
Abstract
A series of small molecules were designed and synthesized based on our previous virtual screening approach, which was performed to discover potent histone deacetylase inhibitors (HDACIs) with novel structures. The derived compounds were tested by Hela cell nucleus extract for enzyme inhibition assay. Tumor cell growth inhibition assays were performed using a series of tumor cell lines. Molecule 4h has the best performance among these compounds with enzyme inhibition IC50 of 0.14 μM and tumor cell growth inhibition IC50 of 1.85 (U937), 2.02 (HL60), 2.67 (K562). Docking studies showed that multiple H-bonds and hydrophobic interactions make 4h binding to the active site of HDAC. 4h has the advantage of low molecular weight, so a variety of structural modifications can be performed in our further studies.
Introduction
Histone deacetylases (HDACs) are a family of 18 enzymes responsible for the regulation of transcription and other nuclear events by regulating the deacetylation of N-terminal tails of histones. They are subdivided into four classes, class I (HDAC1, 2, 3 and 8), class II (IIa: HDAC4, 5, 7 and 9; IIb: HDAC6 and 10) and class IV (HDAC11) are zinc-dependent enzymes. Class III (known as sirtuins, Sirt1-7) consists of NAD-dependent enzymesCitation1–3.Overexpression and aberrant recruitment of HDACs have a significant role in tumorigenesisCitation4–6.
Histone deacetylase inhibitors (HDACIs) have been proved to have the effects of apoptosis, cell cycle arrest and differentiationCitation7–9. Suberoyl anilide hydroxamic acid (SAHA)Citation10 and FK228Citation11 have been approved by FDA for treatment of advanced cutaneous T-cell lymphoma (CTCL). Many HDACIs are still under investigation in various stages of clinical trials for treatment of cancer by administration individually or in combination.
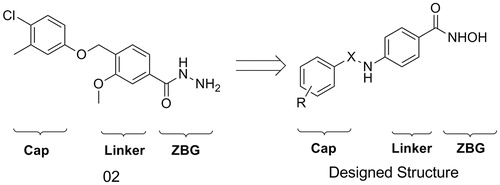
In order to discover new scaffold of HDACIs, a virtual screening approach was performed in our previous workCitation12. A potent small molecule HDAC inhibitor (02) was derived as a lead structure. Although 02 has similar enzyme inhibition activity compared with SAHA, it has no activity in the tumor cell growth inhibition assays. Thus, structural modification was performed to discover more potent HDACIs (). Hydroximic acid group was used to bind to zinc ion, sulphonamide and amide groups were introduced to the linker to improve H-bond interactions. A series of novel hydroximic acid-based compounds were synthesized and tested by enzyme inhibition and tumor cell growth inhibition activity assay. A phenylglycine containing structure (4h) was discovered to have good performance in these activity assays.
Figure 1. Design of new scaffold from the lead structure.
Materials and methods
Chemistry
1H NMR spectra were recorded on a Bruker DRX spectrometer at 600 MHz, δ in parts per million and J in Hertz, using TMS as an internal standard. High-resolution mass spectra were conducted by Shandong Analysis and Test Center in Ji’nan, China. ESI-MS were determined on an API 4000 spectrometer (AB SCIEX, Framingham, MA). Melting points were determined uncorrected on an electrothermal melting point apparatus. Optical rotation values were measured at room temperature using a modular circular polarimeter 200 operating at λ = 589 nm.
Methyl 4-aminobenzoate hydrochloric acid (2)
4-aminobenzoic acid (1) (13.7 g, 100 mmol) was dissolved in 100 mL of MeOH, acetyl chloride (23.6 g, 300 mmol) was added under ice bath. The solution was stirred at 75 °C for 5 h. Then, the solvent was evaporated with the desired compound 2 (14.4 g, 77% yield) being recrystallized as white powder by ester. ESI-MS: m/z: 152.1 [M + H]+.
(S)-2-((tert-butoxycarbonyl)amino)-2-phenylacetic acid (h2)
(S)-2-amino-2-phenylacetic acid (h1) (15.1 g, 100 mmol) was dissolved in 200 mL of MeOH/H2O (3:1), Et3N (15.1 g, 150 mmol) and (Boc)2O (32,7 g, 150 mmol) were added in turn. After stirred at room temperature for 8 h, the solvent was evaporated with the residue being dissoved in saturated citric acid (100 mL). The solution was extracted with EtOAc (3 × 60 mL). The EtOAc solution was washed with saturated brine (3 × 50 mL), dried over MgSO4, and evaporated under vacuum. The desired compound h2 (21.6 g, 86% yield) was derived by crystallization in hexane as white powder. ESI-MS: m/z: 252.1 [M + H]+.
N-hydroxy-4-(4-nitrophenylsulfonamido)benzamide (3a)
To a solution of compound 2 (0.56 g, 3.0 mmol) in THF (50 mL)/H2O (1.0 mL), NaHCO3 (1.01 g, 12 mmol) was added. After 10 min, 4-nitrobenzene-1-sulfonyl chloride (0.73 g, 3.3 mmol) was added. The reaction solution was stirred at room temperature for 8 h. Then, the solvent was evaporated with the residue being taken up in EtOAc (50 mL). The EtOAc solution was washed with saturated citric acid (3 × 20 mL), NaHCO3 (3 × 20 mL) and brine (3 × 20 mL), dried over MgSO4, and evaporated under vacuum. The desired compound 3a (0.68 g, 70% yield) was derived by crystallization in EtOAc as white powder. ESI-MS: m/z: 337.1 [M + H]+.
The other compounds (3b–3g) were prepared in the same procedure as described above.
(S)-methyl 4-(2-((tert-butoxycarbonyl)amino)-2-phenylacetamido)benzoate (3h)
To a solution of h2 (1.26 g, 5 mmol) in DCM (50 mL), Et3N (0.55 g, 5.5 mmol) and TBTU (1.8 g, 5.5 mmol) were added in turn. After 20 min, compound 2 (0.94 g, 5 mmol) and Et3N (0.50 g, 5 mmol) were added. The reaction solution was stirred at room temperature for 8 h. Then, the solvent was evaporated with the residue being taken up in EtOAc (50 mL). The EtOAc solution was washed with saturated citric acid (3 × 20 mL), NaHCO3 (3 × 20 mL) and brine (3 × 20 mL), dried over MgSO4, and evaporated under vacuum. The desired compound 3h (1.44 g, 75% yield) was derived by crystallization in EtOAc as white powder. ESI-MS: m/z: 385.2 [M + H]+.
N-hydroxy-4-(4-nitrophenylsulfonamido)benzamide (4a)
Compound 3a (0.5 g, 1.6 mmol) was dissolved in 14 mL of NH2OK (0.56 g, 24 mmol) methanol solution. After 2 h, the solvent was evaporated under vacuum. The residue was acidified with saturated citric acid, and then extracted with EtOAc (3 × 20 mL). The organic layers were combined, washed with brine (3 × 20 mL) and dried over MgSO4. The desired compound 4a (0.33 g, 62% yield) was derived by crystallization in EtOAc as white powder; m.p. = 190–191 °C; 1H NMR (600 MHz, (CD3)2SO) δ 11.08 (s, 1H), 10.63 (s, 1H), 8.95 (s, 1H), 8.38 (d, J = 9 Hz, 2H), 8.05 (d, J = 8.4 Hz, 2H), 7.65 (d, J = 8.4 Hz, 2H), 7.16 (d, J = 6 Hz, 2H). HRMS (AP-ESI) m/z calcd for C13H12N3O6S [M + H]+ =337.0447, found = 338.0442. Retention time: 2.9 min.
The other compounds (4b–4h and h3) were prepared in the same procedure as described above.
4-(4-acetamidophenylsulfonamido)-N-hydroxybenzamide (4b)
Title compound was obtained as an amorphous white solid (0.35 g, 64% yield); m.p. = 214–216 °C; 1H NMR (600 MHz, (CD3)2SO) δ 11.02 (s, 1H), 10.68 (s, 1H), 10.30 (s, 1H), 8.91 (s, 1H), 7.82 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 10.8 Hz, 2H), 7.71 (d, J = 5.4 Hz, 2H), 7.19 (d, J = 6 Hz, 2H), 2.06 (s, 3H). HRMS (AP-ESI) m/z calcd for C15H16N3O5S [M + H]+ = 350.0810, found = 350.0805. Retention time: 2.9 min.
4-(4-chlorophenylsulfonamido)-N-hydroxybenzamide (4c)
Title compound was obtained as an amorphous white solid (0.32 g, 61% yield); m.p. = 202–204 °C; 1H NMR (600 MHz, (CD3)2SO) δ 11.06 (s, 1H), 10.71 (s, 1H), 8.94 (s, 1H), 7.80 (d, J = 8.4 Hz, 2H), 7.65 (d, J = 7.2 Hz, 2H), 7.63 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 9 Hz, 2H). HRMS (AP-ESI) m/z calcd for C13H12ClN2O4S [M + H]+ = 327.0200, found = 327.0202. Retention time: 2.9 min.
4-(4-fluorophenylsulfonamido)-N-hydroxybenzamide (4d)
Title compound was obtained as an amorphous white solid (0.28 g, 57% yield); m.p. = 181–183 °C; 1H NMR (600 MHz, (CD3)2SO) δ 11.05 (s, 1H), 10.66 (s, 1H), 8.93 (s, 1H), 7.87 (d, J = 8.4 Hz, 2H), 7.63 (d, J = 8.4 Hz, 2H), 7.42 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H). HRMS (AP-ESI) m/z calcd for C13H12FN2O4S [M + H]+ = 311.0502, found = 311.0496. Retention time: 2.9 min.
N-hydroxy-4-(naphthalene-2-sulfonamido)benzamide (4e)
Title compound was obtained as an amorphous white solid (0.36 g, 66% yield); m.p. = 237–239 °C; 1H NMR (600 MHz, (CD3)2SO) δ 11.06 (s, 1H), 10.97 (s, 1H), 8.88 (s, 1H), 8.72 (d, J = 8.4 Hz, 1H), 8.28 (d, J = 7.2 Hz, 1H), 8.23 (d, J = 8.4 Hz, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.75 (t, J = 7.8 Hz, 1H), 7.68 (d, J = 7.2 Hz, 1H), 7.65 (t, J = 7.8 Hz, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.08 (d, J = 9 Hz, 1H). HRMS (AP-ESI) m/z calcd for C17H15N2O4S [M + H]+ = 343.0752, found = 343.0746. Retention time: 2.9 min.
4-(2,6-difluorophenylsulfonamido)-N-hydroxybenzamide (4f)
Title compound was obtained as an amorphous white solid (0.35 g, 63% yield); m.p. = 210–212 °C; 1H NMR (600 MHz, (CD3)2SO) δ 11.24 (s, 1H), 11.05 (s, 1H), 8.94 (s, 1H), 7.20 (d, J = 8.4 Hz, 1H), 7.65 (d, J = 9 Hz, 2H), 7.30 (d, J = 9 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H). HRMS (AP-ESI) m/z calcd for C13H11F2N2O4S [M + H]+ = 329.0407, found = 329.0404. Retention time: 2.9 min.
N-hydroxy-4-(4-(trifluoromethyl)phenylsulfonamido)benzamide (4g)
Title compound was obtained as an amorphous white solid (0.34 g, 62% yield); m.p. = 249–250 °C; 1H NMR (600 MHz, (CD3)2SO) δ 11.06 (s, 1H), 10.87 (s, 1H), 8.94 (s, 1H), 8.01 (d, J = 8.4 Hz, 2H), 7.98 (d, J = 9 Hz, 2H), 7.64 (d, J = 8.4 Hz, 2H), 7.16 (d, J = 9 Hz, 2H). HRMS (AP-ESI) m/z calcd for C14H12F3N2O4S [M + H]+ = 361.0470, found = 361.0463. Retention time: 2.9 min.
(S)-tert-butyl (2-((4-(hydroxycarbamoyl)phenyl)amino)-2-oxo-1-phenylethyl)carbamate (4h)
Title compound was obtained as an amorphous white solid (0.37 g, 63% yield). = −6.26 (c = 1.0, MeOH); m.p. = 200–202 °C; 1H NMR (600 MHz, (CD3)2SO) δ 11.10 (s, 1H), 10.44 (s, 1H), 8.93 (s, 1H), 7.70 (d, J = 9 Hz, 2H), 7.63 (d, J = 9 Hz, 2H), 7.56 (d, J = 8.4 Hz, 1H), 7.49 (d, J = 7.2 Hz, 2H), 7.37–7.35 (m, 2H), 7.31 (d, J = 7.2 Hz, 1H), 5.36 (d, J = 7.2 Hz, 1H), 1.40 (s, 9H). HRMS (AP-ESI) m/z calcd for C20H23N3NaO5 [M + Na]+ = 408.1536, found = 408.1529. Retention time: 2.9 min.
(S)-tert-butyl (2-(hydroxyamino)-2-oxo-1-phenylethyl)carbamate (h3)
Title compound was obtained as an amorphous white solid (0.34 g, 61% yield); m.p. = 154–155 °C; = −6.78 (c = 1.0, MeOH). 1H NMR (600 MHz, (CD3)2SO) δ 10.88 (s, 1H), 8.96 (s, 1H), 7.40 (d, J = 7.8 Hz, 2H), 7.37 (d, J = 8.4 Hz, 1H), 7.33 (d, J = 7.2 Hz, 2H), 7.27 (t, J = 7.2 Hz, 1H), 5.05 (d, J = 8.4 Hz, 1H), 1.38 (s, 9H). HRMS (AP-ESI) m/z calcd for C13H18N2NaO4 [M + Na]+ = 289.1165, found = 289.1158. Retention time: 2.9 min.
(S)-4-(2-amino-2-phenylacetamido)-N-hydroxybenzamide hydrochloride (4hb)
To a solution of compound 4h (0.38 g, 1 mmol) in EtOAc (10 mL), a solution of EtOAc (15 mL) saturated by dry HCl gas was added. The reaction solution was stirred at room temperature for 5 h. Precipitates appeared and were filtered, with the filter being washed with ether, to give desired compound 4hb (0.19 g, 68% yield). Title compound was obtained as an amorphous white solid; m.p. = 187–188 °C; = −6.53 (c = 1.0, MeOH). 1H NMR (600 MHz, (CD3)2SO) δ 11.45 (s, 1H), 10.31 (s, 1H), 8.92–8.91 (m, 3H), 7.73 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 8.4 Hz, 2H), 7.69 (d, J = 7.2 Hz, 2H), 7.46 (d, J = 7.8 Hz, 2H), 7.43 (t, J = 7.2 Hz, 1H), 5.33 (d, J = 5.4 Hz, 1H). HRMS (AP-ESI) m/z calcd for C15H16N3O3 [M + H]+ = 286.1191, found = 286.1186. Retention time: 2.9 min.
Compound h4 was prepared in the same procedure as described above.
(S)-2-amino-N-hydroxy-2-phenylacetamide hydrochloride (h4)
Title compound was obtained as an amorphous white solid (0.21 g, 70% yield); m.p. = 209–210 °C; = −7.21 (c = 1.0, MeOH). 1H NMR (600 MHz, (CD3)2SO) δ 11.10 (s, 1H), 9.30 (s, 1H), 8.87 (s, 3H), 7.56 (d, J = 6.6 Hz, 2H), 7.45 (d, J = 7.2 Hz, 2H), 7.43–7.41 (m, 5H), 4.85 (s, 1H). HRMS (AP-ESI) m/z calcd for C8H11N2O2 [M + H]+ = 167.0820, found = 167.0810. Retention time: 2.9 min.
Enzyme inhibition assay
The HDAC enzyme was extracted from Hela cells. Boc-Lys (acetyl)-AMC was used as the substrate of HDAC. SAHA which is the HDAC inhibitor on market was used as a positive control. The compounds were diluted to six concentrations (25, 5, 1, 0.2, 0.04 and 0.008 uM/L) to investigate their ability of inhibiting HDAC activity.
Tumor cell inhibition assay
HCT116, K562, MDA-MB-231, ES-2, U937, HL60, MCF7 and PC3 cells were used in the cell proliferation assays. A total of 5000 cells were seeded into each well of 96-well plates which were incubated at 37 °C, 5% CO2 for overnight. Then cells (HCT116, K562, MDA-MB-231, ES-2) were treated with 5 μM of all these molecules. In order to calculate the IC50 values, cells (U937, HCT116, K562, HL60, MCF7 and PC3) were treated with compound 4h and SAHA at final concentrations ranging from 0.25 to 20 μM. After 48 h of incubation, 0.5% MTT (Amresco, Solon, OH) solution was added to each well. Then a further 4 h of incubation was performed. After that the culture medium was removed, and 200 μL of DMSO was added to dissolve the formazan. After mixing for 5 min, optical density values were detected at 570 nm on a microplate reader (Thermo Fisher Scientific, Waltham, MA).
Molecular docking
The molecular docking process was performed using Glide (www.schrodinger.com). Crystal structure of HDAC2 (PDB Entry: 3MAX) was selected as a receptor in the docking study. Structural modifications were performed to make the protein suitable for docking. The water molecules and the ligand crystallized in the protein structure were removed. OPLS 2005 force field was assigned to the refined structure. The ligands used in the docking approach were sketched by maestro and prepared by LigPrep (www.schrodinger.com). The active site was defined as a cubic box containing residues around Zn ion at a distance of 20 Å. Extra precision was applied in the docking process, other parameters were set as default.
Results and discussion
Chemistry
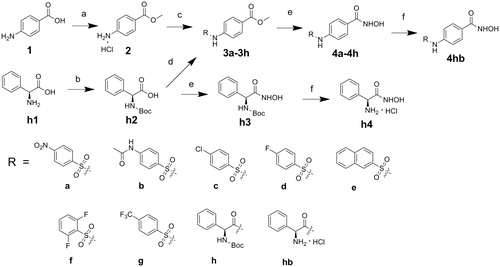
The target compounds were synthesized following the route shown in . The starting material 4-aminobenzoic acid was converted to be compound 2 by esterification. The key intermediates (3a–3g) were generated by nucleophilic substituation of compound 2 with corresponding sulfuryl chlorideCitation13–18. L-phenylglycine was protected by Boc, followed by coupling with compound 2 to yield 3h. Compounds 4a–4h were derived by treating the corresponding intermediates (3a–3h) with NH2OK in methanolCitation19. Compound 4hb was derived by deprotection of the amino group of 4h. Compounds h3 and h4 were prepared via the same route as 4hb.
Scheme 1. Reagents and conditions: (a) CH3COCl, CH3OH; (b) (Boc)2O, Et3N, MeOH/H2O; (c) RSO2Cl, NaHCO3, THF/H2O; (d) TBTU, Et3N, THF; (e) NH2OK, MeOH; (f) EtOAc, HCl.
Activity assay
The derived compounds were assayed by Hela cell nucleus extract using SAHA as a positive control (). Molecule 4h has the best performance in the enzyme inhibition assay. Among compounds 4a–4g, 4e has the best enzyme inhibition activity. The result means bulky R groups are beneficial for these molecules binding to HDACs. Protection of the amino group by Boc group in the phenylglycine improved the activity, which was proved by comparing the IC50 values between 4h (0.14 μM) and 4hb (0.18 μM), h3 (1.60 μM) and h4 (3.79 μM). The results indicate that substituent in the amino group improves the interaction between the ligands and the receptor. Molecules h3 and h4 were subsequently synthesized to find out if the phenylglycine structure has enzyme inhibition activity without a linker. The results suggest that removal of the linker results in the reduction of the activity.
Table 1. The inhibitory activities of derived molecules.*
The results of the tumor cell growth inhibition assay have correlation with the results of the enzyme inhibition assay (). K562 cells are more sensitive to these molecules than other cells. In these assays, 4h is also the most potent inhibitor. Deprotection of the amino group in the phenylglycine and removal of the linker have significant influence on the inhibitory activity.
The IC50 values of 4h in inhibition of U937, HCT116, K562, HL60, MCF7 and PC3 cell growth were derived compared with SAHA (). The results showed that 4h could efficiently inhibit the growth of these cell lines. In inhibition, the growth of K562, HL60 and PC3 cells, 4h even has similar performance with SAHA. Our results also indicate that 4h can selectively inhibit the growth lymphoma cells and hematological tumor cells.
Table 2. The tumor cell growth inhibitory activities of 4h compared with SAHA.*
Binding pattern analysis
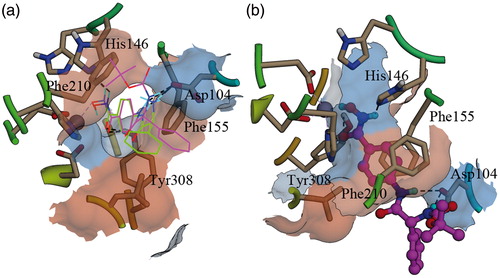
Docking study was performed to simulate the binding pattern between 4h and HDAC. In order to seek evidence for the difference of the activity between 4h and 4hb, 4hb was also docked to the active site of HDAC2. The docking result shows that 4hb has similar binding pattern with 4h (). The hydroximic acid groups of both molecules play a significant role in binding the ligands to the receptor by chelating to the zinc ion and forming H-bond interactions with residues around zinc ion (). The ketonic oxygen of the hydroximic acid group has H-bond interaction with the phenolic hydroxyl group of Tyr308. Nevertheless, the amino group binds to the NE2 of His164 by forming H-bond interactions. The hydroxyl group of Asp104 has H-bond binding with the other two amino groups, which also make important contributions to the binding. The benzene ring of the linker has π–π stacking interactions with the phenyl groups of Phe155 and Phe210, which improves the binding of these two molecules to the active site of HDAC.
Figure 2. Binding patterns of 4h and 4hb in the active site of HDAC2: (a) 4h and 4hb were shown using the wire model, the residues were displayed as sticks; carbon atoms of 4h were colored magenta, carbon atoms of 4hb were colored green, the gray ball is zinc ion, the H-bonds were represented as dash lines (in black--white mode, 4h is the structure containing Boc group); (b) 4h was shown using the ball–stick model.
Conclusion
In order to develop potent HDAC inhibitors with efficient tumor cell inhibitory activities, a series of small molecules were synthesized based on our previous virtual screening results. The enzyme inhibition assay and tumor cell growth inhibition assay results show that 4h is the most potent HDAC inhibitor. Moreover, 4h also has good performance in inhibiting the growth of a series of tumor cell lines compared with SAHA. An important finding is introduction of bulky group to the amino group of the phenylglycine part that can obviously improve the inhibitory activity. Our work emphasizes the prospect and necessity of development HDAC inhibitors with various substituent groups in the amino group of phenylglycine in our further work.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
This work was supported by National Nature Science Foundation of China (Grant No. 90713041), National High Technology Research and Development Program of China (Grant No. 2007AA02Z314 and 2009ZX09103-118) and Graduate Innovation Foundation of Shandong University (GIFSDU) (No. yyx10020).
References
- Bernstein BE, Tong JK, Schreiber SL. Genomewide studies of histone deacetylase function in yeast. P Natl Acad Sci USA 2000;97:13708–13
- De Ruijter AJM, Van Gennip AH, Caron HN, et al. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 2003;370:737–49
- Foglietti C, Filocamo G, Cundari E, et al. Dissecting the biological functions of Drosophila histone deacetylases by RNA interference and transcriptional profiling. J Biol Chem 2006;281:17968–76
- Bode AM, Dong ZG. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer 2004;4:793–805
- Chen LF, Greene WC. Shaping the nuclear action of NF-kappa B. Nat Rev Mol Cell Bio 2004;5:392–401
- Kovacs JJ, Murphy PJM, Gaillard S, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell 2005;18:601–7
- Marks PA, Rifkind RA, Richon VM, et al. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer 2001;1:194–202
- Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov 2002;1:287–99
- Insinga A, Monestiroli S, Ronzoni S, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med 2005;11:233
- Richon VM, Emiliani S, Verdin E, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. P Natl Acad Sci USA 1998;95:3003–7
- Ueda H, Nakajima H, Hori Y, et al. Fr901228, a novel antitumor bicyclic depsipeptide produced by chromobacterium-violaceum no-968.1. Taxonomy, fermentation, isolation, physicochemical and biological properties, and antitumor-activity. J Antibiot 1994;47:301–10
- Zhang L, Li MY, Feng JH, et al. Discovery of a novel histone deacetylase 8 inhibitor by virtual screening. Med Chem Res 2012;21:152–6
- D'Ambrosio K, Smaine F-Z, Carta F, et al. Development of potent carbonic anhydrase inhibitors incorporating both sulfonamide and sulfamide groups. J Med Chem 2012;55:6776–83
- Akdemir A, Vullo D, Luca VD, et al. The extremo-alpha-carbonic anhydrase (CA) from Sulfurihydrogenibium azorense, the fastest CA known, is highly activated by amino acids and amines. Bioorg Med Chem Lett 2013;23:1087–90
- Allouche F, Chabchoub F, Carta F, Supuran CT. Synthesis of aminocyanopyrazoles via a multi-component reaction and anti-carbonic anhydrase inhibitory activity of their sulfamide derivatives against cytosolic and transmembrane isoforms. J Enzym Inhib Med Chem 2013;28:343–9
- Alp C, Maresca A, Alp NA, et al. Secondary/tertiary benzenesulfonamides with inhibitory action against the cytosolic human carbonic anhydrase isoforms I and II. J Enzym Inhib Med Chem 2013;28:294–8
- Maresca A, Scozzafava A, Vullo D, Supuran CT. Dihalogenated sulfanilamides and benzolamides are effective inhibitors of the three beta-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzym Inhib Med Chem 2013;28:384–7
- Supuran CT, Maresca A, Gregan F, Remko M. Three new aromatic sulfonamide inhibitors of carbonic anhydrases I, II, IV and XII. J Enzym Inhib Med Chem 2013;28:289–93
- Su L, Cao J, Jia Y, et al. Development of synthetic aminopeptidase N/CD13 inhibitors to overcome cancer metastasis and angiogenesis. ACS Med Chem Lett 2012;3:959–64
- Zhang YJ, Feng JH, Liu CX, et al. Design, synthesis and preliminary activity assay of 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid derivatives as novel histone deacetylases (HDACs) inhibitors. Bioorg Med Chem 2010;18:1761–72