
Abstract
Human mitochondrial 5′(3′)-deoxyribonucleotidase (mdN) catalyzes dephosphorylation of nucleoside monophosphates, and thus helps maintain homeostasis of deoxynucleosides required for mitochondrial DNA synthesis. Mature mdN is a 23-kDa dimeric protein with highest expression levels in the heart, brain and skeletal muscle. We have identified an alternative splice variant of the mdN gene containing an 18-nucleotide insertion encoding 6 amino acids (GKWPAT) at the 3′-end of the penultimate exon 4. We recombinantly expressed this enzyme variant and characterized its biochemical and kinetic properties as well as its three-dimensional structure. Our high-resolution (1.27 Å) crystal structure revealed that the insertion forms a loop located in the vicinity of the active site pocket and affects enzyme kinetic parameters as well as protein thermal stability.
Introduction
The monophosphate 5′-nucleotidases are ubiquitous enzymes that catalyze the dephosphorylation of nucleoside monophosphates and regulate nucleotide and nucleoside pools in cellsCitation1,Citation2. In addition to their physiological role, these enzymes also are involved in regulating the activation of nucleoside analogues and thus affect the efficacy of anti-cancer and anti-viral treatmentsCitation3. To interfere with DNA synthesis, nucleoside analogues must be activated by intracellular kinases. Both clinical and in vitro studies have suggested that an increase in nucleotidase activity can inhibit nucleoside analogue activation and result in drug resistance. Both cytosolic and mitochondrial human deoxynucleotidases appear to be attractive targets for development of specific inhibitors to circumvent drug resistance.
To date, seven human 5′-nucleotidases with different substrate specificities, cellular locations and tissue-specific expression patterns have been cloned and characterized (reviewed in Ref. 1). Among these enzymes, cytosolic 5′(3′)-deoxyribonucleotidase (cdN) and mitochondrial 5′(3′)-deoxyribonucleotidase (mdN) specifically dephosphorylate the deoxyribo form of nucleoside monophosphates, with dUMP and dTMP being their preferred substrates. These two enzymes share 52% sequence homology and similar gene organization, suggesting that their genes originated from gene duplication. The preference of these enzymes for deoxyribonucleosides, which several drugs are designed to mimic, makes 5′(3′)-deoxyribonucleotidases good candidates for mediating drug resistance. Several nucleoside analogue drugs have proven to be substrates for cdNCitation3, and thus this cytosolic enzyme is considered a target for development of inhibitors to increase the efficacy of clinically used nucleoside analogues.
Some cancer treatments target mitochondriaCitation4–6, where mdN is localized. Mitochondria or changes in cancer-cell mitochondria are thought to be responsible for the resistance of cancer cells to apoptosis and to certain chemotherapeutic agents. Selective inhibition of mdN could have fatal consequences for cancer cells, because mdN protects mitochondria from excessive dTTP levels, which are mutagenic for mitochondrial DNA replicationCitation7.
The gene for mdN, located on chromosome 17p11.2, comprises 5 exons and 4 intronsCitation7 and encodes a 228-amino-acid protein with a 31-amino-acid mitochondrial leader sequence that is cleaved off during import into mitochondria. The mature protein forms dimers, with each monomer weighing 23 kDaCitation7. Expression of mdN is highest in the heart, brain and skeletal muscle, with much lower expression in the pancreas and kidneysCitation7.
The crystal structure of mdN has been determined in free form (PDB code 1MH9Citation8) and in complex with two inhibitors (PDB codes 1Q91 and 1Q92Citation9). In addition, nine structures of the mdN active site mutant (D41N) in complex with various substratesCitation8,Citation10 and nucleoside analogues have been determinedCitation11. Based on these structures, an enzyme mechanism common to all intracellular nucleotidases was proposedCitation8.
Here, we describe cloning and kinetic and structural characterization of an alternatively spliced variant of mdN. This variant (designated mdNi) contains an insertion at the 3′-end of exon 4. This insertion encodes six amino acids that form a loop, which is located at the active site entrance and affects enzyme stability and activity.
Materials and methods
Cloning
The mdN coding sequence was amplified from the Clontech cDNA QuickClone (Universal II) human cDNA library using primers F2 (5′-gatacatatgggAGGCCGCGCCCTACG) and R2 (5′-CCCCACAGAGGAGCCCGAATTCCAGTC), which contain NdeI and EcoRI restriction sites (italicized), respectively (). The amplified fragments were cloned into pUC18 SmaI and sequence verified. In addition to the canonical mdN sequence (GenBank AF210652), we obtained an 18-bp-longer sequence that shows agreement with GenBank entry DQ323995 and encodes a protein-denominated mdNi. To clone constructs for expression of mdN and mdNi in fusion with a hexahistidine tag (His6), the vector pMCSG7 (T7 promoter driven, originally designed for LIC cloningCitation12) was first modified so that the N-terminal His6 tag is followed by a TEV protease recognition site with amino acid residues SNAAS following the cleavage site. The coding sequence for AS corresponds to an NheI site. The original mdN and mdNi coding sequences obtained by PCR from the human cDNA library and contained in pUC18 were reamplified using primers 5′-TTTGCTAGCGGAGGCCGCGCCCTACGG (NheI site is italicized) and R2 (see above) to introduce NheI site at their 5′ termini and then cloned into the modified pMCSG7 vector as NheI-EcoRI inserts.
Figure 1. Sequence alignment of mdN and mdNi. The mitochondrial leader sequence, which is cleaved during protein processing, is hatched. The insertion in mdNi is highlighted in black. The positions of primers used for cloning and PCR analyses are shown as arrows above the sequences. For primers F2 and R2, which introduced restriction sites (see “Materials and methods” section for primer sequences), regions of noncomplementarity to the template sequence are indicated.

PCR analyses
Primers Fint (5′-CTGACCTTCTCATAGACGACCGGCC) and R1 (5′-CACAGAGGAGCCCGAAGCACAGTCC) (), encompassing the site of insertion, were used in PCR with several human oligo-dT-primed cDNA libraries prepared from total RNA isolated from various tissues (kind gift of Dr. Matous Hrdinka of the Institute of Molecular Genetics). The reactions contained Phusion polymerase (1U, Finnzymes) and primers at 0.4 µM final concentration. The thermal cycling conditions were as follows: 35 cycles of 10 sec denaturation at 98 °C and 25 sec annealing/elongation at 72 °C, followed by a final elongation at 72 °C for 7 min. The amplification products were separated on a gel made from a 3% mix (3:1, w/w) of MetaPhor (Lonza) and BioRad agarose. For size standards, PCR was performed with cloned mdN sequences lacking or containing the 18-nt insertion.
To clone full-length mdN, PCR with primers F1 (5′-GGGCCAGCGCGCTGGGCCATGATCC) and R1 (5′-CACAGAGGAGCCCGAAGCACAGTCC) was performed using Clontech human skeletal muscle, heart and brain Quick-Clone cDNA libraries (catalog numbers 637234, 637213 and 637242, respectively). The reactions contained Phusion polymerase (1U, Finnzymes) and primers at 0.5 µM final concentration. Thermal cycling conditions were as follows: 45 cycles of 10 sec denaturation at 98 °C and 35 sec annealing/elongation at 72 °C, followed by a final 7-min elongation at 72 °C. PCR products were gel purified, cloned into pCR-Blunt vector and sequenced.
Protein preparation
Recombinant proteins were expressed in Escherichia coli BL21 (DE3) grown in LB broth (Sigma) supplemented with 0.8% (v/v) glycerol and 100 µg ml−1 ampicillin. Bacterial culture was grown at 37 °C, and protein expression was induced with 150 µM isopropyl-β-d-thiogalactopyranoside when cells reached an OD600 of approximately 0.6. Cultivation then proceeded for additional 4 h at 15 °C.
For purification of the His6-tagged protein, bacteria were suspended in TBS (50 mM Tris-HCl, pH 8, 150 mM NaCl) and disrupted with an EmulsiFlex C3 (Avestin) homogenizer using 2 passes at 1200 psi. The lysate was clarified by centrifugation (48 000 g, 30 min) and subjected to affinity chromatography over a His-select Nickel Affinity Gel (Sigma) column equilibrated with TBS. The His6-tagged protein was eluted with 250 mM imidazole in TBS. The affinity tag was removed by incubation with recombinant His6-tagged TEV protease overnight at 25 °C while dialyzing against buffer containing 20 mM Tris-HCl, pH 7.5, 2 mM DTT and 20 mM MgCl2. TEV protease cleavage yielded mature protein preceded by SNAAS at the N-terminus. The sample was subjected to a second round of Ni-NTA metal-affinity chromatography to remove the TEV protease and cleaved His6 tag, extensively dialyzed against 20 mM Tris-HCl, pH 7.5, 20 mM MgCl2, 2 mM DTT and 1 mM EDTA and concentrated to 1 mg ml−1 using an Amicon Ultra Concentrator (Millipore). Protein solutions were stored at −20 °C.
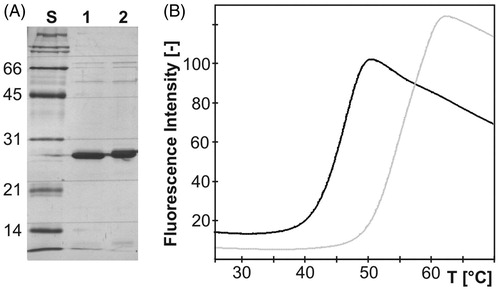
Yields of >95% pure proteins, as judged by silver-stained SDS-PAGE (), were 2.0 mg and 0.96 mg l−1 of culture media for mdNi and mdN, respectively.
Enzyme activity assay
Enzyme activity was determined by high-performance liquid chromatography with spectrophotometric detection of dUMP substrate and dU product at 262 nm. Typically, 2–4 nM enzyme in 20 mM Tris-HCl, pH 7.5, 20 mM MgCl2, 1 mM EDTA and 2 mM DTT were mixed with various concentrations of dUMP in a final volume of 100 µl. The reactions were started by addition of enzyme into the premixed substrate solution, proceeded for 8 min at 20 °C and were stopped by addition of 3 µl of 30% (v/v) TCA. A 5 µl aliquot of reaction mixture was applied to a Zorbax C18 column (150 mm × 3 mm, 2.5 µm particle size, Agilent) connected to an Agilent 1100 system. Separation was performed using isocratic elution in 75 mM KH2PO4. After each run, the column was washed with 100% methanol supplemented with 0.05% (v/v) TFA. The substrate conversion was kept under 10%.
To determine KM values, series of activity assays were performed at different substrate concentrations varying from 20 µM to 3 mM. Each reaction was performed in duplicate. Data were fitted using non-linear regression with the classical Michaelis–Menten equationCitation13.
Differential scanning fluorimetry
Protein solutions (0.1, 0.3 and 0.75 mg ml−1) were prepared in 20 mM Tris-HCl, pH 7.5, 20 mM MgCl2, 1 mM EDTA and 2 mM DTT. Proteins were assayed in the presence of 625-fold diluted Sypro Orange (Invitrogen) in a total volume of 25 µl. All measurements were performed in duplicate in a LightCycler 480 Multiwell Plate 96 (Roche) sealed with LightCycler 480 Sealing Foil (Roche). Using Roche LightCycler 480 II, a temperature gradient from 20 to 95°C was applied at a rate of 0.83 °C min−1. The excitation wavelength was 465 nm, and fluorescence intensity was recorded at 580 nm. Melting temperature values corresponding to inflection points of melting curves were determined as the minima of negative first derivatives using Roche LightCycler 480 SW 1.5 software.
Protein crystallization
For crystallization, proteins were concentrated by ultrafiltration in Centricon YM-10 filters (Millipore). A crystallization drop containing 1.33 µl of protein solution (3.4 mg ml−1) and 0.66 µl of precipitating solution [20 mM KH2PO4, 8% (w/v) PEG 8000, 10% (v/v) glycerol] was equilibrated over a reservoir containing 500 µl of precipitating solution in EasyXtal plates (Qiagen). Crystals grew to a final size of 200 × 150 × 150 µm within five days. For cryoprotection, the crystals were soaked for 10 sec in reservoir solution supplemented with 30% (v/v) glycerol. Crystals were flash cooled by plunging into liquid nitrogen and stored in liquid nitrogen until used for X-ray diffraction experiments.
Data collection and structure determination
Diffraction data at −173°C were collected at the MX14.2 beamline of BESSY, Berlin, GermanyCitation14, at a wavelength of 0.978 Å. The mdNi crystals diffracted to 1.27 Å resolution. Data were integrated and reduced using XDSCitation15 with graphical extension XDSAPPCitation16. Crystals exhibited the P43212 space group symmetry and contained one molecule in each asymmetric unit with a solvent content of approximately 60%. Crystal parameters and data collection statistics are given in .
Table 1. Crystal data and diffraction data collection and refinement statistics.
The structure of mdNi was determined by difference-Fourier techniques using coordinates from the isomorphous structure of canonic mdN protein (PDB code 4L6A)Citation17. Refinement was carried out using the program REFMAC 5.5Citation18 from the CCP4 suiteCitation19 with anisotropic atomic displacement parameters.
The quality of the final crystallographic model was validated with MolprobityCitation20. Refinement statistics are given in . All figures showing structural representations were prepared with the program PyMOLCitation21. Atomic coordinates and experimental structure factors have been deposited into the Protein Data Bank under code 4MUM.
Results and disscussion
Cloning and identification of an mdN splice variant.
We used PCR with primers F2/R2 () to amplify the gene encoding mdN from a Clontech cDNA QuickClone (Universal II) human cDNA library. In addition to the canonical mdN sequence (GenBank AF210652)Citation7, we also obtained an mdNi variant containing an 18-nt insertion, which encodes 6 amino acids (GKWPAT), at the 3′ end of the penultimate exon 4 (). To our knowledge, nothing concerning this variant has been published, although an entry in the GenBank database (DQ323995) exists.
To explore the mRNA expression pattern of mdNi in individual tissues described to have the highest levels of mdN expression, we performed PCR on cDNA libraries from brain, heart and skeletal muscle using primers Fint/R1 (see for primer location and “Materials and methods” section for primer sequences). This PCR was designed to yield diagnostic fragments with sizes of 206 bp and 224 bp that are specific for mdN and mdNi, respectively. Fragments of both sizes were detected in all libraries tested ().
Figure 2. PCR analyses of tissue-specific cDNA libraries with primers encompassing the site of insertion. PCR products were analyzed on a 3% agarose gel: lane 1, brain; lane 2, heart; lane 3, skeletal muscle; + and − denote PCR products obtained from plasmids containing coding sequences of mdNi with (+) or without (−) the 18-nt insertion; S denotes size markers.
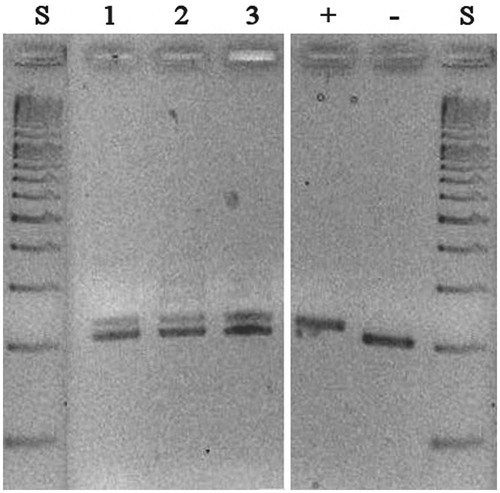
Figure 3. (A) Silver-stained SDS-PAGE of purified recombinant mdN (lane 1) and mdNi (lane 2) with molecular weight standards in kDa (lane S). Each lane contains 3 µg of protein. (B) Thermal unfolding transition of mdN (gray) and mdNi (black) proteins. After reaching the plateau, the fluorescence intensity started to decrease due to denaturation of the protein–dye complex.
To verify that a given tissue contains full-length mRNAs capable of coding for the corresponding variant enzyme, we amplified the full-length mdN gene including the 5′ untranslated regions required for efficient mRNA translation. Tissue-specific cDNA libraries from brain, heart and skeletal muscle were used as templates with F1 and R1 primers (see for primer location and “Materials and methods” section for primer sequences). Primer F1 includes a Kozak sequence and an initiation codon, as present in the sequence of mdN; R1 spans the 3′ untranslated sequence after the stop codon. PCR products were cloned into a pCR-Blunt vector, and a total of 50 clones were sequenced. Only 1 non-canonical mdNi variant was found among 10 clones from the skeletal muscle library, and none were found among 40 clones from the heart and brain libraries (20 clones from each). This analysis confirmed that the variant sequence was present in the cDNA libraries in low abundance but did not attempt to quantify its relative abundance. Interestingly, the mdNi variant with the 3′-extended penultimate exon 4 conforms to the so-called GT-AG rule for the relevant intron boundariesCitation22, whereas in the case of the more prevalent canonical variant, the splicing of the 18-bp-longer intron utilizes GC rather than GT at the donor site of the intron.
Our qualitative PCR analysis revealed that the mdNi variant gene was of low abundance and that it contains all signals necessary for translation into a conceptual protein. To shed some light on the potential biological role of this variant enzyme, we recombinantly expressed the protein and characterized it in terms of biochemical and kinetic properties as well as three-dimensional structure.
mdNi characterization
Both mdN and mdNi were expressed in E. coli and purified to homogeneity (). The recombinant mdN variants contained a cloning artefact (SNAAS) that remained on their N-termini upon cleavage of His-tag by the TEV protease. The presence of this cloning artefact does not affect the enzymatic activity of mdN; the kinetic constant KM of dUMP hydrolysis at 37 °C for mdN with the cloning artefact (KM = 85 ± 14 µM) corresponds to the value published for native mdN (KM =100 µM)Citation3.
During enzyme kinetic characterization, we observed that the mdNi variant loses activity at 37 °C, and we therefore performed the activity assays at 20°C. The Michaelis constant with dUMP as a substrate was more than twofold higher for mdNi than mdN (KM for mdN = 106 ± 21 µM, while KM for mdNi = 262 ± 56 µM.). Also, the turnover number (kcat) was lower for mdNi; kcat values were calculated as 9.4 ± 0.3 s−1 and 6.1 ± 0.4 s−1 for mdN and mdNi, respectively.
To further investigate the stability of mdN variants, we followed temperature-induced protein unfolding using differential scanning fluorimetry (DSF, also known as Thermofluor assay). In DSF experiments, a fluorescent dye is used to monitor the protein unfolding in response to increasing temperature. The unfolding process exposes the hydrophobic region of proteins and results in a large increase in fluorescenceCitation23. Both proteins displayed a melting profile that can be approximated by a two-state model of thermal unfolding (). The melting profiles were used to estimate the midpoint temperature of transition, also called the unfolding transition temperature (Tm) as it correlates with the melting temperatures determined by other methodsCitation24. The mdNi variant proved to be significantly less stable than mdN. The Tm values for mdN and mdNi were 59.5°C and 46.7 °C, respectively.
Crystal structure of mdNi reveals a solvent-exposed loop close to active site
The structure of mdNi was determined at 1.27 Å resolution. The final crystallographic model contains one molecule in the asymmetric unit containing 199 amino acids. Nine amino acid residues could not be traced in electron density; specifically, these included five N-terminal residues (A29-G33) and four residues belonging to the region inserted in mdNi (K183-A186).
The overall structure of mdNi is very similar to that of mdN (PDB code 4L6A)Citation17; the root mean square deviation (RMSD) for superposition of 1315 corresponding atoms is 0.075 Å, a value which is less than those typically observed for identical structuresCitation25. Also, like the mdN structure, the structure of mdNi includes an active site catalytic magnesium ion and a phosphate ion that originated from the crystallization solution ().
Figure 4. Superposition of the crystal structures of mdN (PDB code 4L6A)Citation17 in light orange and mdNi (PDB code 4MUM) in deep purple. Magnesium and phosphate ions bound in the active site are shown in sphere and stick representations, respectively. (A) View of the overall structures. The partially disordered insertion is represented with a black dotted line. (B) Details of the mdNi insert region with 2Fo − Fc electron density map contoured at 1.0 σ. Residues missing in the crystallographic model are indicated. Catalytic residues (D41, D43, D176, D177) are shown in sticks. (C) Surface representation of the active site. mdN is presented as a solid surface, while the surface of mdNi is shown as mesh.
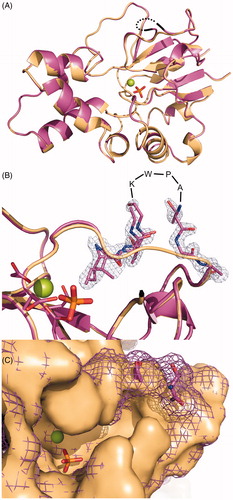
The mdNi has a fold characteristic of the haloacid dehalogenase enzyme superfamily, in which the active site is located in the cleft between the core (α/β Rossman fold) and the cap (short four helix bundle) domains (). The six amino acid residues GKWPAT, inserted between residues 181 and 182 of the mdN sequence, are located in the core domain, specifically within an extended loop connecting the S4 and S5 β-strands and lining the enzyme active site cavity close to the pyrimidine base binding pocket. The insertion forms a solvent-exposed loop that is inherently flexible and thus partially disordered in the crystal structure (). Although four residues in the insertion loop could not be traced in the electron density map, it is apparent that the loop changes the shape of the distal parts of the enzyme active site (). The presence of an inserted loop at the edge of the active site is the probable reason for the change in enzyme kinetic parameters that we observed for mdNi compared to mdN. The lower kcat value for mdNi compared to mdN might indicate that the release of product is obstructed by the insertion loop. To explain the observed lower stability of mdNi, we may speculate that insertion makes the region between S4 and S5 (residues 180–191) more flexible and prone to thermal unfolding. Indeed, values of atomic displacement parameters (ADP) in this region are much higher for mdNi structure than for mdN. While the ADP values for residues 180–190 in mdN are below the average value for the structure (18.5 Å2), in mdNi the ADP values for amino acid residues 179–187 are on average higher by 47% when compared to the average value for all residues (18.6 Å2). No major differences in ADP values were observed for mdN and mdNi structure except for flexible N-terminus. The RMSD values for superposition of individual residues 180–191 are below 0.4 Å with the exception of the two residues preceding and following the inserted region.
In conclusion, we have identified and characterized an alternative splice variant of human mitochondrial 5′(3′)-deoxyribonucleotidase that has a six amino acid insertion. This insertion forms a loop located in the vicinity of the active site pocket. Although the presence of the insertion does not significantly change the overall protein structure, it affects enzyme kinetic parameters as well as protein thermal stability. The tissue localization and possible biological relevance of this enzyme variant remain to be uncovered.
Acknowledgements
The authors thank Dr. P. Šácha of IOCB AS CR for discussions and the gift of Clontech Quick-Clone cDNA libraries. Diffraction data were collected on BL14.1 and BL14.2 operated by the Helmholtz-Zentrum Berlin (HZB) at the BESSY II electron storage ring (Berlin-Adlershof, Germany)Citation14.
Declaration of interest
The authors have no conflicts of interest to declare. This work was supported by the Grant Agency of the Czech Republic (research project No. GA 203/09/0820), the Ministry of Education of the Czech Republic – LK11205 (programme “NAVRAT”), and in part by research projects RVO 61388963 and 68378050 awarded by the Academy of Sciences of the Czech Republic.
References
- Hunsucker SA, Mitchell BS, Spychala J. The 5′-nucleotidases as regulators of nucleotide and drug metabolism. Pharmacol Ther 2005;107:1–30
- Bianchi V, Pontis E, Reichard P. Interrelations between substrate cycles and denovo synthesis of pyrimidine deoxyribonucleoside triphosphates in 3t6 Cells. Proc Natl Acad Sci USA 1986;83:986–90
- Mazzon C, Rampazzo C, Scaini MC, et al. Cytosolic and mitochondrial deoxyribonucleotidases: activity with substrate analogs, inhibitors and implications for therapy. Biochem Pharmacol 2003;66:471–9
- Fantin VR, Leder P. Mitochondriotoxic compounds for cancer therapy. Oncogene 2006;25:4787–97
- Galluzzi L, Larochette N, Zamzami N, Kroemer G. Mitochondria as therapeutic targets for cancer chemotherapy. Oncogene 2006;25:4812–30
- Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria in cancer cells: what is so special about them? Trends Cell Biol 2008;18:165–73
- Rampazzo C, Gallinaro L, Milanesi E, et al. A deoxyribonucleotidase in mitochondria: involvement in regulation of dNTP pools and possible link to genetic disease. Proc Natl Acad Sci USA 2000;97:8239–44
- Rinaldo-Matthis A, Rampazzo C, Reichard P, et al. Crystal structure of a human mitochondrial deoxyribonucleotidase. Nature Struct Biol 2002;9:779–87
- Rinaldo-Matthis A, Rampazzo C, Balzarini J, et al. Crystal structures of the mitochondrial deoxyribonucleotidase in complex with two specific inhibitors. Mol Pharmacol 2004;65:860–7
- Wallden K, Ruzzenente B, Rinaldo-Matthis A, et al. Structural basis for substrate specificity of the human mitochondrial deoxyribonucleotidase. Structure 2005;13:1081–8
- Wallden K, Rinaldo-Matthis A, Ruzzenente B, et al. Crystal structures of human and murine deoxyribonucleotidases: insights into recognition of substrates and nucleotide analogues. Biochemistry 2007;46:13809–18
- Stols L, Gu M, Dieckman L, et al. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif 2002;25:8–15
- Copeland RA. Enzymes: a practical introduction to structure, mechanism, and data analysis. New York: VCH Publishers, Inc.; 1996
- Mueller U, Darowski N, Fuchs MR, et al. Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J Synchrotron Radiat 2012;19:442–9
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr 2010;66:125–32
- Krug M, Weiss MS, Heinemann U, Mueller U. XDSAPP: a graphical user interface for the convenient processing of diffraction data using XDS. J Appl Crystallogr 2012;45:568–72
- Pachl P, Fabry M, Rosenberg I, et al. Crystal structures of human cytosolic and mitochondrial nucleotidases: implications for structure-based design of selective inhibitors. Acta Crystallogr D Biol Crystallogr 2014; D70. doi: 10.1107/S1399004713030502
- Murshudov GN, Skubak P, Lebedev AA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 2011;67:355–67
- Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 2011;67:235–42
- Lovell SC, Davis IW, Adrendall WB, et al. Structure validation by C alpha geometry: phi, psi and C beta deviation. Proteins Struct Funct Genet 2003;50:437–50
- DeLano WL. PyMOL molecular viewer: updates and refinements. Abstracts of Papers of the American Chemical Society. The PyMOL Molecular Graphics System, Version 1.4.1. Schrödinger, LLC; 2009:238
- Mount SM. A catalog of splice junction sequences. Nucleic Acids Res 1982;10:459–72
- Lo MC, Aulabaugh A, Jin G, et al. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem 2004;332:153–9
- Ericsson UB, Hallberg BM, Detitta GT, et al. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem 2006;357:289–98
- Betts MJ, Sternberg MJE. An analysis of conformational changes on protein-protein association: implications for predictive docking. Protein Eng 1999;12:271–83
- Diederichs K, Karplus PA. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat Struct Biol 1997;4:269–75
- Brunger AT. Free R-value – a novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992;355:472–5
- CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 1994;50:760–3
- Vaguine AA, Richelle J, Wodak SJ. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr D Biol Crystallogr 1999;55:191–205