
Abstract
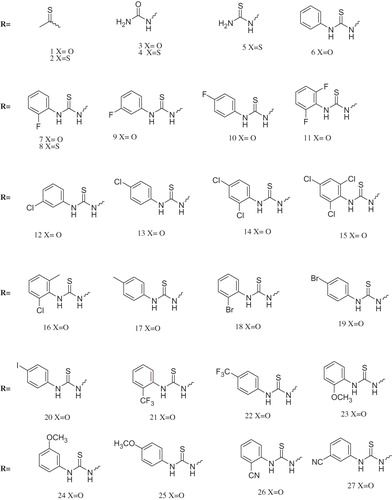
A series of 27 new barbiturates and thiobarbiturates have been synthesized by a convenient multi-component reaction in overall excellent yields (87–96%). All the synthesized compounds were characterized by 1H, 13C NMR, EIMS and elemental analysis (C, H, N and S). Furthermore, all compounds were screened for in vitro antioxidant (DPPH radical scavenging), lipoxygenase, chymotrypsin, α-glucosidase and anti-urease activities. Out of the series, 23 in DPPH, 14 in lipoxygenase, 2 in chymotrypsin have shown appreciable IC50 values.
Introduction
Barbiturates and thiobarbiturates are cheap and readily available synthon used in the manufacturing of diverse pharmaceutical productsCitation1 due to their remarkable biological potentials for various medical conditions. Barbiturates are well-known anesthetic and sleep inducing agentsCitation2 by acting through central nervous system. These are employed as hypnotic, sedative, anticonvulsant and for anesthesiaCitation3,Citation4. Furthermore, some are used for the treatment of psychiatric disorders such as anxiety and epilepsy by posing effects on motor and sensory functionsCitation5,Citation6 and are also reported as bactericidal agentCitation3,Citation4,Citation7.
Previously we have reported and identified various pharmacophores responsible for potent activity of barbiturate/thiobarbiturate nucleus via molecular modeling studiesCitation8–11. It is evident that the imide or β-amino α, β-unsaturated amide functionality is the basic structural moiety in these compounds which has multidimensional array of interaction sites resulting in either potential urease inhibitors (PUI), potential antibacterial (PAB), antifungal Inhibitors (PAF) and potential kinase inhibitors (PKI; ). In case of PUI, PAB and PAF the mode of action is believed to be mainly involving the scavenging or encapsulation of the metal ion whereas in PKI the presence of various donors/receptors interactions via hydrogen bonding, electronic interactions and metal ion encapsulation seems to be the cause. Further studies are still required to strengthen and verification of these findings.
Figure 1. Pharmacophore site combination. (X = O, S). PUI: Potential urease inhibitors (Y, Z = O, N, S); (PAB)/(PAF): potential antibacterial and antifungal inhibitors (Y, Z = O, N, S); (PKI): potential kinase inhibitorsCitation8.
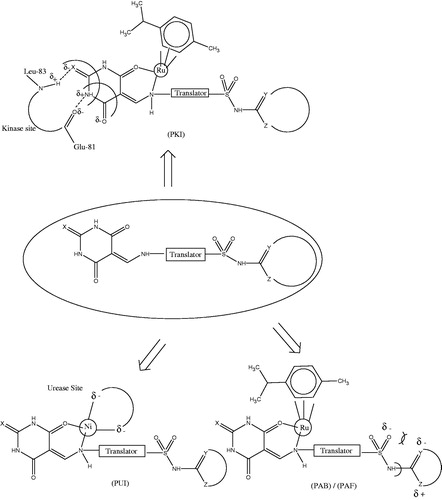
Emphasizing on these valuable pharmacophores, various new libraries of barbiturates/thiobarbiturates were identified and are synthetically afforded in the present series of compounds (1–27) via an efficient one pot multi-component reaction in an excellent yield. All the synthesized compounds were extensively characterized and screened for in vitro antioxidant (DPPH radical scavenging), lipoxygenase, chymotrypsin, α-glucosidase and anti-urease activities.
Material and methods
General
All reagents and solvents were used as supplied and/or recrystallized/redistilled as necessary. Melting points were taken on a Fisher–Johns melting point apparatus and are uncorrected. Elemental analyses were performed on a Leco CHNS-9320 elemental analyzer. The NMR spectra were recorded in DMSO-d6 on Bruker (Rhenistetten-Forchheim, Germany) AM 300 spectrometers operating at 300 MHz, using TMS as an internal standard. 1H chemical shifts are reported in (ppm) and coupling constants in Hz. The electron impact mass spectra (EIMS) were determined with a Finnigan MAT-312 and a JEOL MS Route mass spectrometer. The progress of the reaction and purity of the products were checked on TLC plates coated with Merck silica gel 60 GF254 and the spots were visualized under ultraviolet light at 254 and 366 nm and/or spraying with iodine vapors. In vitro enzyme inhibition and antibacterial assays of the synthesized compounds were carried out at the Department of Biotechnology, The Islamia University of Bahawalpur.
Synthesis
General procedure for the preparation of compounds (1–27)
To a hot stirred solution of barbituric/thiobarbituric acid (2.00 mmoles) and ethylorthoformate (2.02 mmoles) in 2-butanol (10 mL) was added the respective amine (2.00 mmoles). Then the reaction mixture was refluxed till the completion of the reaction (for ≈3 h). The precipitates formed during refluxing were collected by suction filtration. Washing with hot ethanol afforded TLC pure products in good to excellent yieldCitation8 (Scheme 1).
Scheme 1. Synthesis of 1–27.
Enzyme inhibition (in vitro)
DPPH radical scavenging activity
Stable 1,1-diphenyl-2-picrylhydrazyl radical (DPPH) was used for determining antioxidant activity according to the reported methodCitation12. Different concentrations of compounds in respective solvents were added at an equal volume (10 µL) to 90 µL of 100 µM methanolic DPPH in a total volume of 100 µL in 96-well plates. After mixing contents, these were incubated at 37 °C for 30 min. Absorbance was then measured at 517 nm. Quercetin was used as standard antioxidants. Decrease in absorbance indicated that the increased radical scavenging activity which was determined by the following formula.
Lipoxygenase
Lipoxygenase (LOX) activity was assayed according to the reported methodCitation13–15 with slight modifications. A total volume of 200 µL assay mixture contained 140 µL sodium phosphate buffer (100 mM, pH 8.0), 20 µL test compound and 15 µL (600 U) purified lipoxygenase enzyme (Sigma, St. Louis, MO). Contents were mixed, pre-read at 234 nm and pre-incubated for 10 min at 25 °C. Reaction was initiated by addition of 25 µL substrate solution. Change in absorbance was observed after 6 min at 234 nm. Baicalein (0.5 mM well−1) was used as a positive control.
Chymotrypsin
α-Chymotrypsin inhibition activity was performed according to slightly modified method of Rehman et alCitation16. A total volume of 100 μL assay mixture contained 60 μL Tris–HCl buffer (50 mM pH 7.6), 10 μL test compound and 15 μL (0.9 units) purified α-chymotrypsin enzyme (Sigma, St. Louis, MO). The contents were mixed and incubated for 20 min at 37 °C and pre-read at 410 nm. The reaction was initiated by the addition of 15 mL (1.3 mM) substrate (N-succinyl phenyl-alanine-p-nitroanilide). The change in absorbance was observed after 30 min at 410 nm. Chymostatin (0.5 mM well−1) was used as a positive control.
α-Glucosidase
The α-glucosidase inhibition activity was performed according to the slightly modified method of Pierre et alCitation17. Total volume of the reaction mixture of 100 µL contained 70 µL 50 mM phosphate buffer saline, pH 6.8, 10 µL (0.5 mM) test compound, followed by the addition of 10 µL (0.057 units) enzyme. The contents were mixed, pre-incubated for 10 min at 37 °C and pre-read at 400 nm. The reaction was initiated by the addition of 10 µL of 0.5 mM substrate (p-nitrophenyl glucopyranoside). Acarbose was used as positive control. After 30 min of incubation at 37 °C, absorbance was measured at 400 nm using Synergy HT microplate reader. All experiments were carried out in triplicates.
Urease
The enzyme assay is the modified form of the commonly known Berthelot assay. A total volume of 85 µL assay mixture contained 10 µL of phosphate buffer of pH 7.0 in each well in the 96-well plate followed by the addition of 10 µL of sample solution and 25 µL of enzyme solution (0.1347 units). Contents were pre-incubated at 37 °C for 5 min. Then, 40 µL of urease stock solution (20 mM) was added to each well and incubation continued at 37 °C for further 10 min. After given time, 115 µL phenol hypochlorite was added in each well (freshly prepared by mixing 45 µL phenol reagent with 70 µL of alkali reagent). For color development, incubation was done at 37 °C for another 10 min. Absorbance was measured at 625 nm using the 96-well plate reader Synergy HT.
Statistical analysis
Statistical analysis was performed by Microsoft Excel 2003. Results are presented as mean ± SEM. The percent inhibition was calculated by the help of following equation.
IC50 values (concentration at which there is 50% enzyme inhibition) of compounds were calculated using EZ-Fit Enzyme kinetics software (Perrella Scientific Inc., Amherst, NH). All the measurements were done in triplicate. Synergy HT (BioTek, Winooski, VT) 96-well plate reader was used in all experiments. The positive and negative controls were included in the assay.
Results and discussion
This study comprises of synthesis and in vitro evaluation of enzyme inhibitory activities of 27 new barbiturates and thiobarbiturates.
Chemistry
For synthesis, to a refluxing solution of thio/barbituric acid and triethylorthoformate in 2-butanol was added respective amine. The precipitates formed were collected and washed with hot ethanol offering pure products in excellent yields (87–96%). The structures of the synthesized compounds were deduced by analytical and spectroscopic (1H-NMR, 13CNMR and EIMS) data (supplementary material). Satisfactory elemental analyses (±0.4% of calculated values) were obtained for all the compounds.
N-[(2,4,6-trioxotetrahydropyrimidin-5(2H)-ylidene)methyl]ethanethioamide (1)
1H-NMR (300 MHZ, DMSO-d6, δ, ppm): 2.94 (3H, s, CH3), 5.31 (1H, s, NH), 8.00 (1H, s, =CH), 11.51 (2H, s, NHBA); 13C-NMR (300 MHZ, DMSO-d6, δ, ppm): 208, 178, 150, 100, 90, 23; EIMS (70 eV) m/z (%): ([M+], 213 (77)), 197 (14), 185 (17), 155 (100), 144 (32), 128 (37), 114 (14), 99 (13), 85 (11), 69 (25), 59 (44), 44 (41); Anal. Calcd. for C7H7N3O3S: C, 39.44; H, 3.29; N, 19.72; S, 15.02; Found: C, 39.43; H, 3.28; N, 19.69; S, 15.05.
N-[(4,6-dioxo-2-thioxotetrahydropyrimidin-5(2H)-ylidene)methyl]ethanethioamide (2)
1H-NMR (300 MHZ, DMSO-d6, δ, ppm): 2.81 (3H, s, CH3), 5.11 (1H, s, NHCS), 8.20 (1H, s, =CH), 11.45 (2H, s, CONH); 13C-NMR (300 MHZ, DMSO-d6, δ, ppm): 208, 178, 158, 91, 23; EIMS (70 eV) m/z (%): ([M+], 229 (35)), 213 (23), 171 (100), 160 (6), 144 (10), 127 (4), 113 (5), 101 (8), 84 (3), 59 (14), 43 (14); Anal. Calcd. for C7H7N2O2S2: C, 39.07; H, 3.26; N, 13.02; S, 29.77; Found: C, 39.09; H, 3.28; N, 13.00; S, 29.79.
2-[(2,4,6-trioxotetrahydropyrimidin-5(2H)-ylidene)methyl]hydrazincarboxamide (3)
1H-NMR (300 MHZ, DMSO-d6, δ, ppm): 6.56 (2H, s, NH2), 8.83 (1H, s, =CH), 9.93 (2H, s, NHNH), 11.12 (2H, s, NHBA); 13C-NMR (300 MHZ, DMSO-d6, δ, ppm): 167, 157, 151, 99; EIMS (70 eV) m/z (%): ([M+], 213 (1)), 199 (3), 128 (100), 100 (10), 85 (49), 69 (9), 42 (85); Anal. Calcd. for C6H7N5O4: C, 33.80; H, 3.29; N, 32.86; Found: C, 33.82; H, 3.28; N, 32.87.
2[(4,6-dioxo-2-thioxotetrahydropyrimidin-5(2H)-ylidene)methyl]hydrazinecarboxamide (4)
1H-NMR (300 MHZ, DMSO-d6, δ, ppm): 6.49 (2H, s, NH2), 8.06 (1H, s, =CH), 9.90 (2H, s, NHNH), 11.38 (1H, s, NHCOBA),11.92 (1H, s, CSNHBA); 13C-NMR (300 MHZ, DMSO-d6, δ, ppm) 177, 174, 163, 157, 155, 90; EIMS (70 eV) m/z (%): ([M+], 229 (1)), 199 (7), 186 (47), 144 (19), 116 (8), 127 (6), 110 (11), 69 (20), 59 (12), 44 (100); Anal. Calcd. for C6H7N5O2S2: C, 29.39; H, 2.86; N, 28.57; S, 26.12; Found: C, 29.37; H, 2.85; N, 28.58; S, 26.11.
2[(4,6-dioxo-2-thioxotetrahydropyrimidin-5(2H)-ylidene)methyl]hydrazinecarbothioamide (5)
1H-NMR (300 MHZ, DMSO-d6, δ, ppm): 7.05 (2H, s, NH2), 8.07 (1H, s, =CH), 11.44 (2H, s, NHNH), 11.97 (2H, s, NHBA); 13C-NMR (300 MHZ, DMSO-d6, δ, ppm) 181, 177, 162, 91; EIMS (70 eV) m/z (%): ([M+], 245 (100)), 186 (61), 158 (48), 144 (22), 136 (22), 127 (6), 115 (20), 101 (5), 89 (25), 77 (11), 63 (14), 50 (8), 44 (17); Anal. Calcd. for C6H7N5O2S2: C, 29.39; H, 2.86; N, 28.57; S, 26.12; Found: C, 29.37; H, 2.87; N, 28.58; S, 26.11).
N-(phenyl-2-[(2,4,6-trioxotetrahydropyrimidin-5(2H)-ylidene)methyl]hydrazinecarbothio amide (6)
1H-NMR (300 MHZ, DMSO-d6, δ, ppm): 5.67 (1H, s, NHAr), 7.01 (1H, d, J 9, ArH), 7.28 (2H, t, J 6,15, ArH), 7.61 (2H, d, J 6, ArH), 8.00 (1H, s, =CH), 10.51 (2H, s, NHNH), 10.94 (2H, s, NHBA); 13C-NMR (300 MHZ, DMSO-d6, δ, ppm) 179, 167, 154, 151, 150, 128, 128, 86; EIMS (70 eV) m/z (%): ([M+], 305 (4)), 231 (100), 187 (3), 170 (67), 154 (58), 128 (90), 118 (22), 77 (49), 44 (31); Anal. Calcd. for C12H11N5O3S: C, 47.21; H, 3.61; N, 22.95; S, 10.49; Found: C, 47.22; H, 3.60; N, 22.94; S, 10.50.
The 1H-NMR spectra of these compounds exhibited three separate singlets at δ = 4.30–6.56, 9.21–11.45 and 10.00–12.81 for the thiosemicarbazones N4–H, thiosemicarbazone N2–H and N3–H and barbiturate NH, respectively. Whereas =CH appeared as a singlet at δ 7.84–9.84. Peaks in APT 13CNMR appeared at δ 175–200 for C = S, 155–170 for C=O, 140–150 for C = CH–NH, 118–140 for aromatic C, 110–70 for C = CH.
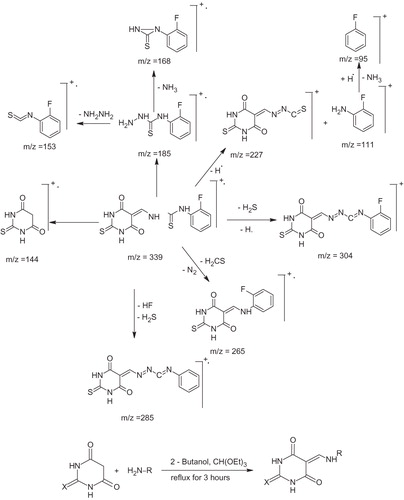
In EI mass spectra, all the compounds showed molecular ions of different intensity, which confirmed their molecular weights. The major fragmentation pathway involved the cleavage of the exocyclic N–N, NH–CS and endocyclic NH–CO bonds. Few of the compounds did not show the molecular ion peaks in their spectra. However, the fragments corresponding to thiosemicarbazone moiety, formed by the cleavage of N–N and NH–CS bonds confirmed their structures.
The proposed fragmentation pattern of 8 is presented in Scheme 2.
Scheme 2. Proposed fragmentation pattern of 8.
Bioactivities
Enzyme assays
Results for enzyme inhibition are presented in . All the compounds are antioxidants except 1, 5, 13 and 19. Compound 10 showed maximum inhibition with IC50 value 63.11 at 0.5 mM and % inhibition of 92.23. 17 Compounds 9, 17, 15, 20, 14, 8, 23, 25, 27, 21, 12, 24, 7 and 11 showed IC50 value of 138.71 at 0.5 mM, 145.25 at 0.5 mM, 196.41 at 0.5 mM, 198.21 at 0.5 mM, 200.31 at 0.5 mM, 204.8 at 0.5 mM, 205.91 at 0.25 mM, 229.1 at 0.25 mM, 303 at 0.25 mM, 329.71 at 0.5 mM, 376 at 0.25 mM, 378.11 at 0.5 mM, 407.11 at 0.5 mM, 407.11 at 0.5 mM, <500 at 0.5 mM, <500 at 0.25 mM and <500 at 0.5 mM against lipoxygenase. Against Chymotrypsin almost all the compounds are active but only 18 and 13 showed reportable IC50 value of 78.4 at 0.25 mM, 12.87 at 0.25 mM and 159.3 at 0.5 mM, respectively. Hence compound 18 found to be most active inhibitor with % inhibition of 78.62.
Table 1. Enzyme assays-Lipoxygenase; 28 quercetin (standard drug), DPPH radical scavenging; 29 baicalein (standard drug), Chymotripsin; 30 chymostatin (standard drug), Alpha-glucosidase; 31 acarbose (standard drug), Antiurease; 32 thiourea (standard drug).
In DPPH activity p-F is most effective inhibitor. Comparing DPPH IC50 values of 4 and 6, it appeared that thiobarbiturate with NH2 is more effective than barbiturate with unsubstituted phenyl ring but less effective than the same with a para Fluro substitution 10. Whereas same Fluro at meta 9, is less inhibitory than 10. Also, from IC50 values of 14 and 15, it is apparent that electron withdrawing substitution at ortho also effects negatively on inhibition activity of such compounds. Order of lipoxygenase activity with respect to substitution position on phenyl ring is m-F > p-CH3 > diortho-p-triCl > p-I > o,p-diCl > o-F > o-OCH3 > p-OCH3 > m-CN > m-OCH3 > m-Cl > o–F. From IC50 values of 9 and 7 (i.e. m-F and o-F) and 9 and 8 (i.e. X = O and X = S), it can be inferred that electron withdrawing fluoro-substituent at meta position inhibits more than at ortho position and also thoibarbiturate is more effective in this typical activity than barbiturate. In chymotrypsin inhibition only o-Br and p-Cl compounds have shown some IC50 values. Therefore, this typical activity can be attributed to the halogens at ortho, para position of such compounds. Comparing IC50 values, it is speculated that electrostatic effects of substituents (translator, ) play an important role in the enzyme inhibitory potential of the synthesized compounds.
Conclusions
This study successfully afforded the synthesis of targeted 27 new barbiturates/thiobarbiturates derivatives in good to excellent yield via an efficient one pot three component reaction. All the synthesized compounds were extensively characterized and screened for biological activities. All the compounds exhibited appreciable % inhibition at each instance strengthening the idea that structural motifs such as imide/β-amino α,β-unsaturated amide play a vital role as pharmacophore–enzyme interaction site and presence of suitable substituents (translator) eventually magnify inhibition of enzymes in general (). In vitro evaluation for antioxidant (DPPH radical scavenging), lipoxygenase, chymotrypsin, α-glucosidase and anti-urease activities yielded 23 compounds in DPPH, 14 in lipoxygenase and two in chymotrypsin exhibiting appreciable IC50 values.
Supplementary material available online
Supplementary materials
Supplementary Material
Download PDF (1.4 MB)Declaration of interest
There is no declaration of interest. One of the authors Saira Mumtaz likes to acknowledge Higher Education Commission of Pakistan for providing funding and support throughout this study.
References
- Kroeger Smith MB, Rouzer CA, Taneyhill LA, et al. Molecular modeling studies of HIV-1 reverse transcriptase nonnucleoside inhibitors: total energy of complexation as a predictor of drug placement and activity. Protein Sci 1995;4:2203–22
- Carolina HA, Kerly FM Pasqualoto, Elizabeth IF, Naton JH. 4D-QSAR: perspectives in drug design. Molecules 2010;15:3281–94
- De Clercq E. The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antiviral Res 1998;38:153–79
- De Clercq E. Novel compounds in preclinical/early clinical development for the treatment of HIV infections. Rev Med Virol 2000;10:255–77
- Kopp EB, Miglietta JJ, Shrutkowski AG, et al. Steady state kinetics and inhibition of HIV-1 reverse transcriptase by a non-nucleoside dipyridodiazepinone, BI-RG-587, using a heteropolymeric template. Nucleic Acids Res 1991;19:3035–9
- Romero DL, Busso M, Tan CK, et al. Nonnucleoside reverse transcriptase inhibitors that potently and specifically block human immunodeficiency virus type 1 replication. Proc Natl Acad Sci USA 1991;88:8806–10
- Pan D, Tseng Y, Hopfinger AJ. Quantitative structure-based design: formalism and application of receptor-dependent RD-4D-QSAR analysis to a set of glucose analogue inhibitors of glycogen phosphorylase. J Chem Inf Comput Sci 2003;43:1591–607
- Rauf A, Liaqat S, Qureshi AM, et al. Synthesis, characterization and urease inhibition studies of 5-substituted-8-methyl-2H-pyrido[1,2-a]pyrimidine-2,4(3H)-diones. J Med Chem Res 2012;21:60–74
- Rauf A, Ahmed F, Qureshi AM, et al. Synthesis and urease inhibition studies of barbituric and thiobarbituric acid derived sulphonamides. J Chin Chem Soc 2011;58:1–10
- Qureshi AM, Qadir M, Rauf A, et al. Antimicrobial efficacy of metal-barbiturate conjugates against pathogenic strains of Escherichia coli and Staphylococcus aureus. Lett Drug Dis Des 2011;8:980–7
- Rauf A, McNab H, Qureshi AM. An efficient one pot three component synthesis of N-(methylene-4-oxo coumarinyl)anilines; their Z,E-isomeric product distribution determination from 1HNMR and their antibacterial studies. Lett Drug Dis Des 2012;9:454–8
- Ellman GL, Courtney KD, Andres V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95
- Tappel AL. The mechanism of the oxidation of unsaturated fatty acids catalyzed by hematin compounds. Arch Biochem Biophys 1953;44:378–95
- Evans AT, Formukong EA, Evans FJ. Actions of cannabis constituents on enzymes of arachidonate metabolism: anti-inflammatory potential. Biochem Pharmacol 1987;36:2035–7
- Baylac S, Racine P. Inhibition of 5-lopoxygenese by essential oils and other natural fragrant extracts. Int J Aromather 2003;13:138–42
- Ghani U, Ng KK, Atta-ur-Rahman, et al. Crystal structure of gamma-chymotrypsin in complex with 7-hydroxycoumarin. J Mol Biol 2001;314:519–25
- Chapdelaine P, Tremblay RR, Dubé JY. P-Nitrophenol-α-d-Glucopyranoside as substrate for measurement of maltase activity in human semen. Clin Chem 1978;24:208–11