
Abstract
A series of arenesulfonyl-2-imidazolidinones incorporating methyl, isopropyl, methoxy, halogen and phenyl moieties were prepared and tested as possible inhibitors of two members of the pH regulatory enzyme family, carbonic anhydrase (CA; EC 4.2.1.1). The inhibitory potencies of the compounds against human isoforms hCA I and hCA II were analyzed by an esterase assay with 4-nitrophenyl acetate as substrate, and the inhibition constants (KI) were calculated. Most compounds investigated here exhibited micromolar inhibition constants against the two isoenzymes. KI values were in the range of 10.2–40.6 μM for hCA I and of 13.1–31.4 μM for hCA II, respectively. Most of the imidazolidinones showed interesting CA inhibitory efficacy, some of them having comparable affinity (for hCA I) as the clinically used sulfonamide acetazolamide (AZA), but their efficacy against hCA II was much lower compared to AZA.
Introduction
The role of metalloenzyme carbonic anhydrase in various physiological processes has been recognized for a long period, being shown that the deregulated expression and/or abnormal performance of the 16 presently known isozymes may have important pathological consequencesCitation1–4. There are several human diseases whose pathopysiological characteristics involve disbalance in the conversion between carbon dioxide and bicarbonate (the two substrates of CA enzymes), resulting in perturbed ion transport, shift in pH and abnormal fluid secretionCitation1–4. Thus, it seems plausible that modulation of CA activity to normal levels either by inhibition or activation offers interesting therapeutic optionsCitation1,Citation2. The first use and clinical testing of CA inhibitors (CAIs) dates back several decades ago, to a period before the recognition of the diversity of isoforms within the CA family and their differential distribution in various human tissues and organsCitation1–4. Traditionally, the use of inhibitors was based on histochemical, biochemical and functional evidences for the presence of active CAs, potentially contributing to disease, and consequences of the treatment were evaluated mainly symptomaticallyCitation1–4. Due to their favorable outcomes, sulfonamides have become widely accepted drugs in the treatment of several CA-based diseases, especially as antiglaucoma agents, diuretics and antiulcer agents among othersCitation2–4. However, systemic and topically administered CA inhibitors regularly showed serious side effectsCitation3–9. It is now clearly known that these undesired effects are due to the existence of at least 12 different active CA isoforms (together with the three inactive isozymes, the CA-related proteins, CARPs VIII, X and XI)Citation3–10, that are indiscriminately inhibited irrespective of whether they play a real role in disease or are just coexpressed in the same tissue and elsewhere in the body. In addition, certain drugs directed primarily against different CA-unrelated targets may also inhibit the activity of CAsCitation1–5. Thus, it is critically important to thoroughly characterize the affinity of different isozymes for different CAIs, and also to better understand the structure activity relationshipsCitation1–10.
Our groups recently investigated the interaction of CA I and II isozymes with several types of phenols, pyrrole derivatized sulfonamides, dopaminergic bromophenolic compounds, antioxidant bisphenols and several of its substituted derivatives, e.g. salicyclates and some of their derivativesCitation8,Citation9. Here, we extend these earlier investigations to series of arenesulfonyl-2-imidazolidinone-based compounds. Diarenesulfonylurea and thiourea functionalities have attracted reasonable attentionCitation11–15, especially after the discovery of sulofenurCitation16. It is generally assumed that the strong cytotoxicity and, as a consequence, the antitumor properties of the diarenesulfonylurea is due to the uncoupling of mitochondriaCitation11,Citation12, however, other mechanisms, such as inhibition of the mitochondrial isozyme V of carbonic anhydrase (CAs) have also been hypothesized, since hydrolysis of the cytotoxic agent leading to the formation of unsubstituted sulfonamides as the principal products has been reported both in vivo and in vitroCitation17.
It is well known that aromatic/heterocyclic sulfonamides (formed after such a hydrolytic process) act as very potent inhibitors of CAsCitation18,Citation19, and that these enzymes are involved in a multitude of crucial physiologic processesCitation20. However, clinical trials of sulofenur have yielded unsatisfactory results because of its high protein binding and limited dosing caused by the appearance of anemia, and methemoglobinemia, a side effect that is likely caused by its aniline-related metabolitesCitation21,Citation22. Trying to overcome the serious side effects of sulofenur, several cyclic diarenesulfonylurea such as 4-phenyl-2-imidazolidinoneCitation23–26, arylidenehydantoin have been synthesized and screened for antitumor activity against various human solid tumorsCitation27,Citation28 ().
Figure 1. Structure of the tested compounds.
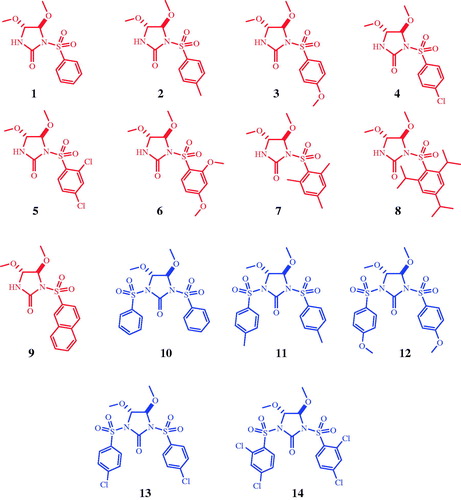
In the present study, we have purified human CA I, II (hCA I, hCA II) isoenzymes and examined the in vitro inhibition effects of some arenesulfonyl-2-imidazolidinones mentioned above on these enzymes.
Materials and methods
CNBr-activated Sepharose 4B, protein assay reagents, p-aminobenzene sulfonamide l-tyrosine, 4-nitrophenylacetate and chemicals for electrophoresis were purchased from Sigma–Aldrich Co. All other chemicals were of analytical grade and obtained from Sigma-Aldrich (Bornem, Belgium).
General method for the synthesis of compounds
A solution containing 0.001 mol 6-(un)substituted 3-formylchromone in 5–7 mL ethanol was stirred with heating until dissolved. Then catalytic amount of p-toluenesulfonic acid (p-TsOH) was added followed by the addition of 0.001 mol of 3- and 4-aminobenzene sulfonamide in equal volumes of ethanol. Reaction mixture was refluxed for 3.5 h and kept overnight. Solid product was obtained by filtration and purified by recrystallization from a mixture of hot ethanol and acetone (1:1).
Kinetic studies
Purification of hCA I and hCA II were previously describedCitation8,Citation9. The activity assay was carried out according to Verpoorte et al.Citation29.
Protein determination
Protein quantity was determined spectrophotometrically at 595 nm according to the Bradford method during the purification steps, using bovine serum albumin as the standardCitation30.
SDS polyacrylamide gel electrophoresis
SDS polyacrylamide gel electrophoresis was performed after purification of the enzymes. It was carried out in 10% and 3% acrylamide for the running and the stacking gel, respectively, containing 0.1% SDS according to Laemmli procedureCitation31.
Results and discussion
Our groups recently investigated the interactions of sulfonamides and some of their substituted derivatives with all mammalian CA enzymesCitation17–24, demonstrating some low micromolar/submicromolar inhibitors as well as the possibility to design isozyme selective CAIs. The inhibition profile of various CAs with this class of agents is very variable, with inhibition constants ranging from the millimolar to the submicromolar rangeCitation10. Thus, it seemed reasonable to us to extend the previous studiesCitation32, including in this investigation a series of arenesulfonyl-2-imidazolidinone compounds.
The purification of the two CA isozymes was performed with a simple one step method by a Sepharose-4B-aniline-sulfanilamide affinity column chromatoghrapyCitation9. hCA I was purified, 108.6-fold with a specific activity of 963.12 EU mg−1 and overall yield of 56%, hCA II was purified, 903.2-fold with a specific activity of 6790 EU mg−1 and overall yield of 66%. Inhibitory effects of compounds 1–14 on enzyme activities were tested under in vitro conditions. The sulfonamide CAI acetazolamide AZA has been used as a negative control in our experiments, and for comparison reasons. Data of show the following regarding inhibition of hCA I and II with compounds 1–14 and AZA (as standard):
Against the slow cytosolic isoform hCA I, the arenesulfonyl-2-imidazolidinone derivatives investigated here showed moderate to effective inhibitory properties. Thus, derivatives 1–14 exhibited weaker inhibition of this isoform, with KI in the range of 37.1–41.3 μM, with a comparable potency as the reference compound AZA (KI: 36.2 μM) (). The compounds 1–7 were more effective inhibitors against hCA I, with KI in the range of 10.2–23.4 μM, being more effective than AZA. However, compounds 4 and 5 acted as the most effective hCA I inhibitor (KI-s: 2.14–2.59 μM) (). These results demonstrate the contribution of the halogen atoms to the inhibition efficacy.
Against the ubiquitous and dominant rapid cytosolic isozyme hCA II, compounds 1–14, acted as weak inhibitors (KI in the range of 12.9–31.4 μM) compared to the clinically used sulfonamide AZA (). Similar to the trend in hCA I, compounds 1–7 were more effective inhibitors than others with KI in the range of 12.9–19.1 μM. However, remaining seven derivatives 8–14 as weaker hCA II inhibitors (KI-s: 21.1–31.4 μM), with much lower KI values than that of the reference compound AZA (). Findings of our study indicates that arenesulfonyl-2-imidazolidinone derivatives show good inhibitory activity on hCA I with comparable or lower KI values than the clinically used sulfonamide AZA, but this trend was not seen for the rapid enzyme hCA II.
Table 1. hCA I and II inhibition data with compounds 1–10, by an esterase assay with 4-nitrophenylacetate as substrateCitation9.
Declaration of interest
The authors report no declarations of interest. The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding the work through the research group project No. RGP-VPP-163.
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Disc 2008;7:168–81
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74
- Supuran CT. Carbonic anhydrases: catalytic mechanism, distribution and physiological roles. In: Supuran CT, Scozzafava A, Conway J, eds. Carbonic anhydrase – its ınhibitors and activators. Boca Raton (FL): CRC; 2004:1–24
- Çavdar H, Ekinci D, Talaz O, et al. α-Carbonic anhydrases are sulfatases with cyclic diol monosulfate esters. J Enzyme Inhib Med Chem 2012;27:148–54
- Senturk M, Ekinci D, Goksu S, Supuran CT. Effects of dopaminergic compounds on carbonic anhydrase isozymes I, II, and VI. J Enzyme Inhib Med Chem 2012;27:365–9
- Ekinci D, Karagoz L, Ekinci D, et al. Carbonic anhydrase inhibitors: in vitro inhibition of α isoforms (hCA I, hCA II, bCA III, hCA IV) by flavonoids. J Enzyme Inhib Med Chem 2013;28:283–8
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229
- Demirdag R, Comakli V, Senturk M, et al. Purification and characterization of carbonic anhydrase from sheep kidney and effects of sulfonamides on enzyme activity. Bioorg Med Chem 2013;21:1522–5
- Durdagi S, Senturk M, Ekinci D, et al. Kinetic and docking studies of phenol-based inhibitors of carbonic anhydrase isoforms I, II, IX and XII evidence a new binding mode within the enzyme active site. Bioorg Med Chem 2011;19:1381–9
- Alterio V, Vitale RM, Monti SM, et al. Carbonic anhydrase inhibitors: X-ray and molecular modeling study for the interaction of a fluorescent antitumor sulfonamide with isozyme II and IX. J Am Chem Soc 2006;128:8329–35
- Ekinci D, Ceyhun SB, Sentürk M, et al. Characterization and anions inhibition studies of an α-carbonic anhydrase from the teleost fish Dicentrarchus labrax. Bioorg Med Chem 2011;19:744–8
- Toth JE, Grindey GB, Ehlhardt WJ, et al. Sulfonimidamide analogs of oncolytic sulfonylureas. J Med Chem 1997;40:1018–25
- Medina JC, Shan B, Beckmann H, et al. Novel antineoplastic agents with efficacy against multidrug resistant tumor cells. Bioorg Med Chem Lett 1998;8:2653–6
- Ekinci D, Cavdar H, Talaz O, et al. NO-releasing esters show carbonic anhydrase inhibitory action against human isoforms I and II. Bioorg Med Chem 2010;18:3559–63
- Ekinci D, Cavdar H, Durdagi S, et al. Structure-activity relationships for the interaction of 5,10-dihydroindeno[1,2-b]indole derivatives with human and bovine carbonic anhydrase isoforms I, II, III, IV and VI. Eur J Med Chem 2012;49:68–73
- Ekinci D, Kurbanoglu NI, Salamci E, et al. Carbonic anhydrase inhibitors: inhibition of human and bovine isoenzymes by benzenesulphonamides, cyclitols and phenolic compounds. J Enzyme Inhib Med Chem 2012;27:845–8
- Alp C, Ekinci D, Gultekin MS, et al. A novel and one-pot synthesis of new 1-tosyl pyrrol-2-one derivatives and analysis for carbonic anhydrase inhibitory potencies. Bioorg Med Chem 2010;18:4468–74
- Balaydin HT, Soyut H, Ekinci D, et al. Synthesis and carbonic anhydrase inhibitory properties of novel bromophenols including natural products. J Enzyme Inhib Med Chem 2012;27:43–50
- Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem 2007;15:4336–50
- Demirdag R, Yerlikaya E, Senturk M, et al. Heavy metal ion inhibition studies of human, sheep and fish α-carbonic anhydrases. J Enzyme Inhib Med Chem 2013;28:278–82
- Ceyhun SB, Senturk M, Yerlikaya E, et al. Purification and characterization of carbonic anhydrase from the teleost fish Dicentrarchus labrax (European seabass) liver and toxicological effects of metals on enzyme activity. Environ Toxicol Pharmacol 2011;32:69–74
- Forouzesh B, Takimoto CH, Goetz A, et al. A phase I and pharmacokinetic study of ILX-295501, an oral diarylsulfonylurea, on a weekly for 3 weeks every 4-week schedule in patients with advanced solid malignancies. Clin Cancer Res 2003;9:5540–9
- Jung SH, Song JS, Lee HS, et al. Synthesis and evaluation of cytotoxic activity of novel arylsulfonylimidazolidinones. Bioorg Med Chem Lett 1996;6:2553–8
- Jung SH, Lee HS, Song JS, et al. Synthesis and antitumor activity of 4-phenyl-1-arylsulfonyl imidazolidinones. Bioorg Med Chem Lett 1998;8:1547–50
- Lee SH, Park KL, Choi SU, et al. Effect of substituents on benzenesulfonyl motif of 4-phenyl-1-arylsulfonylimidazolidinones for their cytotoxicity. Arch Pharm Res 2000;23:579–84
- Kim I, Lee C, Kim H, Jung, S. Importance of sulfonylimidazolidinone motif of 4-phenyl-1-arylsulfonylimidazolidinones for their cytotoxicity: synthesis of 2-benzoyl-4-phenyl[1,2,5]thiazolidine-1,1-dioxides and their cytotoxcity. Arch Pharm Res 2003;26:9–14
- Zuliani V, Carmi C, Rivara M, et al. 5-Benzylidene-hydantoins: synthesis and antiproliferative activity on A549 lung cancer cell line. Eur J Med Chem 2009;44:3471–9
- Abdel-Aziz AA, El-Azab AS, El-Subbagh HI, et al. Design, synthesis, single-crystal and preliminary antitumor activity of novel arenesulfonylimidazolidin-2-ones. Bioorg Med Chem Lett 2012;22:2008–14
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–9
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–51
- Laemmli DK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680–5
- Scozzafava A, Mastrolorenzo A, Supuran CT. Carbonic anhydrase inhibitors and activators and their use in therapy. Expert Opin Ther Pat 2006;16:1627–44