
Abstract
A series of 1-aroyl-3-(4-aminosulfonylphenyl)thioureas containing free sulfonamide group has been evaluated for their ability to inhibit bovine carbonic anhydrase II (bCA, EC 4.2.1.1). All compounds in the series were able to inhibit bCA II, the most active inhibitor had IC50 value of 0.26 ± 0.01 µM. Molecular docking studies and detailed structure–activity relationship studies were carried out. The absorption, distribution, metabolism, excretion (ADME) properties, as a predictor of oral absorption, were computationally calculated and compared with the clinically used drug acetazolamide.
Introduction
Carbonic anhydrase (CA, EC 4.2.1.1) is a zinc containing ubiquitous enzyme found in a variety of organisms. In higher vertebrates including humans, 16 different isozymes of CA have been found. These include cytosolic isozymes (CA I–III, CA VII and CA XIII), mitochondrial isozymes (CA VA and CA VB), membrane bound isozymes (CA IV, CA IX, CA XII, CA XIV and CA XV) as well as one secreted isozyme (CA VI)Citation1. This classification is based on different sub-cellular localization and tissue distribution of these CA isozymes. CA catalyzes the reversible hydration of carbon dioxide and bicarbonate ion with the release of protons, it is therefore important in maintaining the homeostasis, acid-base balance, removing carbon dioxide from cells and tissues, maintaining electrolytic secretion, bone resorption and calcification and many other physiologic or pathologic processesCitation2–4. Certain physiological disorders are associated with up-regulation or over expression of certain CA isozymes, common among them are glaucoma, gastric ulcers, hypoxic tumors and obesityCitation5,Citation6. Accordingly role of CA inhibitors has found applications as important therapeutic agents. The most well known, clinically established sulfonamide CA inhibitors include acetazolamide (AZM), dorzolamide, brinzolamide, methazolamide, ethoxzolamide and dichlorophenamide. Dorzolamide and brinzolamide are topically acting drugs, whereas all others (mentioned above) are administered systemically and therefore have many undesirable side effects associated with them due to lack of sufficient selectivity towards CA isozymesCitation3,Citation7,Citation8. Recently the role of CA inhibitors as anticancer agents is rapidly gaining popularityCitation9–11, due to the observation that membrane bound CA isozymes CA IX and CA XII are over expressed in hypoxic tumorsCitation12–19. Hypoxic tumors are typically characterized by an acidic environment (due to over expression of these tumor associated CA isozymes), as a result such tumors become non-responsive towards routine chemotherapyCitation20–22. The use of selective CA IX and CA XII inhibitors has been suggested to bring the acidic environment of hypoxic tumors to physiological pH thus making it responsive towards already established methods of chemotherapyCitation23.
It is therefore essential that new compounds be screened as CA inhibitors in hopes of finding isozyme selective CA inhibitorsCitation24,Citation25. Over the years a variety of sulfonamide based inhibitors of CA have been reportedCitation26, in order to extend these efforts and find new class of sulfonamide CA inhibitors with favorable absorption, distribution, metabolism, excretion (ADME) properties, a series of 1-aroyl-3-(4-aminosulfonylphenyl)thioureas containing a free sulfonamide group (the main zinc binding function) were designed and synthesized by our research groupCitation27.
Materials and methods
Carbonic anhydrase inhibition assay
The compounds evaluated for bCA II inhibition activity were previously synthesized by our research group by reacting equimolar amounts of substituted benzoyl/acyl isothiocyanates with sulfanilamideCitation27. Compounds were evaluated for their inhibition activity against bovine carbonic anhydrase II (bCA-II EC 4.2.1.1). After standardization of the enzymatic reaction conditions the activity of bCA II was determined by the method of Pocker and StoneCitation28 with slight modification. The hydrolysis of p-nitrophenyl acetate to p-nitrophenol catalyzed by bCA II was monitored spectrophotometrically at 405 nm, using a 96-well microplate reader (Bio-Tek ELx 800TM, Instruments, Inc., Winooski, VT). Reaction mixture contained 50 mM Tris-SO4 buffer, pH 7.6 containing 0.1 mM ZnCl2, 10 µL of test compound in enzyme assay buffer and 10 µL (0.35 U, with activity units of 700 U/g) bovine enzyme per well. The reaction mixture was pre-incubated for 10 min at 25 °C, then 20 µL of 6 mM p-nitrophenyl acetate having 5% acetonitrile was added to each well (total reaction volume was 100 µL) and incubated for 30 min at 25 °C. The contents were mixed and read at 405 nm. Suitable controls and known inhibitor AZM were included in the assay. The control (blank) was without inhibitor (compound) present. AZM was used as the standard for the enzymatic assays. Each concentration was analyzed in triplicate. The linear regression parameters were determined for each curve and the IC50 values were measured using the non-linear curve fitting program PRISM 5.0 (GraphPad, San Diego, CA).
Molecular docking methodology
Two experimentally determined crystal structures of human and bCA II with PDB IDs 3K34 and 1V9E, respectively, were downloaded from the RCSB protein data bank. Our research group has already reported the structural comparison and percent similarity (87.3%) and percent identity (80%) between these two enzymes. The root-mean-square deviation (RMSD) with respect to the active site residues of the two proteins is 0.22 ÅCitation29. Method validation was done by re-docking the bound ligand extracted from the active site of hCA II (3K34). The docking method was able to reproduce the experimentally bound conformation of ligand in the active site with an RMSD of 0.65. On this basis hCA II was selected to carry out molecular docking studies. Molecular docking studies were carried out for compounds which had IC50 values in the range 0.261–3.40 µM. Before docking the structures of all molecules were drawn using ACD/ChemSketchCitation30. Gasteiger charges were added on each ligand using ANTECHAMBERCitation31 and the energy of each molecule was minimized through 100 steepest descents and 100 conjugate gradient steps using a step size of 0.02 each using ChimeraCitation32.
For molecular docking studies, AutoDock 4.2 was usedCitation33. For visualization of docked results, Discovery Studio Visualizer 3.5 was usedCitation34. AutoDock uses an empirical scoring function based on free energy of binding. The receptor file of enzyme (hCA II) for docking was prepared by deleting all solvents and hetero molecules, missing hydrogen atoms were added using MolprobityCitation35,Citation36. The generated Ramachandran plots (given in supporting information) were analyzed to assess the quality of enzyme. The appropriate files (.pdbqt) for docking of both receptor (hCA II) and ligand were generated using ADTCitation33. Calculation of affinity maps was done using AutoGrid, where each point in the grid has pre-calculated affinity potentials for all atom types of the ligand. By using these grid maps AutoDock allows flexibility to the ligand, whereas the macromolecule or receptor is kept fixed during docking. Grid dimensions selected were 60 × 90 × 60 with a resolution of 0.375 Å. The grid box was centered at the enzyme’s catalytic active site and was large enough to allow free movement of the ligand. For docking search parameters, Lamarckian Genetic Algorithm (LGA) was used. Maximum number of evaluations was 2 500 000 and docking generations were 27 000, the number of GA runs was 100. Genetic algorithms apply multiple cycles of genetic operators (mutation and crossover, for this study, set at rate of 0.02 and 0.8, respectively) to a population of initially randomly placed ligand conformations giving rise to new generations with improved “solution” that is, binding free energyCitation37.
Results and discussion
The aim of the study was to investigate the effect of substitution pattern on bCA II inhibition activity; accordingly compounds with o-, m- and p-substituents were selected for this study. Further effect of di- and tri-substitution was also explored (, ). bCA II inhibition data are given in . Similar compounds have also been reported as inhibitors of carbonic anhydraseCitation38. Molecular docking studies were carried out for compounds which had IC50 values in the range 0.261–3.40 µM. For molecular docking studies hCA II was selected which has 87.3% percent similarity with the bCA II enzymeCitation29. The molecular docking method was validated using “self-docking” by re-docking the bound ligand extracted from the active site of hCA II. The docking method was able to reproduce the correct binding conformation for the ligand as found in experimentally determined crystal structure with an RMSD of 0.65. shows the overlap of ligand extracted from 3K34 with its docked conformation using AutoDock.
Figure 1. Overlap of bound ligand (green) extracted from crystal structure of hCA II (3K34) and its docked conformation (pink).
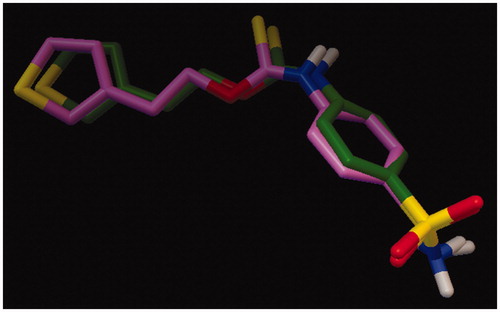
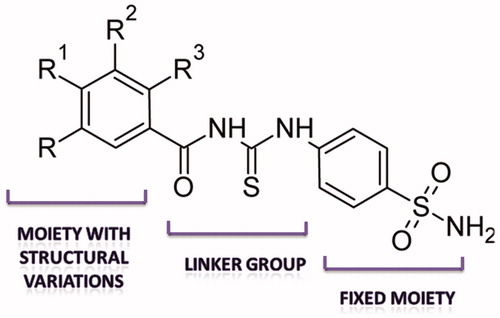
Figure 2. Modulation of structural elements for SAR of bCA II inhibition.
Scheme 1. Structures of thiourea derived sulfonamide based inhibitors of bCA II.
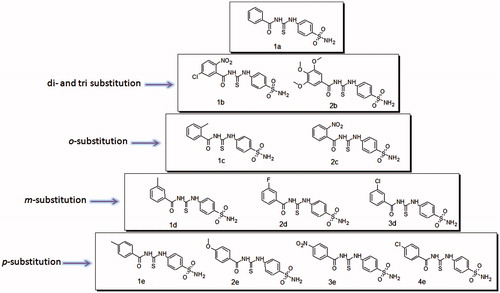
Table 1. bCA II inhibition data for thiourea derived sulfonamides.
Molecular docking studies revealed the most favorable binding modes for these compounds as well as important hydrogen bond interactions with the amino acid residues lining the active site. Different binding conformations of same ligand differing by less than 2 Å RMSD are by default grouped together in one cluster. Careful inspection of cluster profile (given in supporting information) provides a good qualitative assessment of the docking results while the generated binding free energy (ΔG) provides a good quantitative assessment of the docking resultsCitation39. In theory, either of the two parameters (lowest energy cluster versus most densely populated cluster) can be used, however best results are obtained when either of these parameters is combined with a “biological metric”, for example a previous knowledge of certain groups or metal centers in the enzyme’s active site that are known to interact or bind with the ligand under investigation, such as binding of sulfonamides through sulfonamide nitrogen atom to the Zn metallic centerCitation40. The calculated binding free energy (docking score) for docked compounds along with their rank and sub rank (according to cluster profile) is given in .
Table 2. Calculated binding free energy (ΔG kcal/mol) along with rank and sub-rank of compounds.
In all compounds reported in this study, the mode of binding of sulfonamide moiety with the Zn center and Thr199 backbone is the same (), however it is the substitution pattern and the nature of substituent group on the phenyl ring that affects the CA inhibition activity. Detailed structure–activity relationship (SAR) and molecular docking studies help to explain the effect of substitution pattern on inhibition activity. For p-substituted compounds (1e–4e), the inhibition activity increased according to the general trend Cl > NO2 > OMe > Me. All p-substituted compounds showed similar mode of binding. The sulfonamide nitrogen atom is in contact with the Zn ion and also interacts with Glu106. One of the sulfonamide oxygen atoms makes a hydrogen bond contact with Thr199. This mode of binding to the zinc center is typical of sulfonamide CA inhibitors and is well documented in literatureCitation29,Citation41,Citation42. Among p-substituted compounds, the CA inhibition activity increases with the increase in electronegativity of the substituent at the para position of the phenyl ring, thus p-methyl substituted 1e is least active while p-chloro substituted 4e is most active. In contrast to m-substituted analogs, the carbonyl oxygen in p-substituted compounds does not make any hydrogen bond contact, and this may explain less inhibition activity exhibited by p-substituted compounds as compared to m-substituted compounds.
Figure 3. Overlap of all docked compounds inside the enzyme’s active site showing similar mode of binding of sulfonamide moiety with the Zn center of the active site.
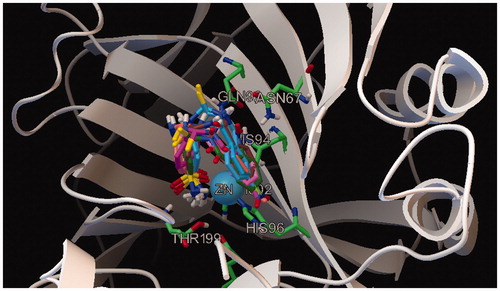
Similarly all m-substituted compounds (1d--3d) were also found to have similar modes of binding. The sulfonamide nitrogen interacts with the Zn ion of the active site and at the same also interacts with Glu106. One of the sulfonamide oxygen atoms is involved in making a contact with Thr199; these interactions stabilize the binding of sulfonamide moiety to the Zn containing active siteCitation29. In all m-substituted compounds, the carbonyl oxygen atom makes a hydrogen bond with Gln92. In this series of compounds, more electronegative substituents at meta position were favored thus m-methyl substituted compound was least active. The most active compound in this series was 3d, in which the nitrogen atom of amide group interacts with Asn67. This interaction is not present in other meta substituted compounds.
The o-substituted compounds 1c and 2c also have similar modes of binding. The sulfonamide nitrogen interacts with the Zn ion of the active site while sulfonamide oxygen interacts with Thr199. Compound 2c is the most active compound in the series. Increased inhibition activity of 2c can be attributed to increased stability of the enzyme inhibitor adduct formed as Thr200 is involved in making two hydrogen bond contacts, one with the nitrogen atom of amide group and the other with oxygen atom of the nitro group. These interactions are absent in compound 1c. Detailed receptor ligand interactions observed by docking studies are given in supporting information (Figures S9–S16).
Evaluation of ADME properties of any molecule is an important step in drug discovery process. The ADME profile has a major impact on the likelihood of success of a molecule as a drug. Keeping this in mind, compounds were computationally screened for Lipinski’s rule of 5 (Rule of 5) and ADME properties. For comparison, the ADME profile of clinically used sulfonamide based CA inhibitor drug AZM was also calculated (). In a famous study by Lipinski et al.Citation43, it was concluded that a compound was more likely to be membrane permeable and hence easily absorbed by the body if it fulfilled following criteria: (1) it had a molecular weight of <500, (2) its log P (the logarithm of the partition coefficient between water and octanol, a measure of lipophilicity of the compound) was less than 5, (3) the number of groups in the molecule that can donate hydrogen atoms to form hydrogen bonds (usually the sum of hydroxyl and amine groups in a drug molecule) was less than 5 and (4) the number of groups that can accept hydrogen atoms to form hydrogen bonds, that is number of hydrogen bond acceptors (estimated by the sum of oxygen and nitrogen atoms) was less than 10. Lipinski’s rule applies only to absorption by passive diffusion of compounds through cell membranes; compounds that are actively transported through cell membranes by transporter proteins are exceptions to the ruleCitation44. The Lipinski’s rule is widely used by medicinal chemists to predict not only the absorption of compounds but also the overall drug-likeness. Accordingly the parameters selected for this study include octanol--water distribution coefficients (S + log P and M log P), the pH-dependent octanol--water distribution coefficient (S + log D), number of hydrogen bond donors (HBDH), hydrogen bond acceptor (sum of nitrogen and oxygen atoms M_NO) and topological polar surface area (T_PSA). The estimation of T_PSA is used as a model for estimation of ability of compounds to cross the blood–brain barrier (BBB). Molecules with T_PSA values <60 Å2 are usually expected to be completely absorbed while those with T_PSA values >140 Å2 are undesirable and cannot be expected to posses sufficient bioavailabilityCitation45. Number of rotatable bonds (nrotb) was calculated using online Molinspiration server (http://www.molinspiration.com). Typically, compounds having molecular weight < 500, number of hydrogen bond acceptor < 10, number of HBDH < 5 and a log P value of < 5 are considered to be orally bio-available with favorable ADME profileCitation46,Citation47.
Table 3. Estimation of “drug-likeness” and ADME properties of compounds.
In all the compounds studied no violation of Lipinski’s rule of 5 was observed. Furthermore, in these compounds no more than 5 HBDH and no more than 10 hydrogen bond acceptors were found, the log P values were significantly lower than 5, although not as low as that of standard drug AZM. All these parameters indicate a favorable ADME profile. Based on T_PSA values, all compounds can be expected to have reasonable cell membrane permeability or bioavailability. Only compounds 1b, 2c and 3e (all containing a highly polar –NO2 substituent) had T_PSA values >140 and therefore cannot be expected to posses sufficient bioavailability. Interestingly, while the substitution by –NO2 group increases the inhibition of CAII, at the same time their relatively high T_PSA values (>140) suggests insufficient cell permeability.
Conclusion
A series of 1-aroyl-3-(4-aminosulfonylphenyl)thioureas containing free sulfonamide group was evaluated as inhibitors of carbonic anhydrase (bCA II). Compounds were found to be sufficiently active; the most active inhibitor in the series (2c) had IC50 value of 0.261 ± 0.011 µM. Molecular docking studies were carried out to indicate the most favorable binding modes of these compounds inside the active site of enzyme. In order to estimate the “drug-likeness” of these compounds, ADME profile, as a predictor of oral absorption, was generated and compared with that of clinically used sulfonamide CA inhibitor AZM. The compounds showed favorable ADME profiles, no violation of the Lipinski’s rule of 5 was observed. Molecular docking and SAR studies suggest that while ortho substitution by bulky electronegative groups (such as –NO2), leads to increased CA inhibition activity, the corresponding increase in T_PSA value, which limits the ability of a compound to cross BBB, is an undesirable feature.
Declaration of interest
The authors report no conflict of interest.
Supplementary material available online
Supplementary Figures S1–S16
Supplementary Material
Download PDF (970.2 KB)References
- Breton S. The cellular physiology of carbonic anhydrases. J Pancrease 2001;2:159–64
- Imtaiyaz Hassan M, Shajee B, Waheed A, et al. Structure, function and applications of carbonic anhydrase isozymes. Bioorg Med Chem 2013;21:1570–82
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–89
- Supuran CT, Scozzafava A. Carbonic anhydrase inhibitors and their therapeutic potential. Expert Opin Ther Pat 2000;10:575–600
- Supuran CT. Carbonic anhydrases as drug targets -- an overview. Curr Top Med Chem 2007;7:825–33
- De Simone G, Supuran CT. Antiobesity carbonic anhydrase inhibitors. Curr Top Med Chem 2007;7:879–84
- Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem 2007;15:4336–50
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229
- Pastorekova S, Vullo D, Casini A, et al. Carbonic anhydrase inhibitors: inhibition of the tumor-associated isozymes IX and XII with polyfluorinated aromatic/heterocyclic sulfonamides. J Enzyme Inhib Med Chem 2005;20:211–17
- Poulsen SA. Carbonic anhydrase inhibition as a cancer therapy: a review of patent literature, 2007–2009. Expert Opin Ther Pat 2010;20:795–806
- Said HM, Hagemann C, Carta F, et al. Hypoxia induced CA9 inhibitory targeting by two different sulfonamide derivatives including acetazolamide in human glioblastoma. Bioorg Med Chem 2013;21:3949–57
- Thiry A, Dogné JM, Masereel B, Supuran CT. Targeting tumor-associated carbonic anhydrase IX in cancer therapy. Trends Pharmacol Sci 2006;27:566–73
- Winum JY, Scozzafava A, Montero JL, Supuran CT. Inhibition of carbonic anhydrase IX: a new strategy against cancer. Anticancer Agents Med Chem 2009;9:693–702
- Guler OO, De Simone G, Supuran CT. Drug design studies of the novel antitumor targets carbonic anhydrase IX and XII. Curr Med Chem 2010;17:1516–26
- Pastorekova S, Zatovicova M, Pastorek J. Cancer-associated carbonic anhydrases and their inhibition. Curr Pharm Des 2008;14:685–98
- Winum JY, Rami M, Scozzafava A, et al. Carbonic anhydrase IX: a new druggable target for the design of antitumor agents. Med Res Rev 2008;28:445–63
- Guler OO, De Simone G, Supuran CT. Drug design studies of the novel antitumor targets carbonic anhydrase IX and XII. Curr Med Chem 2010;17:1516–26
- De Simone G, Supuran CT. Carbonic anhydrase IX: biochemical and crystallographic characterization of a novel antitumor target. Biochim Biophys Acta 2010;1804:404–9
- Gieling RG, Williams KJ. Carbonic anhydrase IX as a target for metastatic disease. Bioorg Med Chem 2013;21:1470–6
- Said HM, Staab A, Hagemann C, et al. Distinct patterns of hypoxic expression of carbonic anhydrase IX (CA IX) in human malignant glioma cell lines. J Neurooncol 2007;81:27–38
- Takacova M, Holotnakova T, Barathova M, et al. Src induces expression of carbonic anhydrase IX via hypoxia-inducible factor 1. Oncol Rep 2010;23:869–74
- Kivelä AJ, Knuuttila A, Räsänen J, et al. Carbonic anhydrase IX in malignant pleural mesotheliomas: a potential target for anti-cancer therapy. Bioorg Med Chem 2013;21:1483–8
- Monti SM, Supuran CT, De Simone G. Anticancer carbonic anhydrase inhibitors: a patent review (2008–2013). Expert Opin Ther Pat 2013;23:737–49
- Winum JY, Temperini C, El Cheikh K, et al. Carbonic anhydrase inhibitors: clash with Ala65 as a means for designing inhibitors with low affinity for the ubiquitous isozyme II, exemplified by the crystal structure of the topiramate sulfamide analogue. J Med Chem 2006;49:7024–31
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229
- Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68
- Saeed A, Zaib S, Pervez A, et al. Synthesis, molecular docking studies, and in vitro screening of sulfanilamide-thiourea hybrids as antimicrobial and urease inhibitors. Med Chem Res 2013;22:3653–62
- Pocker Y, Stone JT. The catalytic versatility of erythrocyte carbonic anhydrase. 3. Kinetic studies of the enzyme-catalyzed hydrolysis of p-nitrophenyl acetate. Biochemistry 1967;6:668–78
- Ul-Haq Z, Usmani S, Mahmood U, et al. In-silico analysis of chromone containing sulfonamide derivatives as human carbonic anhydrase inhibitors. Med Chem 2013;9:608–16
- ACD/ChemSketch, version 12.01, Advanced Chemistry Development, Inc., Toronto, ON, Canada; 2013. Available from: www.acdlabs.com
- Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 2006;25:247–60
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–12
- Morris GM, Huey R, Lindstrom W, et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comput Chem 2009;16:2785–91
- Accelrys Software Inc., Discovery Studio Modeling Environment, Release 4.0, San Diego (CA): Accelrys Software Inc.; 2013
- Chen VB, Arendall WB 3rd, Headd JJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr 2010;D66:12–21
- Davis IW, Leaver-Fay A, Chen VB, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 2007;35:W375–83
- Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comp Chem 1998;19:1639–62
- Vullo D, Franchi M, Gallori E, et al. Carbonic anhydrase inhibitors. Inhibition of mitochondrial isozyme V with aromatic and heterocyclic sulfonamides. J Med Chem 2004;47:1272–9
- Ricci CG, Netz PA. Docking studies on DNA-ligand interactions: building and application of a protocol to identify the binding mode. J Chem Inf Model 2009;49:1925–35
- Cosconati S, Forli S, Perryman AL, et al. Virtual screening with AutoDock: theory and practice. Expert Opin Drug Discov 2010;5:597–607
- Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68
- Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem 2007;15:4336–50
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 1997;23:3–25
- Leeson P. Drug discovery: chemical beauty contest. Nature 2012;481:455–6
- Zakeri-Milani P, Tajerzadeh H, Islambolchilar Z, et al. The relation between molecular properties of drugs and their transport across the intestinal membrane. DARU 2006;14:164–71
- Asokkumar K, Prathyusha LT, Umamaheshwari M, et al. Design, admet and docking studies on some novel chalcone derivatives as soluble epoxide hydrolase enzyme inhibitors. J Chil Chem Soc 2012;57:1442–6
- Clark DE, Pickett SD. Computational methods for the prediction of drug likeness. Drug Discov Today 2000;5:49–58