
Abstract
New class of FGFR1 kinase inhibitors with naphthostyril heterocycle has been identified. A series of N-phenylnaphthostyril-1-sulfonamides has been synthesized and tested in vitro. It was revealed that the most active compound N-(4-hydroxyphenyl)naphthostyril-1-sulfonamide inhibited FGFR1 with IC50 of 2 µM. In our preliminary studies, N-phenylnaphthostyril-1-sulfonamides demonstrated selectivity of FGFR1 inhibition and antiproliferative activity on cancer cell line. N-phenylnaphthostyril-1-sulfonamides have a good potential for further development as anticancer agents.
Introduction
The fibroblast growth factor receptors (FGFRs) are a family of four similar in structure and function transmembrane proteins that belong to receptor tyrosine kinases (RTKs). The FGFR protein consists of extracellular immunoglobulin-like receptor domain, transmembrane helix domain and intracellular catalytic kinase domain. The natural ligands of FGFR are fibroblast growth factors (FGFs). The binding of this small protein to the extracellular domain induces receptor dimerization, autophosphorylation of tyrosine residues in the intracellular kinase domain and recruitment of signaling proteins. This process activates downstream pathways Ras/MAPK, PI3K/Akt and PLCγ leading to proliferation stimulation and cell survivalCitation1.
FGFR signaling plays important role in embryonic development of mesenchymal tissue, formation of nervous system, lung, breast and other organs. In the adult organism, FGFR1 is involved in reparative processes, inflammation, angiogenesis and differentiation of muscle and fat cells. FGFR gene amplification, activated mutation or aberrant overexpression described in a variety of cancers such as squamous cell lung cancerCitation2, glioblastomaCitation3, breast cancerCitation4, prostate cancerCitation5, etc. Other FGFRs family members also take part in oncogenic disorders such as bladder cancer, myeloma, sarcoma, stomach cancer and melanomaCitation6. FGFR1 also plays an indirect role in the development of solid tumors, participating in the process of oncogenic angiogenesisCitation7.
Thus, inhibition of FGFR1 activity represents an attractive therapeutic strategy for tumors with abnormal activity of this kinase. Most of the known FGFR1 inhibitors target the ATP binding site. Several FGFR1 inhibitors reveal high antitumor potency in tests in vivo and now in clinical trialsCitation8.
This article is devoted to development of naphthostyril FGFR1 inhibitors. Naphthostyril core is a close structural analog of oxindole heterocycle and differs by fused benzene ring. Oxindoles are known class of RTK inhibitors, including FGFR1Citation9. The oxindole multitargeted RTK inhibitor Sunitinib () is used for treatment of several solid tumorsCitation10. We have compared pharmacophore features of oxindole RTK inhibitors with naphtostyriles. Similar to oxindole naphthostyril heterocycle contains 2-pyrrolidone ring. This moiety is responsible for formation of hydrogen bonds with kinase hinge region in oxindole-based inhibitors. Flat aromatic nature of naphthostyril core potentially permits formation of hydrophobic contacts with amino acids surrounding adenine binding subsite of FGFR1 as in case of oxindole inhibitors. Based on these assumptions, we suggested that naphthostyrils potentially have high affinity to FGFR1 ATP-binding site similarly to oxindole FGFR1 inhibitors. Successful oxindole substitution for naphthostyril was reported for CDK2 inhibitors ()Citation11. X-ray complex of naphthostyril inhibitor with CDK2 described in Ref. [11] reveal hydrogen bonds between carbonyl and amide groups of naphthostyril with kinase hinge region, similarly to hydrogen bonds formed by oxindole kinase inhibitors.
Figure 1. Chemical structures of kinase inhibitors from oxindole and naphthostyril classes.
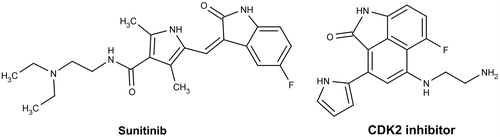
Thus, naphthostyril is a promising scaffold for development of inhibitors for receptor tyrosine protein kinases, including FGFR1. The aim of this article is identification and optimization of naphthostyril FGFR1 inhibitors as potential anticancer agents.
Experimental
All appropriate anilines and 1,8-naphtalic anhydride and other chemicals were commercially available (Acros, Fairlawn, NJ). 1H NMR spectra were recorded with a Varian Mercury VXR400 spectrometer (Varian Inc., Palo Alto, CA) operating at 400 MHz in a DMSO-d6 solution, with tetramethylsilane (TMS) as an internal standard. Chemical shifts are shown as d values (ppm). Signal multiplicities are represented as s (singlet), d (doublet), t (triplet), q (quartet) or m (multiplet).
Benz[cd]indol-2(1H)-one (8)
About 0.3 mol of 1,8-naphthalic anhydride, 0.3 mol of hydroxylamine hydrochloride and 330.64 g of pyridine were charged in the reactor and heated while stiffing. The reaction was conducted under reflux for 1 h, followed by cooling to 80 °C. Then 0.6 mol of p-toluenesulfonyl chloride was added to the reaction system. After addition, the temperature was raised and the reaction was carried out under reflux for 1 h, followed by cooling. The reaction mixture was poured into 1.25 l of water and stirred to precipitate crystals, which were collected by filtration. The crystals were transferred to a beaker and washed successively with 1 L of a sodium hydrogencarbonate aqueous solution and 1 l of water, followed by filtration. The crystals were washed with water and dried to give an intermediate in a yield of 81.1%. The whole amount of the intermediate, 175 ml of ethanol and 200 ml of water were put in a reactor and stirred. Then 325 ml of a 2.7 mol/L aqueous solution of sodium hydroxide was added dropwise to the mixture. Thereafter, the mixture was heated to refluxing temperature, at which the reaction was carried out for 3 h while distilling off ethanol. After completion of the reaction, the reaction mixture was cooled to 75 °C, and 105 ml of concentrated hydrochloric acid was added dropwise. In the meantime, crystals precipitated at 60 °C. After completion of the dropwise addition, the mixture was further cooled. The precipitated crystals were collected by filtration, washed with ion-exchanged water, and dried to give 38.67 g (yield: 94.0%) of the title compound. 1H NMR (DMSO-d6), δ: 7.42 (d, 1H), 7.63 (t, 1H), 7.69 (t, 1H), 7.87 (d, 1H), 7.91 (d, 1H), 8.42 (d, 1H), 10.64 (s, 1H).
6-Nitrobenz[cd]indol-2(1H)-one (9)
Nitric acid (18.76 g, 0.297 mol) was added dropwise to the solution of benz[cd]indol-2(1H)-one 8 (38.7 g, 0.229 mol) in 170 ml of glacial acetic acid. At first, there was a very minor exotherm and then, over the course of 1 h, the reaction temperature raised to 50 °C. The reaction mixture was gradually cooled to room temperature with a cold water bath. A thick dark green paste resulted. This mixture was filtered, washed with 50% aqueous acetic acid, and pulled as dry as possible. The resulting wet filter cake was refluxed in 600 ml methanol and then cooled to 0 °C. The mixture was filtered, washed with cold methanol and dried in vacuo to yield 25.4 g (51%) of 6-nitrobenz[cd]indol-2(1H)-one 9. 1H NMR (DMSO-d6), δ: 7.82 (t, 1H), 7.94 (d, 1H), 8.55 (d, 1H), 8.59 (d, 1H), 8.93 (d, 1H), 10.65 (d, 1H).
6-Aminobenz[cd]indol-2(1H)-one (10)
About 0.15 g of 10% Pd/C was added to the mixture of 25.4 g compound 9 in 200 ml of ethanol. The reaction vessel was degasificated until the solvent begins to bubble, then vessel was carefully backfilled with hydrogen. This operation was repeated twice and then the reaction vessel was allowed to stir overnight with attached source of hydrogen. Then, hydrogen was evacuated, reaction vessel was backfilled with argon and Pd/C was filtered off. Ethanolic solution was evaporated and obtained compound 10 was used without further purification. Yield 20.5 g (94%). 1H NMR (DMSO-d6), δ: 5.63 (s, 2H), 7.19 (d, 1H), 7.30 (d, 1H), 7.46 (t, 1H), 7.76 (d, 1H), 8.42 (d, 1H), 10.64 (s, 1H).
2-oxo-1,2-dihydrobenz[cd]indol-6-sulfonyl chloride (11)
About 20.5 g of compound 10, 82 ml of concentrated hydrochloric acid and 40 ml water were mixed and heated to 40 °C and stirred for 30 min keeping that temperature. Then the reaction mixture was cooled to 0 °C and the solution of 7.83 g sodium nitrite in 15 ml water was slowly added dropwise maintaining temperature between 0 and 5 °C. After the addition, reaction mixture was stirred for 30 min at 0 °C.
In total, 105 ml of 2.5 M solution of SO2 in acetic acid and solution of 3.9 g copper(II) chloride dehydrate in 10 ml water was mixed in another flask and this mixture was cooled to 10 °C and solution of diazonium compound was added in one portion. The temperature was raised and gas was started bubbling. The reaction mixture was stirred for 1 h and then mixture was poured to 250 ml of ice-chilled water. Resulting mixture was stirred to precipitate crystals, which were collected by filtration and washed with water twice. Yield 19.7 g (66%); 1H NMR (DMSO-d6), δ: 7.63 (d, 1H), 7.80 (t, 1H), 8.11 (d, 1H), 8.26 (d, 1H), 8.69 (d, 1H), 10.65 (s, 1H).
General procedure of 2-oxo-1,2-dihydrobenz[cd]indol-6-sulfanilanilides 12–28 synthesis
Two equivalent of pyridine was added to the solution of 1 eq. of 11 and 1,2 eq. corresponding aniline in THF (3 ml/g 11). Resulting mixture was stirred for 20 h at room temperature and then crystals of targeted compound were collected by filtration. Precipitate was washed with 20% aqueous methanol, dried on filter, washed with hot ethanol and dried in the open air. Yields and NMR spectra data are presented in .
Table 1. Chemical characteristics of synthesized compound.
Kinase assays
The FGFR1 kinase assays with recombinant cytoplasmic domain of the FGFR1 tyrosine kinase (Millipore, Billerica, MA, Cat. N. 14-582) were performed in a total volume of 30 µl containing 10 mM MOPS (pH 7.2), 0.1 mM sodium orthovanadate, 0.2 mM EDTA, 0.002% Brij 35, 0.2 mg/ml BSA, 0.02 % β-mercaptoethanole, 250 mM of peptide substrate (KKKSPGEYVNIEFG, GenScript, Piscataway, NJ), various concentrations of inhibitor dissolved in DMSO (final DMSO concentration in probe less than 1%) and 7 ng of enzyme. The reaction was initiated by the addition of ATP (50 µM ATP, 25 mM MgAc containing 0.1 mCi of [γ-32 P]ATP per incubation) and samples incubated at 30 °C for 25 min. The reaction was terminated by the addition of 5% phosphoric acid and the precipitation of material onto phosphocellulose filter mats “Whatman P81”. Filters were washed thrice with 0.75% phosphoric acid and the incorporation of [32P] into the peptide substrate was determined by counting the radioactivity retained on the filters in a PerkinElmer (Waltham, MA) scintillation counter. Kinase residual activity was expressed in percentages with respect to DMSO control. The concentration of compound that inhibited enzymatic activity by 50% (IC50) was determined graphically.
Inhibition of selectivity panel kinases was performed according to enzyme provider protocols (Millipore, Billerica, MA). ATP concentration in reaction mixture was 100 µM.
Molecular modeling
Ligands were prepared for docking with conformation minimization and converted in pdbqt format with Vega ZZ (Milano, Italy)Citation12. Docking was carried out in crystal structure of FGFR1 (pdb bank code 2FGI) by AutoDock 4.2 (La Jolla, CA) with default parameters. Docking results were analyzed with AutoDockTools-1.5.6 (La Jolla, CA)Citation13.
Cellular proliferation assay
KG1 cell viability was examined using a standard MTT assayCitation14. Cells were grown in DMEM containing 10% fetal bovine serum (FBS), 100 mg/mL penicillin and 100 mg/mL streptomycin in humidified air at 37 °C with 5% CO2. Viable cell were seeded into 96 ell tissue plate at 2 × 105 cells/mL pre-incubated for 24 h and treated with the synthesized compounds (compounds in DMSO solution, final DMSO concentration less than 0.5%) at various concentrations. After 72 h treatment, the cells were incubated with 15 µl MTT (3 -(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl-tetrazolium bromide from Sigma, St. Louis, MO) solution (5 mg/mL) for 4 h at 37 °C, 5% CO2. The formazan precipitates were dissolved in 100 µl DMSO and the absorbance at 495 nm of each well was measured by spectrofluorometer MR 700 (Dynatech, Chantilly, VA). The cell viability was expressed as a percentage relative to the untreated control cells.
Results and discussion
Screening of naphthostiriles
Search of compounds with naphthostyril scaffold was performed in our in-house collection. In total, 397 compounds with naphthostyril core were found by substructure search. This set of compounds was clustered using ECFP_4 fingerprints and subjected to diversity sorting, resulting in selection of 18 compounds for FGFR1 inhibition tests. For compounds reducing the kinase activity more than 50% at concentration 33 µM IC50 was determined and their close structural analogs were tested. Totally, it was found six active FGFR1 inhibitors (1–6). Their chemical structures and activity are shown in .
Table 2. Chemical structure and in vitro activities of the naphthostyril derivatives.
The most active compound (1) identified during screening has N-(3-chlorophenyl)sulfonamide substituent at the position 6 of naphthostyril ring and IC50 value of 4.2 µM. Substituted in 6 and 8 positions naphthostyriles 2 and 3 effectively reduce kinase activity at screening concentration 33 µM but have unexpectedly high IC50 values. A possible reason for this effect is the low solubility of these compounds. Compound 4 with 6-methylacetamide substituent has IC50 of 22 μM. Several structural analogs of this compound were inactive for kinase inhibition (data not shown). Compound 5 has lower FGFR1 inhibition activity than structurally close compound 1 implying the significant role of phenyl group in the structure of compound 1 for the interaction with kinase. Compound 6, 6-Methyl-1H-indolo[5,4,3-def]isoquinoline-2,5,7-trione with IC50 of 4.4 µM was slightly less active than compound 1. But analogs of compound 6 with bulkier pyridine or butyl substituents instead of methyl at position 6 were inactive for FGFR1 inhibition (data not shown).
Thus, the most promising inhibitor from screening of naphthostyriles was compound 1 with a single digit micromolar activity, therefore N-phenylnaphthostyril sulfonamide was chosen as scaffold for further optimization.
Molecular modeling
Docking of compound 1 in the FGFR1 catalytic site was carried out to clarify the relationship between chemical structure and inhibition activity of kinase by N-phenylnaphthostyril sulfonamide compounds (). The naphthostyril core of compound 1 occupies the adenine binding portion of the ATP binding site and forms two hydrogen bonds with kinase hinge region. The NH moiety of naphthostyril is involved in hydrogen bond formation with Glu562 residue while the carbonyl group forms a hydrogen bond with the backbone of Ala564. The hydrophobic interactions of the naphthostyril core with Leu484, Leu630, Val492, Ala512 (not labeled) stabilize the position of inhibitor in binding site. Obtained docking binding pose well correlates with X-ray position of CDK2 naphthostyril inhibitors from Ref. [11].
Figure 2. Docking model of compound 1 bound to the ATP-binding site of the FGFR1. Hydrogen bonds marked as dashed lines.
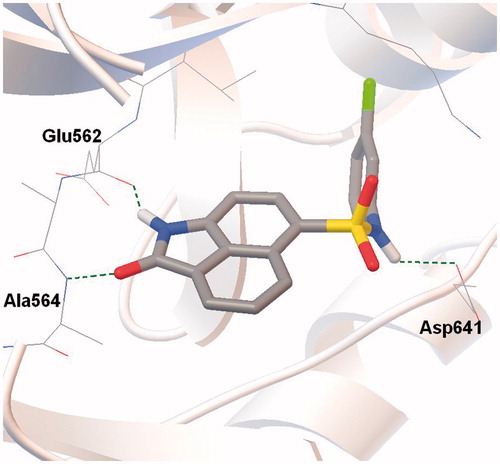
The N-(3-chlorophenyl)sulfonamide substituent at the position 6 of compound 1 extends in depth of ATP binding site. The amide group of sulfonamide linker forms a hydrogen bond with carboxyl group of Asp641 side chain. The chlorophenyl group of N-(3-chlorophenyl)sulfonamide substituent is involved in hydrophobic interactions with amino acid residues Lys514, Met535, Val559, Ile545, Ala640 and Val561 in hydrophobic region I. Meta chlorine on the phenyl group of N-phenylnaphthostyril-1-sulfonamide interact with amino acid residues in the hydrophobic pocket depth, and possibly have a significant impact on binding affinity in our interaction model of N-phenylnaphthostyril-1-sulfonamides with FGFR1. Based on these considerations, phenyl group was chosen for chemical optimization of N-phenylnaphthostyril-1-sulfonamides as FGFR1 inhibitors.
Chemistry
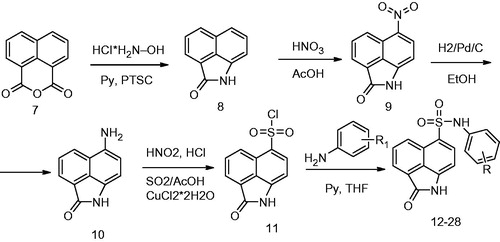
Taking into consideration, the prospects of search of FGFR1 kinase inhibitors among N-phenylnaphthostyril-1-sulfonamide derivatives, a number of compound 1 structural analogs with different substituted phenyl were synthesized according to Scheme 1. First, 2- hydroxybenzo[de]isoquinolin-1,3-dione was obtained from 1,8-naphtalic anhydride through the reaction with hydroxylamine hydrochloride. Then the oxygen of hydroxyl of 2-hydroxybenzo[de]isoquinolin-1,3-dione was acylated one-pot by p-toluenesulfonyl chloride and benz[cd]indol-2(1H)-one (8) was obtained through rearrangementCitation15. Then compound 8 was nitrated by concentrated nitric acid in glacial acetic acid and obtained compound 9 was hydrogenated on 10% Pd/C. Sulfonyl chloride 11 was obtained through diazotation of obtained on previous stage aniline 10 and following reaction resulting diazonium compound with solution of SO2 in acetic acid and copper(I) as a catalyst. Targeted compounds 12–28 were obtained by reaction of sulfonyl chloride 11 with a number of corresponding anilines.
Scheme 1. Synthesis of N-phenylnaphthostyril-1-sulfonamides.
Structural activity relationship (SAR) of N-phenylnaphthostyril-1-sulfonamides
A series of newly synthesized N-phenylnaphthostyril-1-sulfonamides (12–28) was tested for inhibition of FGFR1 using P32 radioassay. The chemical structures and biological activity of these compounds are shown in .
Table 3. Chemical structure and in vitro activities of the N-phenylnaphthostyril-1-sulfonamide derivatives. 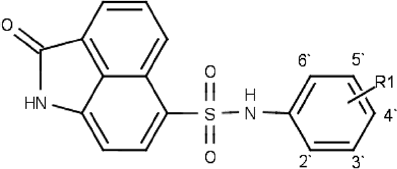
Relationship between substituents in phenyl group of N-phenylnaphthostyril-1-sulfonamid and FGFR1 inhibitory activity was investigated. Compound 19 with unsubstituted phenyl group was a weak inhibitor of FGFR1. Compound 28 with ortho-carboxyl group and compounds with methoxy groups located in different positions of phenyl (23–27) also displayed poor inhibitory activity and their IC50 values were not determined. But compounds with methoxy group in ortho position (25, 27) were slightly more active than N-phenylnaphthostyril-1-sulfonamides with meta and para methoxy groups in kinase residual activity test. Compounds with halogen substituents in phenyl group demonstrated higher activity than the methoxy derivatives and have IC50 values below 10 µM. Compounds with halogen substituent in the para position of the phenyl were generally more active inhibitors than compounds with halogen substituent in the ortho position (16 versus 17, 13 versus 14). The most active halogenated compound 13 has two chlorine atoms in the para and meta positions (IC50 = 2.3 µM). Activity of this compound was significantly higher in comparison with compounds 1 and 12 containing only para and meta chloro phenyl and having twofold and threefold lower activity, respectively. From investigated N-phenylnaphthostyril-1-sulfonamide derivatives the highest inhibitor activity was observed in compounds containing hydroxyl group positioned at the para position (20, 21, 22). Compound 20 with hydroxyl group in para position of phenyl and without any others substituents in phenyl was the most active inhibitor in examined compound series. The introduction of chlorine in the meta position (21) reduced activity compared to compound 20; presence of methyl in the ortho position (22) also slightly decreased activity. These results indicated that hydrogen bonding donor group introduced to the phenyl group might results in an increased activity. According to molecular modeling studies, hydroxyl group in the para position of the phenyl can form a hydrogen bond with the carbonyl group of Ile545 deeply in hydrophobic pocket I. This fact can explain increasing of compound 20 activity.
Selectivity study
Two active compounds 20 and 22 were taken for kinase selectivity analysis with in-house panel of 6 kinases. The results, presented in , show that studied N-phenylnaphthostyril-1-sulfonamides have good selectivity profile in the panel of tested kinases, including receptor tyrosine protein kinases Tie-2 and c-MET. Thereby N-phenylnaphthostyril-1-sulfonamides are potentially selective class of FGFR1 inhibitors.
Table 4. Kinase selectivity of 20 and 22.
Inhibition of proliferation in cancer cell lines
Three the most active compounds (compounds 13, 20 and 22) with IC50 below 3 μM were tested for antiproliferative activity against KG1 myeloma cell line at concentrations 10 and 100 μM. Constitutive activity of FGFR1 was shown in this cancer cell lineCitation16 that used to investigate FGFR1 inhibitors cytotoxicityCitation17. Results of proliferation inhibition tests of N-phenylnaphthostyril-1-sulfonamides are shown in . The antiproliferative activity of compounds 13 and 20 was weak but dose dependent, compound 22 was inactive. The lack of compound 22 activity was possibly established with its low membrane permeability caused by the presence of methyl group in the ortho position of phenyl ring.
Table 5. Antiproliferative activity of compounds 13, 20 and 22 at KG1 cancer cell line.
Conclusions
A new series of N-phenylnaphthostyril-1-sulfonamide compounds with low micromolar FGFR1 inhibition activity was identified. The SAR indicated that introducing of hydroxyl group in para position of phenyl group contributed much to the inhibition activity. In parallel, molecular docking analysis helped to rationalize SAR observations, led to a proposal of the preferred binding conformation. The most potent inhibitor in the series (20) with IC50 of 2 μM showed good selectivity and some antiproliferative effect. N-phenylnaphthostyril-1-sulfonamides are perspective for further development of FGFR1 inhibitors.
Declarations of interest
The authors report no declarations of interest.
References
- Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 2005;16:139–49
- Heist RS, Mino-Kenudson M, Sequist LV, et al. FGFR1 amplification in squamous cell carcinoma of the lung. J Thorac Oncol 2012;7:1775–80
- Yamada SM, Yamaguchi F, Brown R, et al. Suppression of glioblastoma cell growth following antisense oligonucleotide-mediated inhibition of fibroblast growth factor receptor expression. Glia 1999;28:66–76
- Turner N, Pearson A, Sharpe R, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res 2010;70:2085–94
- Yang F, Zhang Y, Ressler SJ, et al. FGFR1 is essential for prostate cancer progression and metastasis. Cancer Res 2013;73:3716–24
- Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J 2011;437:199–213
- Murakami M, Elfenbein A, Simons M. Non-canonical fibroblast growth factor signalling in angiogenesis. Cardiovasc Res 2008;78:223–31
- Greulich H, Pollock PM. Targeting mutant fibroblast growth factor receptors in cancer. Trends Mol Med 2011;17:283–92
- Mohammadi M, McMahon G, Sun L, et al. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 1997;276:955–60
- Gan HK, Seruga B, Knox JJ. Sunitinib in solid tumors. Expert Opin Invest Drugs 2009;18:821–34
- Liu JJ, Dermatakis A, Lukacs C, et al. 3,5,6-Trisubstituted naphthostyrils as CDK2 inhibitors. Bioorg Med Chem Lett 2003;13:2465–8
- Pedretti A, Villa L, Vistoli G. VEGA – an open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J Comput Aided Mol Des 2004;18:167–73
- Morris GM, Huey R, Lindstrom W, et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009;16:2785–91
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;16:55–63
- ADEKA Corporation; US Patent 2011/152538; 2011; (A1) English
- Gu TL, Goss VL, Reeves C, et al. Phosphotyrosine profiling identifies the KG-1 cell line as a model for the study of FGFR1 fusions in acute myeloid leukemia. Blood 2006;108:4202–4
- Ren M, Qin H, Ren R, Cowell JK. Ponatinib suppresses the development of myeloid and lymphoid malignancies associated with FGFR1 abnormalities. Leukemia 2013;27:32–40