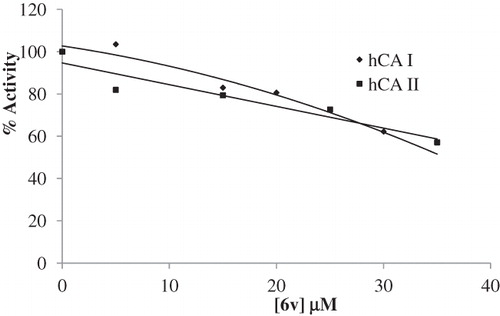
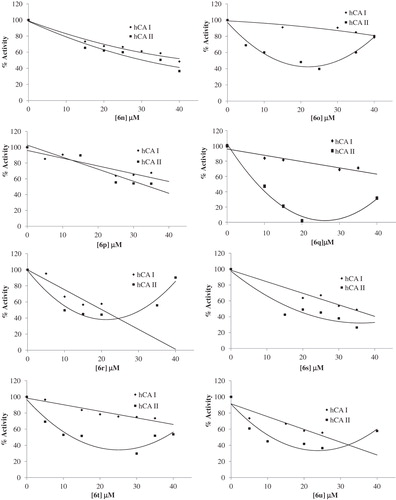
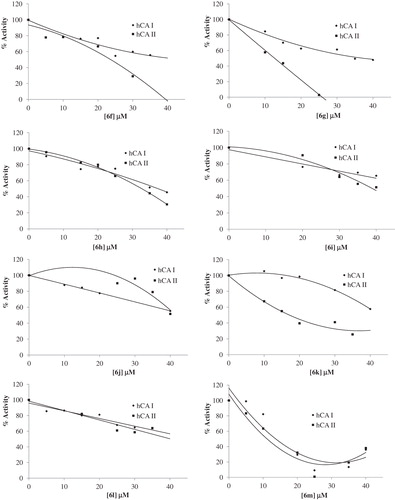
Table 1. Inhibitory effects of saccharin derivatives on hCA I and hCA II.
We have determined the IC50 values of 13.57–74.90 µM for the inhibition of hCA I and 6.54–49.00 µM for the inhibition of hCA II. Among all compounds, 6m (IC50 = 13.67 µM) was found to be the most active one for hCA I inhibitory activity and 6b (IC50 = 6.54 µM) showed the highest hCA II inhibitory activity. 6d (IC50 = 30.81 µM) was found to be the most active one for hCA I inhibitory activity and 6b (IC50 = 6.54 µM) showed the highest hCA II inhibitory activity for the urea derivatives. Among the thiourea derivatives, 6m (IC50 = 13.67 µM) showed the highest hCA I inhibitory activity and 6q (IC50 = 8.10 µM) showed the highest hCA II inhibitory activity.
It was reportedCitation7 that saccharin most likely coordinates in a deprotonated state through its nitrogen atom to the catalytically active zinc ion. Additionally, ureido-substituted benzenesulfonamide moieties were evidenced when the inhibitor was bound within the enzyme active siteCitation10,Citation11. We believe that the synthesized saccharin urea/thiourea derivatives inhibited hCA I and II in the same way.
Structure–activity relationships
Generally, we have seen that all compounds (excluding 6c and 6r) have a higher inhibitory activity on hCA II than hCA I in the SARs study. When same substituents bonded to phenyl ring, most of the thiourea derivatives (6l--v) exhibited higher hCA I and hCA II inhibitory activity than urea derivatives (6a--k). Additionally, the following results were obtained.
For urea derivatives:
Although electron-withdrawing groups (nitro and halogens) bonded to meta position of the phenyl ring (6e, 6g and 6h) increased the inhibitory activity on hCA I, electron-donating groups (methoxy) bonded to meta position of phenyl ring (6b) had the highest hCA II inhibitory activity (IC50 = 6.54 µM).
Electron-donating groups (methoxy, methyl) bonded to para position of the phenyl ring (6c and 6d) inhibited hCA I activity more than halogens and electron-withdrawing groups bonded. On the other hand, halogen groups bonded to the para position of the phenyl ring (6f, 6g and 6k) increased the inhibitory activity on hCA II.
For thiourea derivatives:
Electron-donating groups bonded to the para position of the phenyl ring (6m and 6n) increased the hCA I inhibitory activity.
Moving-F group on the phenyl ring from ortho (6o, IC50 = 30.23 µM) to meta (6p, IC50 = 14.55 µM) and para (6q, IC50 = 8.10 µM) positions led to major enhancement of hCA II inhibitory activity.
In same moving for hCA I activity, 6p (IC50 = 46.70 µM) was more inhibited than 6o (IC50 = 74.90 µM) and 6q (IC50 = 55.87 µM).
Halogen series on the meta position of the phenyl ring showed a linear relationship for higher hCA I inhibitory activity with increasing size and polarizability (for size and polarizability, I > Cl > F, for hCA I inhibitory activity, 6r (IC50 = 20.21 µM) > 6s (IC50 = 33.41 µM) > 6p (IC50 = 46.70 µM). Interestingly, this series showed an inverse relationship for hCA II inhibitory activity with increasing size and polarizability (for hCA II inhibitory activity, 6r (IC50 = 30.70 µM) < 6s (IC50 = 17.19 µM) < 6p (IC50 = 14.55 µM).
Conclusions
In conclusion, we evaluated 6-(phenylurenyl/thiourenyl) saccharin derivatives (6a--v) effects on hCA I and hCA II purified from human erythrocytes and SARs were examined. All compounds inhibited both hCA I and hCA II enzyme activities. Most of the compounds had higher hCA II inhibitory activity than hCA I activity. Most of the thiourea derivatives (6l--v) exhibited higher hCA I and hCA II inhibitory activities than urea derivatives (6a--k). The present study revealed that activity could also be influenced by the type and position of the substituent on the phenyl ring. Among all compounds, 6m showed the highest hCA I inhibitory activity and 6b showed the highest hCA II inhibitory activity.
In summary, enzyme inhibition is the most important issue for drug design and biochemical applicationsCitation15–27. Therefore, our results suggested that saccharin derivatives are likely to be adopted as candidates to treat glaucoma and may be taken for further evaluation in in vivo studies.
Declaration of interest
The authors report no conflicts of interest.
This work was supported by Sakarya University Scientific Research Project (Project No. 2011–50-02-020).
References
- Supuran CT, Scozzafava A. Carbonic anhydrase inhibitors. Curr Med Chem Imm Endoc Metab Agents 2001;1:61–97
- Supuran CT, Scozzafava A. Carbonic anhydrase inhibitors and their therapeutic potential. Exp Opta Ther Pat 2000;10:575–9
- Hewett-Emmet D. The carbonic anhydrase – new horizons. In: Chegwidden WR, Edwards Y, Carter N, eds. Evolution and distribution of the carbonic anhydrase gene families. Basel: Birkhauser Verlag; 2000:29–78
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Disc 2008;7:168–81
- Poulsen SA. Carbonic anhydrase inhibition as a cancer therapy: a review of patent literature 2007–2009. Exp Opin Ther Pat 2010;20:795–806
- Yeung KS, Meanwell NA, Li Y, Gao Q. A facile construction of 4-hydroxymethylbenzisothiazolone-1,1-dioxide. Tetrahedron Lett 1998;39:1483–6
- Köhler K, Hillebrecht A, Wischeler JS, et al. Saccharin inhibits carbonic anhydrases: possible explanation for its unpleasant metallic aftertaste. Angew Chem Int Ed 2007;46:7697–9
- Kasımoğulları R, Bülbül M, Arslan BS, Gökçe B. Synthesis, characterization and antiglaucoma activity of some novel pyrazole derivatives of 5-amino-1,3,4-thiadiazole-2-sulfonamide. Eur J Med Chem 2010;45:4769–73
- Sugrue MF. Pharmacology and ocular hypotensive properties of topical carbonic anhydrase inhibitors. Prog Ret Eye Res 2000;19:87–112
- Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureidobenzenesulfonamides and correlate to inhibitor potency. Chem Commun 2010;46:8371–3
- Pacchiano F, Carta F, McDonald PC, et al. Ureido-Substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902
- Gencer N, Demir D, Sonmez F, Kucukislamoglu M. New saccharin derivatives as tyrosinase inhibitors. Bioorg Med Chem 2012;20:2811–21
- Arslan O, Nalbantoglu B, Demir N, et al. A new method for the purification of carbonic anhydrase isozymes by affinity chromatography. Turk J Med Sci 1996;26:163–6
- Maren TH. A simplified micromethod for the determination of carbonic anhydrase and its inhibitors. J Pharm Exp Ther 1960;130:2629–34
- Aydemir T, Kavrayan D. Purification and characterization of Glutathione-S-Transferase from chicken erythrocyte. Artif Cell Blood Sub Biotechnol 2009;37:92–100
- Gencer N, Ergün A, Demir D. In vitro effects of some anabolic compounds on erythrocyte carbonic anhydrase I and II. J Enzyme Inhib Med Chem 2012;27:208–10
- Sinan S, Gencer N, Turan Y, Arslan O. In vitro inhibition of the carbonic anhydrase from saanen goat (Capra hircus) with pesticides. Pest Biochem Phys 2007;88:307–11
- Kiranoglu S, Sinan S, Gencer N, et al. In vivo effects of oral contraceptives on paraoxonase, catalase and carbonic anhydrase enzyme activities on mouse. Biol Pharm Bull 2007;30:1048–51
- Sinan S, Kockar F, Gencer N, et al. Amphenicol and macrolide derived antibiotics inhibit paraoxonase enzyme activity in human serum and human hepatoma cells (HepG2) in vitro. Biochem Moscow 2006;71:46–50
- Demir D, Gençer N, Er A. Purification and characterization of prophenoloxidase from Galleria mellonella L. Artif Cell Blood Sub Biotechnol 2012;40:391–5
- Gokce B, Gencer N, Arslan O, et al. Evaluation of in vitro effects of some analgesic drugs on erythrocyte and recombinant carbonic anhydrase I and II. J Enzyme Inhib Med Chem 2012;7:37–42
- Senturk M, Alici HA, Beydemir S, Kufrevioglu OI. In vitro and in vivo effects of some benzodiazepine drugs on human and rabbit erythrocyte carbonic anhydrase enzymes. J Enzyme Inhib and Med Chem 2012;27:680–4
- Arslan M, Gençer N, Arslan O, Guler OO. In vitro efficacy of some cattle drugs on bovine serum paraoxonase 1 (PON1) activity. J Enzyme Inhib Med Chem 2012;27:722–9
- Sayin D, Cakir DT, Gencer N, Arslan O. Effects of some metals on Paraoxonase activity from shark Scyliorhinus canicula. J Enzyme Inhib Med Chem 2012;27:595–8
- Cankaya M, Aktas M, Kuzucu M, et al. Effects of some drugs on human cord blood erythrocyte carbonic anhydrases I and II: an in vitro study. J Enzyme Inhib Med Chem 2012;27:641–5
- Senturk M, Ekinci D, Goksu S, Supuran CT. Effects of dopaminergic compounds on carbonic anhydrase isozymes I, II, and VI. J Enzyme Inhib and Med Chem 2012;27:365–9
- Ekinci D, Al-Rashida M, Abbas G, et al. Chromone containing sulfonamides as potent carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:744–7