
Abstract
EGFR tyrosine kinase has been reported mainly in 40–80% of non-small lung cancers, in addition to colon and breast cancers. In this study, we illustrate the synthesis of a highly potent antitumor agent. The synthesized compound 4 was screened at NCI, USA, for antitumor activity against non-small lung cancer, colon cancer and breast cancer cell lines. Results indicated that this compound is more potent antitumor agent compared to erlotinib against all tested cell lines except breast cancer (MDA-MB-468) cell line. In addition, it was tested initially at a single dose concentration of 100 µM over 11 different kinases. At this concentration, 94.45% inhibition of the enzymatic activity of EGFR kinase was observed, while the inhibition in activity was below 55% in all other kinases. Compound 4 was further tested in a 10-dose IC50 mode and showed IC50 value of 0.239 µM for EGFR kinase. In vivo acute toxicity of this compound was also tested.
Introduction
The epidermal growth factor receptor (EGFR) is a transmembrane glycoprotein belonging to the human epidermal receptor (HER) familyCitation1. It plays a vital role in signal transduction pathways, regulating key cellular functions such as cell proliferation, survival, adhesion, migration and differentiationCitation2. The binding of a ligand to EGFR induces conformational changes within the receptor which increase its intrinsic catalytic activity of a tyrosine kinase and result in autophosphorylation, which is necessary for biological activityCitation3,Citation4. Mutations that lead to EGFR overexpression or over activity have been associated with a variety of human cancers, including lungCitation5, colonCitation6 and breastCitation7 cancers. Therefore, inhibitors of EGFR-inhibiting EGFR kinase activity by competing with its cognate ligands-may potentially constitute a new class of effective drugs in clinical use or cancer therapyCitation8,Citation9.
Anilinoquinazolines are the most developed class of drugs that inhibit EGFR tyrosine kinase intracellularlyCitation10,Citation11. Anilinoquinazoline-containing compounds, erlotinib (Tarceva®)Citation12,Citation13 and gefitinib (Iressa®)Citation14 have been approved for chemotherapeutic treatment of patients with advanced non-small cell lung cancer. Our strategy is directed toward designing ligands which are structurally similar to the basic skeleton, 4-anilinoquinazoline of erlotinib through replacing quinazoline ring with benzothiazole since both are isosteric with adenine portion of ATP and can mimic the ATP competitive binding regions of EGFR tyrosine kinase. With this aim, a 3D pharmacophore model was created based on erlotinib binding pose to the active site of EGFR tyrosine kinase comprising two hydrophobic features, two hydrogen bond acceptors and one hydrogen bond donor. Herein, we report the discovery of a potent and highly selective EGFR tyrosine kinase inhibitor 4. The structure based design strategy developed utilizing QSAR techniques, enabled the understanding of the pharmacophoric requirements of erlotinib as EGFR tyrosine kinase inhibitor (Supplementary Figure S1).
Experimental
Molecular modeling and computational studies
Protein structure preparation
The crystal structure of EGFR kinase domain (PDB ID: 1M17) in complex with an irreversible inhibitor was obtained from the protein data bank (PDB; http://www.rcsb.org/pdb/home/home.do). Refinement of crude PDB structure of receptor was performed. Polar hydrogens were added, Kollman charges were assigned and atomic solvation parameters were added, the internal degrees of freedom and torsions were set for all the designed small molecules. The optimized receptor was then saved as mol file and used for docking simulation.
Ligand structure preparation
The 2D structure of the compound 4 was built and then converted into the 3D with the help of vLife MDS 3.0 software. The 3D structure was then energetically minimized up to the rms gradient of 0.01 using CHARMM22 force field. All conformers were then energetically minimized up to the rms gradient of 0.01 and then saved in separate folder.
Docking protocol
Docking simulation was done by SwissDock softwareCitation15. All the conformers were virtually docked at the defined cavity of the receptor. The number of placements was fixed at 30 placements and the rotation angle was fixed at 30°. By rotation angle, the ligand gets rotated for different poses. By placements, the method will check all the 30 possible placements into the active site pocket and results out few best placements out of 30. For each ligand, all the conformers with their best placements and their dock score will be saved in output folder. The method also highlights the best placement for the best conformer of one particular ligand which is having best (minimum) dock score. After docking simulation, the best docked conformer of compound 4 and receptor was merged and its complex was then energetically optimized by defining the radius of 10 Å measured from the docked ligand. Stepwise energy optimization was done by first hydrogen, second side chains and finally the backbone of receptor.
Synthesis
Melting points (°C) were recorded on Fisher–Johns melting point apparatus and are uncorrected. The infrared spectra were recorded using Nicolet Magna-IR Fourier-Transform 560 Spectrometer (v in cm−1) at the Department of Chemistry, Georgia State University, USA. Nuclear magnetic resonance (1H and 13C NMR) spectra were obtained on Bruker Avance 400 MHz spectrometer using CDCl3, and Acetone-d6 as solvents at the Department of Chemistry, Georgia State University, USA. The chemical shifts are expressed in δ ppm using tetramethylsilane (TMS) as internal reference. Mass spectra were recorded on nano LC-Q-TOF micro (Waters Micromass) spectrometer in negative ion mode as necessary at the Department of Chemistry, Georgia State University, USA. Reaction times were monitored using TLC plates, Silica gel 60 F254 pre-coated (E. Merck) and the spots were visualized by UV(366 nm). Chloroform:methanol (9:1) was adopted as an elution solvent. 2-Amino-6-chlorobenzothiazole (1) was purchased from Richest Co., China.
A general approach to synthesize compound 4 is outlined in Scheme 1, started with the synthesis of 2-chloro-N-(6-chlorobenzothiazol-2-yl)acetamide (2). Chloroacetyl chloride (1.13 g, 0.01 mol) was added slowly with stirring to a mixture of 2-amino-6-chlorobenzothiazole (1) (1.85 g, 0.01 mol) and triethylamine (0.1 mL) in carbon tetrachloride (20 mL). The mixture was heated at reflux temprature for 12 h, then the solvent was evaporated under reduced pressure. The remaining solid was crystallized from ethanol. Yield 61%, m.p. 140–142 °CCitation16.
Scheme 1. Reaction conditions and reagents: (i) chloroacetyl chloride, CCl4, 12 h, 61%; (ii) hydrazine hydrate, C2H5OH, reflux, 14h, 58%; (iii) diethyl ethoxymethylenemalonate, anhydrous K2CO3, CH3CN, reflux, 12h, 45%.
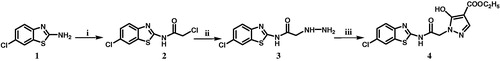
Synthesis of N-(6-chlorobenzothiazol-2-yl)-2-hydrazinylacetamide (3)
A mixture of compound 2 (2.6 g, 0.01 mol) and hydrazine hydrate 99% (5 g, 0.1 mol) in ethanol (30 mL) was heated at reflux temprature for 14 h. The reaction mixture was cooled and the precipitated solid was collected by filtration, washed with water, dried and crystallized from ethanol.
Yield 58%, m.p. 162–163 °C. IR spectrum (KBr, v, cm−1): 3255, 3210 (NH2), 3115 (2NH), 1655 (C = O). 1H NMR spectrum: (Acetone-d6, δ ppm): 2.96 (s, 2H, NH2), 4.45 (s, 2H, CH2), 6.96 (s, 2H, 2NH), 7.24 (d, 1H, Ar–H), 7.37 (d, 1H, Ar–H), 7.71 (s, 1H, Ar–H). HRMS: m/z (ESI) Calcd for C9H8ClN4OS−, [M–H]−: 255.0186; found: 255.0189.
Synthesis of ethyl 1-[2-((6-chlorobenzothiazol-2-yl)amino)-2-oxoethyl]-5-hydroxy-1H-pyrazole-4-carboxylate (4)
A mixture of compound 3 (0.257 g, 0.001 mol), diethyl ethoxymethylenemalonate (0.216 g, 0.001 mol) and anhydrous potassium carbonate (0.207 g, 0.0015 mol) in acetonitrile (15 mL) was heated at reflux temperature for 12 h. After cooling, the solution was acidified with dilute hydrochloric acid and the precipitated dark yellow solid was collected by filtration, dried and crystallized from ethanol.
Yield 45%, m.p. 195–197 °C. IR spectrum (KBr, v, cm−1): 3260 (OH), 3150 (NH), 1710 (COOC2H5), 1685 (C = O). 1H NMR spectrum: (CDCl3, δ ppm): 1.30 (t, 3H, CH2CH3), 3.55 (s, 2H, CH2), 4.05–4.35 (q, 2H, CH2CH3), 7.20–7.80 (m, 4H, Ar–H, pyrazole–H), 10.40 (s, 1H, OH), 11.50 (s, 1H, NH). 13C NMR: spectrum: (CDCl3, δ ppm): 14.6, 56.9, 61.0, 95.8, 121.0, 125.8, 128.0, 132.0, 135.5, 145.8, 157.2, 161.4, 167.2. HRMS: m/z (ESI) Calcd for C15H12ClN4O4S−, [M–H]−: 379.0346; found: 379.0352. CHN Analysis for C15H13ClN4O4S: Found (Calculated): C: 47.56 (47.31), H: 3.25 (3.44), N: 14.47 (14.71).
Tyrosine kinase assay
Initial screening over 11 kinases
The tested compound 4 was dissolved in DMSO and tested at a single concentration of 100 µM with a final DMSO concentration of 2%. Compound 4 was then added to reaction plates containing the particular kinase in assay buffer [20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.5, 10 mM MgCl2, 1 mM ethylene glycol tetra-acetic acid (EGTA), 0.02% Brij35, 0.02 mg/mL bovine serum albumin (BSA), 0.1 mM Na3VO4, 2 mM dithiothreitol (DTT), 1% DMSO]. Reactions were initiated by the addition of a mixture of ATP (Sigma, St. Louis, MO) and 33P ATP (Perkin Elmer, Waltham, MA) to a final concentration of 10 μM. Reactions were carried out at room temperature for 2 h, followed by spotting of the reactions onto P81 ion exchange filter paper (Whatman Inc., Piscataway, NJ). Unbound phosphate was removed by extensive washing of filters in 0.75% phosphoric acidCitation17. Kinase activity data was reported as the percentage remaining enzyme activity after subtraction of enzyme inhibitory activity of DMSO control reactions as background ().
Table 1. Percentages of enzymatic inhibition exerted by compound 4 on 11 human protein and lipid kinases.
Testing against EGFR kinase
Compound 4 was tested in a 10-dose IC50 mode with threefold serial dilutions starting at 20 µM. Staurosporine was used as a control compound in a 10-dose IC50 mode with fivefold serial dilutions starting at 20 µM. Reaction was carried out at 10 µM ATP concentrationCitation17. IC50 value of 4 was calculated.
Antitumor screening
Full in vitro five-dose antitumor assay
The human tumor cell lines of the cancer screening panel were grown in RPMI 1640 medium containing 5% fetal bovine serum and 2 mM l-glutamine. For a typical screening experiment, cells were inoculated into multiwell microtiter plates in 100 mL at plating densities ranging from 5000 to 40 000 cells/well depending on the doubling time of individual cell lines. After cell inoculation, the microtiter plates were incubated at 37 °C, 5% CO2, 95% air and 100% relative humidity for 24 h and then two plates of each cell line were fixed in situ with trichloroacetic acid (TCA) to represent a measurement of the cell population for each cell line at the time of compound addition. The tested compound was solubilized in DMSO at 400-fold the desired final maximum test concentration and stored frozen prior to use. At the time of compound addition, an aliquot of frozen concentrate was thawed and diluted to twice the desired final maximum test concentration with complete medium containing 50 mg/mL gentamicin. Additional 4-, 10-fold or 1/2 log serial dilutions were made to provide a total of five compound concentrations plus control. Aliquots of 100 mL of these dilutions were added to the appropriate microtiter wells already containing 100 mL of medium, resulting in the required final compound concentrations. Following compound addition, the plates were incubated for additional 48 h at 37 °C, 5% CO2, 95% air and 100% relative humidity. For adherent cells, the assay was terminated by the addition of cold TCA. Cells were fixed in situ by gentle addition of 50 mL of cold 50% (w/v) TCA (final concentration, 10% TCA) and incubated for 60 min at 4 °C. The supernatant was discarded, and the plates were washed five times with tap water and air dried. Sulforhodamine B (SRB) solution (100 mL) at 0.4% (w/v) in 1% acetic acid was added to each well, and plates were incubated for 10 min at room temperature. After staining, unbound dye was removed by washing five times with 1% acetic acid and the plates were air dried. Bound stain was subsequently solubilized with 10 mM trizma base and the absorbance was read on an automated plate reader at a wavelength of 515 nmCitation18–20. GI50 values (µM) of compound 4 and erlotinib are shown in .
Table 2. GI50 values (µM) of compound 4 and erlotinib over cell lines of non-small lung cancer, colon cancer and breast cancer subpanels.
In vivo acute toxicity testing
Adult male Swiss albino mice (weighing 20–25 g) were obtained from animal house, Department of Pharmacology, Faculty of Pharmacy, Mansoura University, Mansoura, Egypt. Animals were housed in microlon boxes in a controlled environment (temperature 25 ± 2 °C) with a regular 12 h light/12 h dark and were allowed free access to standard laboratory food and water.
Animals were divided into groups each consisting of six mice. One of these groups served as control and received normal saline. The other groups were subjected to acute intraperitoneal toxicity study using the tested compound in different concentrations and mice were observed for toxic symptoms, signs of poisoning and mortality continuously after dosingCitation21. Finally, the number of survivors in each group was noted after 24 h. The arithmetical method of Karber was used for the determination of LD50 values (mg/kg) of the selected compoundsCitation22.
Results and discussion
Molecular modeling and computational studies
With the software LigandScoutCitation23, a 3D interaction map was generated from the ligand (erlotinib) co-crystallized in the protein binding site (PDB ID: 1M17; http://www.rcsb.org/pdb/home/home.do). The generated structure-based pharmacophore showed that two main hydrophobic features accommodating the hydrophobic residues of the ligand in the binding pocket (Supplementary Figure S2). Compound 4 forms impressive alignment with the 3D pharmacophore of erlotinib binding pose which may indicate that both compounds possess similar arrangements of pharmacophoric characters required for activity (Supplementary Figure S3).
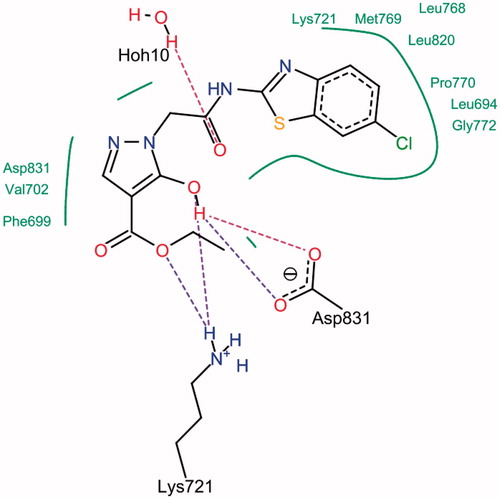
The SwissDock softwareCitation15 was used to find plausible docking poses for compound 4 into the crystal structure of EGFR kinase domain (PDB ID: 1M17; http://www.rcsb.org/pdb/home/home.do). Evaluation was done using two scoring functions; simple fitness and full fitness (Supplementary Table S1). Compound 4 was deeply embedded into the ATP-binding cleft of EGFR tyrosine kinase (Supplementary Figure S4). Analysis of the docking pose of compound 4 using Lead IT software showed good complementarities between the docked ligand and the hydrophobic subsites of the enzymatic cavity. In the first subsite, the benzothiazole ring interacted with the residues Lys-721, Met-769, Leu-768, Leu-820, Pro-770, Leu-694 and Gly-772, while the pyrazole moiety interacted with the residues Asp-831, Val-702 and Phe-699 of the second enzyme hydrophobic subsite. Furthermore, it was involved in three hydrogen bonds with the active site of EGFR tyrosine kinase ().
Figure 1. 2D Interaction of compound 4 with the binding site of EGFR-TK. Dashed lines represent hydrogen bonds. Hydrophobic interactions are shown by green solid lines.
Chemistry
The synthetic protocol for compound 4 is outlined in Scheme 1. The starting compound, 2-amino-6-chlorobenzothiazole (1) was allowed to react with chloroacetyl chloride in carbon tetrachloride to produce the acetamide derivative 2Citation16. Reaction of 2 with hydrazine hydrate in ethanol gave the hydrazinylacetamide analog 3. Condensation of 3 with diethyl ethoxymethylenemalonate in acetonitrile and in the presence of anhydrous potassium carbonate gave the targeted ethyl pyrazole-4-carboxylate derivative 4.
Tyrosine kinase assay
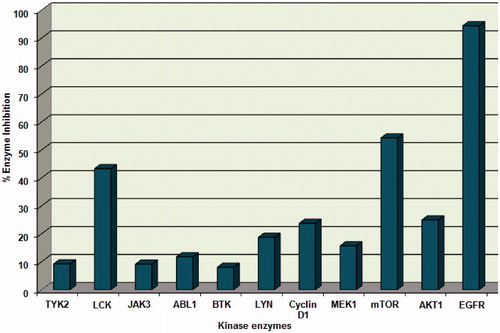
In vitro profiling of compound 4 was performed at Reaction Biology Corporation to assess its inhibitory activity of EGFR tyrosine kinase. Kinase activity was assessed using HotSpot technology, a miniaturized radioisotope-based filter binding assayCitation17. Kinase activity data was reported as the percent remaining enzyme activity after subtraction of enzyme inhibitory activity of the negative control (DMSO). Results are presented as percentage enzyme inhibition and compared to staurosporine as a reference EGFR tyrosine kinase inhibitor. Compound 4 displayed 94.4% inhibition of the enzymatic activity of EGFR kinase at 100 µM ( and ).
Figure 2. Kinase profile assay of compound 4 against different human protein and lipid kinases.
Compound 4 was further tested over EGFR kinase in order to determine its IC50 value, where a 10-dose IC50 mode with threefold serial dilutions starting at 20 µM concentration was applied against staurosporineCitation24–26 as a reference kinase inhibitor. Compound 4 showed IC50 value of 0.239 µM, while the IC50 value for the non-selective kinase inhibitor staurosporine was 0.0533 µM.
Compound 4 was also examined over other 10 different human kinases containing SH2 or both SH2 and SH3 domains and other human protein or lipid kinases adopting the kinase profile assayCitation17 using staurosporine and LY294002 as control compounds. Results showed that compound 4 exhibited weak to moderate inhibitory activity in the range of 8.1–54.3% with the highest inhibitory activity against mTOR kinase with percentage inhibition value of 54.3% ( and ).
The high selectivity of compound 4 toward EGFR kinase might be attributed to difference in the geometry of the binding pocket of this enzyme that enables the fitting and interaction of compound 4. It is worth mentioning that the mechanism of its unique inhibitory activity is still unclear.
Antitumor screening
Comparison of the antitumor activity of compound 4 with the activity of erlotinib was performed against non-small lung cancer (NCI-H522) cell line which harbors EGFR wild typeCitation27. Moreover, colon cancer (HCT-116, HCT-15 and HT-29) cell lines were selected for the comparison since they express high levels of EGFRCitation28–30, e.g. HCT-116 and HT-29 cell lines express approximately 100 000–150 000 EGFRs per cellCitation29. Breast cancer (MDA-MB-468 and MDA-MB-231) cell lines were also selected for the same comparison, whereas, MDA-MB-468 cell line was proved by Western blot to express the highest EGFR level compared to colon cancer (HCT-116, HT-29, IEC-6 and Caco-2) and breast cancer (SKBR-3) cell linesCitation31. In addition, MDA-MB-231 cell line expresses high levels of EGFR compared with MCF-10 A cellsCitation32.
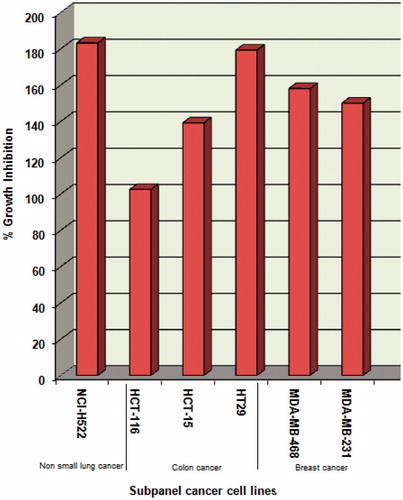
Compound 4 was evaluated for its antitumor activity in accordance with the current protocol of the National Cancer Institute (NCI), USACitation18–20. The data was reported as mean-graph of the percent growth of the treated cells, and presented as percentage growth inhibition (GI%). It exhibited lethal effects (>100% inhibition) at a single dose (10 µM) against non-small lung cancer (NCI-H522), colon cancer (HCT-116, HCT-15 and HT29) and breast cancer (MDA-MB-468 and MDA-MB-231) cell lines in which EGFR is overexpressed in varying levels (). Therefore, it was carried over by the NCI for the five-dose screening.
Figure 3. % Inhibition expressed by compound 4 at a single-dose concentration of 10 µM over the selected cancer cell lines.
Non-small lung cancer (NCI-H522), colon cancer (HCT-116, HCT-15, and HT29) and breast cancer (MDA-MB-468 and MDA-MB-231) cell lines were incubated with five concentrations (0.01–100 µM) of compound 4 and were used to create log concentration – % growth inhibition curves. The GI50 values of compound 4 and erlotinib against the selected cancer cell lines are listed in . Results indicated that compound 4 is more potent than erlotinib against all selected cell lines except MDA-MB-468. It showed the lowest GI50 value (57.3 nM) against non-small lung (NCI-H522) cancer cell line.
Further investigation of antitumor activity of compound 4 against additional cancer cell lines belonging to non-small lung cancer, colon cancer and breast cancer subpanels is presented in . Compound 4 was proved to be highly selective against non-small lung cancer (NCI-H522) cell line compared to the other cell lines belonging to the same subpanel. Moreover, it showed moderate selectivity against colon cancer (HT29) cell line compared to the other cell lines belonging to the same subpanel. With regard to the breast cancer subpanel, compound 4 showed GI50 values in the submicromolar range only against breast cancer (MDA-MB-468 and MDA-MB-231) cell lines.
In vivo acute toxicity testing
LD50 is a measurement used in toxicological studies to determine the potential impact of toxic substances on different types of test animals typically mice, rabbits, guinea pigs or even larger animals such as dogs. It provides an objective measure to compare and rank the toxicity of substances. The LD50 is defined as the amount of the substance required to kill 50% of a given test population, it is usually expressed as the amount of substance per kg of body weight. Compound 4 was screened for potential acute toxicity in miceCitation21 and the dose (mg/kg) required to kill 50% of animals (LD50) within 24 h was calculated. Compound 4 showed LD50 value of 365 mg/kg, while the LD50 value for the reference antitumor agent, 5-fluorouracil was 115 mg/kg.
Conclusion
In conclusion, a highly potent, safe and selective antitumor agent as well as EGFR tyrosine kinase inhibitor has been synthesized and can be used as a promising lead for new selective inhibitors for EGFR tyrosine kinase. Also, it is worth mentioning that the development of new selective inhibitors for such kinase might open the way for new selective therapeutics for non-small lung, colon and breast cancers.
Supplementary material available online
Supplementary Figures S1–S4 and Table S1
IENZ_887707_supplemental_information.pdf
Download PDF (286.8 KB)Acknowledgements
The authors would like to express their sincere thanks to the staff members of the Department of Health and Human Services, National Cancer Institute (NCI), Bethesda, Maryland, USA for performing the antitumor testing of the newly synthesized compound. Thanks to Reaction Biology Corporation, USA for performing the kinase profile assay.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Holbro T, Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu Rev Pharmacol Toxicol 2004;44:195–217
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001;2:127–37
- Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 2002;110:669–72
- Voldborg BR, Damstrup L, Spang-Thomsen M, Poulsen HS. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann Oncol 1997;8:1197–206
- Tanno S, Ohsaki Y, Nakanishi K, et al. Small cell lung cancer cells express EGFR and tyrosine phosphorylation of EGFR is inhibited by gefitinib (“Iressa”, ZD1839). Oncol Rep 2004;12:1053–7
- Van der Veeken J, Oliveira S, Schiffelers RM, et al. Crosstalk between epidermal growth factor receptor- and insulin-like growth factor-1 receptor signaling: implications for cancer therapy. Curr Cancer Drug Targets 2009;9:748–60
- Nguyen D, Yang SX. Epidermal growth factor expression in breast cancer. J Clin Oncol 2005;23:8118–41
- Ma XH, Wang R, Tan CY, et al. Virtual screening of selective multitarget kinase inhibitors by combinatorial support vector machines. Mol Pharm 2010;7:1545–60
- Boschelli DH. Small molecule inhibitors of receptor tyrosine kinases. Drug Future 1999;24:515–37
- Xu YY, Li SN, Yu GJ, et al. Discovery of novel 4-anilinoquinazoline derivatives as potent inhibitors of epidermal growth factor receptor with antitumor activity. Bioorg Med Chem 2013;21:6084–91
- Bridges AJ. Chemical inhibitors of protein kinases. Chem Rev 2001;101:2541–72
- Reck M, Zandwijk NV, Gridelli C, et al. Erlotinib in advanced non-small cell lung cancer: efficacy and safety findings of the global phase IV tarceva lung cancer survival treatment study. J Thorac Oncol 2010;5:1616–22
- Smith J. Erlotinib: small-molecule targeted therapy in the treatment of non-small cell lung cancer. Clin Ther 2005;27:1513–34
- Tamura K, Fukuoka M. Gefitinib in non-small cell lung cancer. Expert Opin Pharmacother 2005;6:985–93
- Grosdidier A, Zoete V, Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. J Comput Chem Nucleic Acids Res 2011;39:270–7
- Mohd A, Asif S, Israr A, Mohd ZH. Synthesis of benzothiazole derivatives having acetamido and carbothioamido pharmacophore as anticonvulsant agents. Med Chem Res 2012;21:2661–70
- Ma H, Deacon S, Horiuchi K. The challenge of selecting protein kinase assays for lead discovery optimization. Expert Opin Drug Discov 2008;3:607–21
- Grever MR, Schepartz SA, Chabner BA. The National Cancer Institute: cancer drug discovery and development program. Semin Oncol 1992;19:622–38
- Boyd MR, Paull KD. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Rev Res 1995;34:91–109
- Monks A, Scudiero D, Skehan P, et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst 1991;83:757–66
- Nalbantsoy A, Karabay-Yavasoglu NU, Sayım F, et al. Determination of in vivo toxicity and in vitro cytotoxicity of venom from the Cypriot blunt-nosed viper Macrovipera lebetina lebetina and antivenom production. J Venom Anim Toxins Incl Trop Dis 2012;18:208–16
- Turner R, ed. Quantal responses: calculation of ED50. In Screening methods in pharmacology. New York: Academic Press; 1965:61–3
- Wolber G, Langer T. LigandScout: 3D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Comp Sci 2005;45:160–9
- Omura S, Sasaki Y, Iwai Y, Takeshima H. Staurosporine, a potentially important gift from a microorganism. J Antibiot (Tokyo) 1995;48:535–48
- Gescher A. Staurosporine analogues – pharmacological toys or useful antitumour agents? Crit Rev Oncol Hematol 2000;34:127–35
- Yang S, Malaviya R, Wilson LJ, et al. Simplified staurosporine analogs as potent JAK3 inhibitors. Bioorg Med Chem Lett 2007;17:326–31
- Kubo T, Yamamoto H, Lockwood W, et al. MET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitors. Int J Cancer 2009;124:1778–84
- Milenic D, Wong K, Baidoo K, et al. Cetumximab: preclinical evaluation of monoclonal antibody targeting EGFR for radioimmunodiagnostic and radioimmunotherapeutic applications. Cancer Biother Radiopharm 2008;23:619–31
- Sahlberg S, Spiegelberg D, Lennartsson J, et al. The effect of a dimeric Affibody molecule (ZEGFR:19072) targeting EGFR in combination with radiation in colon cancer cell lines. Int J Oncol 2012;40:176–84
- Ryan A, Wedge S. ZD6474, an inhibitor of VEGFR and EGFR tyrosine kinases, blocks VEGF-C-induced activation of VEGFR-3 and cell proliferation in human colon cancer cell lines. Br J Cancer 2005;92:S6–13
- Xu H, Yu Y, Marciniak D, et al. Epidermal growth factor receptor (EGFR)-related protein inhibits multiple members of the EGFR family in colon and breast cancer cells. Mol Cancer Ther 2005;4:435–42
- Reginato MJ, Mills KR, Paulus JK, et al. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat Cell Biol 2003;5:733–40