
Abstract
The role of progesterone in women’s cancers as well as the knowledge of the progesterone receptor (PR) structure has prompted the design of different therapies. The aim of this review is to describe the basic structure of PR agonists and antagonists as well as the recent treatments for illness associated with the progesterone receptor. The rational design for potent and effective drugs for the treatment of female cancer must consider the structural changes of the androgen and progestogen skeleton which are an indicator of their activity as progestins or antiprogestins. The presence of a hydroxyl group at C-17 in the progesterone skeleton brings about a loss of progestational activity whereas acetylation induces a progestational effect. The incorporation of an ethynyl functional group to the testosterone framework results in a loss of androgenic activity with a concomitant enhancement of the progestational effect. On the other hand, an ester function at C-3 of dehydroepiandrosterone skeleton induces partial antagonism to the PR.
Introduction
Long-acting orally effective steroids that competitively interact with the progesterone receptor (PR) are essential for the optimal development of new drugs suitable for the treatment of progesterone-dependent cancers and gynecological disorders as well as contraceptive drugs.
Progesterone (P) binds to both isoforms of the PRs, PR-A and PR-B, which belong to the transcription factors family; this complex induces several physiological effects by the well-known steroids genomic pathway (). These two isoforms of the PR are produced by a single gene and have two transcriptional sites. The PR-B isoform has 164 amino acids at the end of the chain which are not present in PR-ACitation1–3. This sequence in PR-B is a transactivation site that connects to co-activators, whereas PR-A binds only to co-repressors with high affinityCitation3. Both isoforms are expressed in target tissues since the PR-A isoform is capable of transrepressing PR-BCitation4–7. In addition, other mechanisms that could modify gene transcriptions mediated by P play a role in the signaling pathway such as the activation of second messengers like MAPkinases ensuing in CREB phosphorylation which regulates the activity of co-activators such as SRC2 ()Citation8. Moreover, this post-transduction modification affects multiple PR functions as has been observed in breast cancer cells phosphorylated on more than 10 sites. Phosphorylation of PR in specific sites is produced in response to P or kinase activation, stimulated by growth factors such as EGF thus inducing a cross-communicationCitation9–12. EGF increases P induction of selected PR gene targets, and enhances progestin-dependent growth of breast cancer cells. This fact confirms the existence of a cross-talk between EGF and P which produces a hyperactivated PRCitation12.
Figure 1. Genomic (slow) and non-genomic (rapid) mechanisms of action of PR complexes (PRA; PRB). Non-genomic signaling requires a membrane progesterone receptor (mPR) that stimulates a cascade which induces CBP phosphorylation and stimulation of co-activators involved in the genomic mechanism.
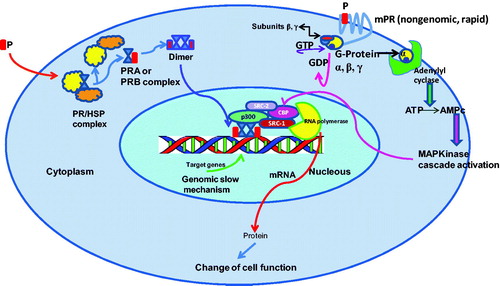
In addition to estrogens, the role of P in the proliferation of mammary gland epithelium had been recognized; P regulates this proliferation controlling the expression of target gene effectors, such as the paracrine factor (RANKL) that facilitates the proliferation of neighboring PR-negative cells. This mechanism involves the recruitment of the co-activator Stat5, which supports the PR-DNA binding to specific sites of the genomeCitation13.
Furthermore, a family of membrane progesterone receptors (mPRs) has been found that mediates rapid actions of P (). This non-genomic mechanism has been described in some cellular functions in females such as: oocyte meiotic maturation, GnRH secretion, reproductive behavior, apoptosis of granulose, breast cancer and neuronal cellsCitation14–17. These mPRs are G-protein-coupled and function following the cAMP cascade (). On the other hand, it has been found that a specific mPRα is associated with PGRMC1-protein. When this complex binds specifically to P their transport to the cell surface is completed; and an apoptotic process in the cell takes placeCitation18–21.
With a lead structure such as P (1, ), a natural progestagen, several progesterone derivatives have been synthesized and used for the treatment of different afflictions as well as contraceptive drugs. Two groups of these progestins have been classified on the basis of their structure: one related to progesterone (P) and the second to testosterone 2 (T).
Figure 2. Progestins structurally related to progesterone. 1 – Progesterone, 2 – testosterone, 3 – 17-α-hydroxyprogesterone, 4 – 17α-acetoxyprogesterone, 5 – medroxyprogesterone acetate, 6 – chlormadinone acetate and 7 – cyproterone acetate.
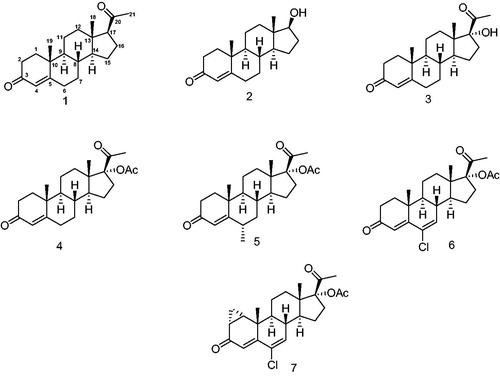
Different structural groups of progestins
The loss of biological activity observed when a hydroxyl group is incorporated into C-17Citation22 in progesterone 1, to form 3 (17-α-hydroxyprogesterone) has encouraged the design of new P-derivatives as synthetic progesterone antagonists. Surprisingly, when compound 3 was acetylated, a new molecule 17α-acetoxyprogesterone 4 was obtained showing an enhanced progestational activity. Furthermore, when a methyl group was added to C-6, the altered structure, medroxyprogesterone acetate 5 (MPA) exhibited a relatively high progestational activityCitation23. The addition of a double bond and a chlorine atom to C-6 in the hydroxyprogesterone scaffold afforded chlormadinone acetate 6 having a higher progestational activity. Chlormadinone acetate was further converted to the highly active progestagen and anticancer drug cyproterone acetate 7.
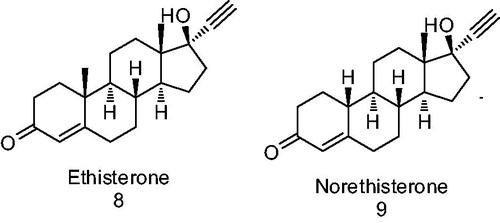
In the search for a lead structure with progestational activity, researchers have also used the testosterone scaffold as a starting point, since both T and P show common features of structure and conformation with the substituent at C-17 being the only difference (hydroxyl or COCH3 group). As a result of these modifications effected on the testosterone molecule, variations in the biological activity have been observed. The addition of an ethynyl group to C-17 on the T skeleton causes a loss of androgenicity as has previously been described for ethisterone 8 ()Citation22. The progestational and oral activities of 8 increase when the methyl group at C-10 is removed (compound 9 norethisterone or norethindrone, ); as a consequence of this a complete elimination of androgenicity is attainedCitation23.
Figure 3. Derivatives of testosterone.
On the other hand, different studies have demonstrated that substitution at C-7 and C-11 on the progesterone 1 skeleton exerted a small effect on the PR recognition and anchorage sites. However, several compounds substituted at C-11 have shown biological activity such as RU486 (mifepristone 12, )Citation23,Citation24. This compound is an 11-β-dimethylaminophenyl derivative of norethisterone having a double bond at C-9 (). It has been determined that steroidal derivatives having a propynyl group at C-17 form a stable complex with glucocorticoid (GR) and progesterone (PR) receptors. RU486 apparently did not show agonistic activity; however, it exhibited antiprogestational and anticortisol properties in several animal modelsCitation23. This steroid has high affinity for the nuclear PR receptorCitation25 nevertheless; some agonistic progestational properties of 12 were later identified, as well as gonadotropin inhibitory activity in post-menopausal womenCitation26.
Figure 4. Agonist, partial agonist and antagonists of PR.
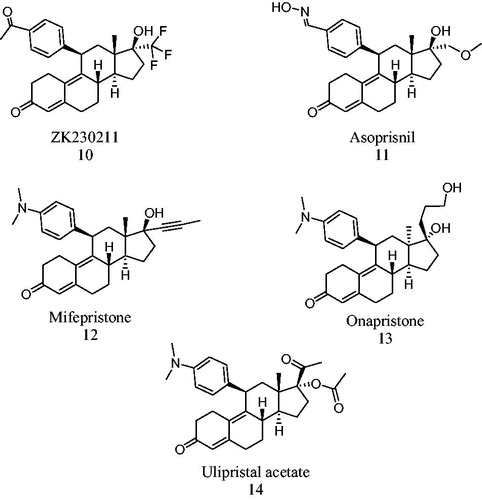
The biological profiles of several antiprogestins have also been studied assessing the agonistic and antagonist effects, selectivity and potency of their metabolites. The metabolism of 12 was characterized by rapid demethylation and hydroxylation. Similar initial metabolic pathways have been proposed for ulipristal acetate 14 (CDB-2914, ) and their putative metabolites have been synthesizedCitation27. These steroids bind to PR and GR but showed lower antiprogestational and antiglucocortocoid activity than 12; however, they did not exhibit any agonistic activityCitation28. In addition, 14 has been studied as an alternative treatment for uterine fibroids (leiomyomata) and the results of a successfully completed phase III clinical trial with 14 have been publishedCitation29. The administration of 5 or 10 mg/daily of this compound induced a rapid decrease of bleeding; it also reduced the size of the fibroids by 50% and restored the quality of women's lifeCitation30.
Crystallographic studies of PR leading to the design of new progestins
On the ground of several X-ray crystallographic studies, advances have been made in the design of metabolically active drugs based on the recently elucidated PR structureCitation31. These results explain also the molecular mechanisms supporting the simultaneous agonistic and antagonistic activities observed with some steroidal molecules that bind to the PR. In this respect, it has been reported that clashes between these molecules with Met909 in helix 12 of the PR reduced agonistic activityCitation31. The presence of a bulky moiety in non-steroidal compounds or at C-11 of the steroidal skeleton prevents agonistic activity, since this group clashes with helix 12, thus inhibiting the linking with co-activatorsCitation31,Citation32. It is well known that agonists bind directly to co-activators, whereas antagonists bind to co-repressors. Therefore, the relationship between co-activators or co-repressors present in target cells will define the effect of different ligand exhibiting mixed activitiesCitation31.
The facility of the complex of antagonist-PR to bind to co-activators or co-repressors is associated with target gene expression. An example of this process is the over expression of co-repressors that suppress the partial agonistic effect of 12; whereas the over expression of co-activators improves the partial agonistic activity of this steroidCitation31–34. Nevertheless, the binding of 13 to the PR () shows different properties, since the antagonistic activity of this steroid is not affected by the expression of co-activators or co-repressors molecules. In this case, the PR ligand complex presents only one different conformation as compared to that of 12. This fact could explain why 10 was more effective than 12 or 13 by considering the presence of co-activatorsCitation35,Citation36.
On the bases of their effect as pure antagonist, different types of PR antagonists were identified and described; this prevents the binding of PR complex to DNA (13, )Citation32–34. Partial antagonists are those which facilitate the binding of the ligand-join PR to the progesterone response element (PRE) present in DNA helix; however, they show also an agonistic activity in the target tissues (12, 11, )Citation35.
The antiproliferative activity of steroid 10 () is explained on the ground that this steroid can attach to the ligand binding domain of PR, a helix 12 conformation. Thus, helix 12 forms a selective surface that binds to co-repressors and; as a result of this, an antiproliferative activity is observedCitation36.
Current use of progestins for cancer treatment, contraception and hormone replacement therapy
Recently a phase II study with PR-antagonist 10 was carried out on breast cancer tumor cells that express PR. This study investigated the efficacy, safety and tolerability of 10 at two different doses (25 and 100 mg) before the initiation of phase III trialsCitation37.
The synthetic steroid onapristone (13, ), functions as a PR antagonist for a first line endocrine therapy in patients with breast cancer. This antagonist was prepared by the Schering Corporation and identified as an antiprogestational and antitumoral drug; however, the clinical trials with this compound were suspended due to its liver toxicityCitation34.
Mifepristone 12, which was synthesized by Roussel–Uclaf Company, is a second-generation progestin with a specific PR antagonistic activity. This steroid has been used as an abortive drug; however, its use can give rise to several collateral effects. This compound induced a metastatic breast cancer in postmenopausal womenCitation38; as a result of its action on the pituitary–adrenal axes. The increase of plasmatic’s androstendione as well as the presence of aromatase enzyme in different tissues effected the conversion of this androgen into estradiol. The presence of this steroid in the plasma could explain this contradictory response in post-menopausal womenCitation37. Nevertheless, the studies carried out with cancer cell lines and animals indicated that 12 did not produce genotoxic effectsCitation39.
Asoprisnil 11 () is a steroidal molecule that was synthesized by Schering and TAP Pharmaceutical Products Laboratories and was used for the treatment of myomata tumor and endometriosisCitation40. Compound 11 is an antagonist of the PR and its effect in endometrium has been described. Asoprisnil decreases glandular and stromal proliferation in endometrium and in leiomyomataCitation41. However, in 2005, 11 was discontinued due to endometrial problems observed in patients in Phase III trials.
It is well known that estrogens stimulate the growth of endometrial cancer cells; this fact has prompted the development of several new ER antagonists (progestins derivatives)Citation42 for the treatment of endometrial cancer. Progestagens (gestagens) are synthetic molecules having a P-action; these molecules inhibit the growth, differentiation of endometrial gland and epithelial cells, thus reducing endometriosis and endometrial cancer. In addition to this, the progestagens decrease the expression of the ER; it is well known that gestagens activate the tumor suppressor gene p21; and as a consequence of this, the growth of endometrial cancer cells is inhibitedCitation43. The most common progestin used for this target includes: norethisterone 15, levonorgestrel 16, desorgestrel 17, dianogest 18, tibolone 19 and dydrogesterone 20 ().
Figure 5. Structure of different progestagens.
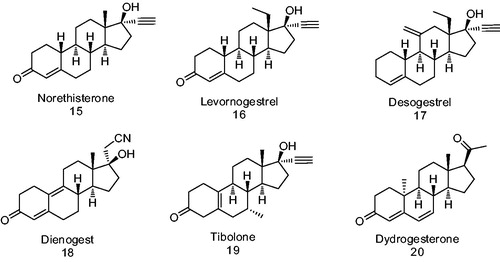
Levonorgestrel 16 is the active form of norgestrel (d-(−)-norgestrel) and is the most potent orally active form as compared to the progestins of the second generation. In addition to its use as a therapeutical tumor suppressor, it can also be used as a contraceptive, emergency contraceptive and in hormonal replacement therapyCitation44.
Several more potent progestin inhibitors for endometrial cancer cell growth such as: medroxyprogesterone acetate 5 (MPA; ) and dienogest 18 () have been synthesizedCitation45,Citation46. Different studies have been carried out using endometrial cancer cell lines such as HEC-88nu and Ishikawa. Ishikawa cell line expresses ER and PR, while HEC-88nu corresponds only to ER. When the Ishikawa cell line was treated with 5 and 18, it showed that a reticence of its growthCitation47; on the other hand, HEC-88nu did not respond to the treatment with compound 5. Nevertheless, compound 18 inhibited proliferation of this cell line; and this fact supports its action via non-PR mediated pathways.
Therefore, compound 18 could be the choice for the treatment of independent PR cancersCitation46. Because the effect of this steroid on endometrial cancer was to inhibit tumor’s vascularization, in this case steroid 18 could be used also as a monotheraphy for the treatment of endometrial cancer and endometriosisCitation46. On the other hand, it has been demonstrated that 18 shows antiandrogenic properties; this fact could be considered as an advantage for the amelioration of some side effects, such as a decrease of bone mineral density. Dienogest (18) appears to have an improved bioavailability in oral administration in women due to its superior elimination as compared to other progestinsCitation48.
In addition to its use as antitumor drugs, steroidal compounds 5 and 18, have been used also as a contraceptive, in hormone replacement therapy and for the treatment of endometriosis as well as several afflictions. However, these progestins tend to produce side effects such as: acne, changes in menstrual flow, drowsiness and birth defects. Other common collateral effects include breast tenderness, increased facial hair, decreased scalp hair, difficulty falling or remaining asleep, stomach pain and weight lossCitation49.
Tibolone 19 () is a versatile progestin that binds to different steroidal receptors and is used as a hormone therapy in post-menopausal womenCitation50. When this synthetic progestin is metabolized, two principal tibolone’s metabolites are formed; these hydroxy metabolites bind and activate ER with a preference for ERα. Delta 4-tibolone and 19 () are both agonists for the androgen receptor (AR) and PR. However, the reduced forms of tibolone, 3α- and 3β-OH are antagonists for AR and PR. Furthermore, 19 and its primary metabolites are GR and MR antagonists with a higher affinity for MR than for GR. Therefore, 19 has a complex effects due to its actions on different receptors and its capacity to convert into different metabolitesCitation42.
Dydrogesterone 20 () is a progestin produced by Abbott Laboratories and distributed in over 100 countries worldwide. This compound is used for the treatment of a variety of gynecological conditions related to progesterone deficiency such as infertility, menstrual disorders, abortion, endometriosis and premenstrual syndrome. This steroid is orally active at low doses and binds to PR with an RBA = 15.9%. Dydrogesterone showed similar agonistic activity than progesterone for PR type B; however, its agonistic activity was weak in the case of PR type A. These data indicated that this steroid showed selectivity for PR type B, and this offers a therapeutical advantage, since each type induces different physiological function in target tissuesCitation4. However, dydrogesterone did not bind to ER (types α and β), AR or glucocorticoid receptor (GR); and as a result of this, it could prevent collateral effectsCitation51. The pharmaceutical effect of dydrogesterone has been described as a neuroendocrine’s modulator of ovarian and endometrial functionsCitation52. On the other hand, dydrogesterone inhibited the activity of the enzymes 5α-reductase type 2 (5α-R2) as well as 17-β-hydroxysteroid dehydrogenase (17-β-HSD) types 3 and 5Citation51. These data are important since the growth of estrogen/progesterone-insensitive breast cancer tumors is due to the presence of a high concentration of 5α-progesterone (5αP) metaboliteCitation53. As a result of this, dydrogesterone could be an alternative for the treatment of this type of breast cancer.
Novel structure of progestins
Since changes at C-17 in the skeleton of androgen and progesterone molecules appear to be of utmost importance for the activity of these derivatives to function as progestins or antiprogestins, our group synthesized different dehydroepiandrosterone derivatives such as: 17-β-methyl, 16-β-phenyl-D-homoandrost-4,6-diene compounds 21, 22 and 23 having a phenylacetic acid ester at C-17-β position (). These steroids are potent antagonists of the PR in endometrial tissue from mice treated with LHRHCitation54.
Figure 6. 17-β-Methyl, 16-β-phenyl-D-homoandrost-4,6-diene derivatives having a phenylacetic acid ester at C-17β position and dehydroepiandrosterone derivatives as an antagonists and partial antagonists. 5α-Reductase enzyme IC50 (5α-R IC50) is the concentration of the synthetic steroid to produce 50% inhibition of 5α-R activity.
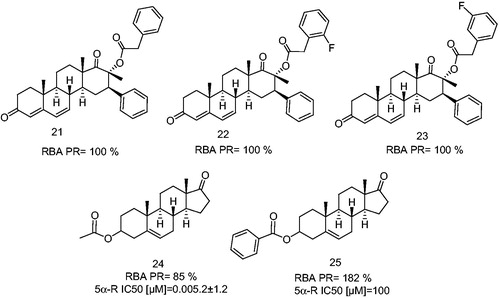
However, the presence of an ester moiety at C-3 on the androstane skeleton appears to be also useful for the enhancement of PR-binding. This fact was supported by the biological activity exhibited by 17-oxaandrost-5-ene-3β-yl acetate (24) and 17-oxaandrost-5-ene-3β-yl-p-bromobenzoate (25). These dehydroepiandrosterone derivatives (24, 25, ) were characterized also as antagonists and partial antagonists, since compound 24 binds to the PR and inhibited the ovulation, endometrial secretion and mammary glands proliferation in cycling mice stimulated with LHRH. In addition to this, 24 inhibited also the 5α-R enzyme and blocked the conversion of progesterone to 5α-dihydroprogesterone. On the other hand, 25 did not inhibit the enzyme 5α-reductase; however, it binds to the PR with high affinity thus, inducing a synergistic activity with P in these tissuesCitation54. Compound 24 inhibited the ovulation, mammary glands proliferation and endometrial secretion, since it blocked the conversion of progesterone to 5α-dihydroprogesterone. As a result of blockage, the ductal and alveolar epithelial cell proliferation in the mammary as well as the secretion in endometrial glands was inhibited. It was also observed that the function of both glands depends on 5α-dihydroprogesteroneCitation54,Citation55. When the acetyl group at C-3 was replaced with a phenyl group, compound 25, the affinity for the progesterone receptor was markedly increasedCitation54,Citation55.
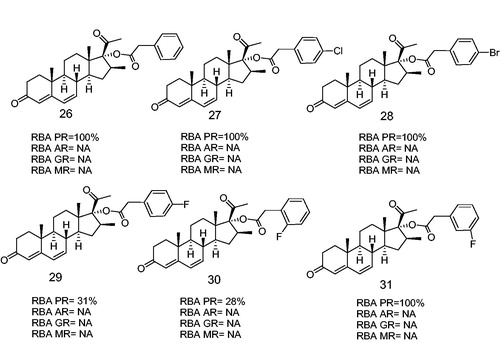
Recently, our group synthesized several new halogen substituted aromatic esters at C-17 of the progesterone skeleton having a potent PR antagonistic activity (). These compounds (26, 27, 28 and 31) bind selectively to the PR with RBAs of 100%, and inhibited the ovulation in mice. These steroidal derivatives showed also an anti-progestational activity in vivo, thus indicating that the presence of fluorine atom in meta position in the phenylacetoxy ester at C-17, improved the binding activity as compared to that for the ortho and para positionsCitation56.
Figure 7. Halogen-substituted phenylacetic acid derivatives antagonists of PR. Androgen receptor (AR), glucocorticoid receptor (GR), mineralocorticoid receptor (MR). RBA, Relative binding affinity; PR, Progesterone receptor; AR, Androgen receptor; GR, glucocoticoid receptor; MR, mineralocorticoid receptor; NA, Non-active.
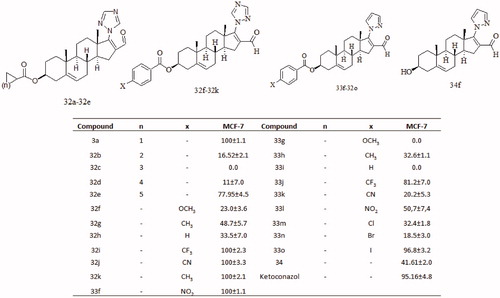
Following with this study, we synthesized several new steroidal derivatives having an azole functional group at C-17 (32a–32k and 33f–33o and 34; ). The triazole derivatives (32a–32k) exhibited considerably higher cytotoxic activity in mammary cancer cell line MCF-7 as compared to that of the pyrazole derivatives (33f–33o). The difference in this activity could be attributed to the ability of the triazole (three nitrogen atoms) to form stronger hydrogen bonds with the active site of some proteins (receptor or enzyme) of the cellCitation57. Compounds 32f–32k having an aromatic ester at C-3 showed that an enhanced cytotoxic activity as compared to their aliphatic counterpart 32a–32e. Apparently, the electronegative phenyl ring increased the polarity of the molecule, thus enhancing the dipole–dipole association of the steroidal molecule with some active protein (receptor or enzyme) of the cell ().
Figure 8. Inhibitory activity of different dehydroepiandrosterone derivates on the growth of mammary cancer cell line (MCF-7) treated with 50 μM of novel compounds.
On the ground of PR-structure knowledge, different types of progesterone antagonists have been describedCitation58. These molecules have been identified by their effect as pure antagonists, which avoid the binding to the PRE (e.g. 13, )Citation10,Citation32,Citation33. Partial antagonists are those that enable binding to PRE; however, they show also an agonistic activity in the target tissues, thus inducing protein kinase A-stimulation (e.g. 12, 11, and )Citation31,Citation33,Citation38,Citation58–60. Antagonists that bind to PR and allow the join to PRE show only antagonistic activities in target tissues, e.g. 10 ()Citation60.
Conclusion
The fear of losing female mammary glands, uterus and ovaries by surgery creates a distressing effect on the mentality of women. At the present time, a free side effect therapy does not exist for the treatment of women’s cancers; therefore, the research is focused on the development and design of new drugs.
Presently, different therapies with potent progestins are being used for the treatment of women’s cancers that depend on ER+, PR− or ER+, PR+. The endometrial cancer ER+, PR− that does not respond to MPA, is currently being treated in experimental models with dienogest. Furthermore, the fact that the growth of Ishikawa cell line, ER+, PR+, was inhibited by MPA and dienogest; suggests a new possibility for the treatment of this cancer with dienogest, which is more potent than MPA for the cell proliferation inhibition.
The understanding of the PR structure has prompted the design of several molecules for the improvement of potential therapeutic methods. On the ground of the results of several studies, we can infer that the progesterone derivatives having an aromatic ester at C-17 showed an enhanced relative binding activity for the progesterone receptor (). In addition, the 5-d-homoandrosterone derivatives () reported in this article exhibited very high values for the relative binding affinity for the PR in the range of 85–182%. These steroids exhibited also an important antiprogestagen activity in mice model.
In addition, the rational design must consider the changes at C-17 of the androgen and progesterone skeletons; this appears to be a suitable scaffold for the activity of these derivatives to function as progestins or antiprogestins. The addition of a hydroxyl group at C-17 of the progesterone skeleton brings about a loss of the progestational activity whereas an acetoxy group at C-17 enhances this effect. Addition of an ethynyl group at C-17 on the testosterone skeleton reduces the androgenic effect, however, it enhances the progestational activity. It is important to emphasize that an ester function at C-3 or C-17 of the steroid skeleton (progesterone and androsterone) could induce a partial antagonism for the progesterone receptor.
Furthermore, triazole steroidal derivatives (three nitrogen atoms in the ring) showed high cytotoxic potency in the mammary cancer cell line MCF-7 as compared to the pyrazole group having two nitrogen atoms. The addition of aromatic ester at C-3 enhanced cytotoxic activity as compared to their aliphatic counterpart. Apparently the presence of an azole ring (triazole or pyrazole) could be a structural requirement for the enhancement of the cytotoxic activity on MCF-7 cancer cell line.
The fact that estrogens stimulate growth of endometrial cancer cells has prompted also the development of several new progestagens as antagonists for the ER in order to ameliorate the endometrial cancer progression. These molecules inhibit the growth, differentiation as well as the expression of the ER of endometrial glands and epithelial cells, thus reducing endometriosis and endometrial cancer. Potent progestins such as medroxyprogesterone acetate (MPA 5, ) have been characterized as inhibitors of cancer cells proliferation (ER+, PR+).
Moreover, compounds 11–13 induced also agonistic activity thus affecting a kinase A-stimulation (). The stimulation of this enzyme and other kinases, could enhance the phosphorylation of PR and its co-activators in the presence of these antagonists (11–13). In fact, progestins bind to the PR, in cells’ cytoplasm or membrane, thus inducing a cell signaling cascades that promotes phosphorylation of different proteins that could be involved in transcriptionCitation7,Citation10,Citation12.
Mifepristone (12) is an antiprogestagen and when administrated in combination with prostaglandin is successfully used to induce abortion in early pregnancy in some countries. Compound 12 is also useful as an anti-implantation agent in emergency contraception. Other potential uses could include a once-a-week contraceptive, an inhibitor of ovulation and as a daily minipillCitation44.
This review summarizes the recent advances in the design of drugs and potential therapies for the treatment of progesterone-dependent diseases.
Declaration of interest
This manuscript represents an original, unpublished material not under editorial consideration elsewhere following ethical guidelines in the conduct of the research. This paper was written also, according to the authors guide and do not have any conflict of interest. This study was supported by a Grant from Consejo Nacional de Ciencia y Tecnología (project number 54853) and DGAPA (project number IN211312).
References
- Graham JD, Clarke C. Progesterone receptors – animal models and cell signaling in breast cancer: expression and transcriptional activity of progesterone receptor A and progesterone receptor B in mammalian cells. Breast Cancer Res 2002;4:187–90
- Wen DX, Xu YF, Mais DE, et al. The A and B isoforms of the human progesterone receptor operate through distinct signaling pathways within target cells. Mol Cell Biol 1994;14:8356–64
- Sartorius CA, Melville MY, Hovland AR, et al. A third transactivation function (AF3) of human progesterone receptors located in the unique N-terminal segment of the B-isoform. Mol Endocrinol 1994;8:1347–60
- Giangrande PH, Kimbrel EA, Edwards DP, McDonnell DP. The opposing transcriptional activities of the two isoforms of the human progesterone receptor are due to differential cofactor binding. Mol Cell Biol 2000;20:3102–15
- Conneely OM. Perspective: female steroid hormone action. Endocrinology 2001;142:2194–9
- Liu Z, Auboeuf D, Wong J, et al. Coactivator/corepressor ratios modulate PR-mediated transcription by the selective receptor modulator RU486. Proc Natl Acad Sci USA 2002;99:7940–4
- Karteris E, Zervou S, Pang Y, et al. Progesterone signaling in human myometrium through two novel membrane G protein-coupled receptors: potential role in functional progesterone withdrawal at term. Mol Endocrinol 2006;20:1519–34
- Hopp TA, Weiss HL, Hilsenbeck SG, et al. Breast cancer patients with progesterone receptor PR-A-rich tumors have poorer disease-free survival rates. Clin Cancer Res 2004;10:2751–60
- Shyamala G, Yang X, Silberstein G, et al. Transgenic mice carrying an imbalance in the native ratio of A to B forms of progesterone receptor exhibit developmental abnormalities in mammary glands. Proc Natl Acad Sci USA 1998;95:696–701
- Daniel AR, Knutson TP, Lange CA. Signaling inputs to progesterone receptor gene regulation and promoter selectivity. Mol Cell Endocrinol 2009;308:47–52
- Hagan CR, Regan TM, Dressing GE, Lange CA. ck2-dependent phosphorylation of progesterone receptors (PR) on ser81 regulates PR-B isoform-specific target gene expression in breast cancer cells. Mol Cell Biol 2011;31:2439–52
- Moore NL, Narayanan R, Weigel NL. Cyclin dependent kinase 2 and the regulation of human progesterone receptor activity. Steroids 2007;72:202–9
- Obr AE, Grimm SL, Bishop KA, et al. Progesterone receptor and Stat5 signaling cross talk through RANKL in mammary epithelial cells. Mol Endocrinol 2013;27:1808–24
- Zhu Y, Rice CD, Pang Y, et al. Cloning, expression, and characterization of a membrane progestin receptor and evidence it is an intermediary in meiotic maturation of fish oocytes. Proc Natl Acad Sci USA 2003;100:2231–6
- Sleiter N, Pang Y, Park C, et al. Progesterone receptor A (PRA) and PRB-independent effects of progesterone on gonadotropin-releasing hormone release. Endocrinology 2009;150:3833–44
- Frye CA, Sumida K, Lydon JP, et al. Mid-aged and aged wild-type and progestin receptor knockout (PRKO) mice demonstrate rapid progesterone and 3α,5α-THP-facilitated lordosis. Psychopharmacology 2006;185:423–32
- Pang Y, Dong J, Thomas P. Characterization, neurosteroid binding and brain distribution of human membrane progesterone receptors δ and ε (mPRδ and mPRε) and mPRδ involvement in neurosteroid inhibition of apoptosis. Endocrinology 2012;154:283–95
- Peluso JJ, Romak J, Liu X. Progesterone receptor membrane component-1 (PGRMC1) is the mediator of progesterone’s antiapoptotic action in spontaneously immortalized granulosa cells as revealed by PGRMC1 small interfering ribonucleic acid treatment and functional analysis of PGRMC1 mutations. Endocrinology 2008;149:534–43
- Dressing G, Alyea R, Pang Y, Thomas P. Membrane progesterone receptors (mPRs) mediate progestin induced antimorbidity in breast cancer cells and are expressed in human breast tumors. Horm Cancer 2012;3:101–12
- Dressing GE, Pang Y, Dong J, Thomas P. Progestin signaling through mPRα in Atlantic Croaker Granulosa/Theca cell cocultures and its involvement in progestin inhibition of apoptosis. Endocrinology 2010;151:5916–26
- Thomas P, Pang Y, Dong J. Enhancement of cell surface expression and receptor functions of membrane progestin receptor alpha (mPRα) by progesterone receptor membrane component 1 (PGRMC1): evidence for a role of PGRMC1 as an adaptor protein for steroid receptors. Endocrinology 2014:155:1107–19
- Stanczyk FZ. All progestins are not created equal. Steroids 2003;68:879–90
- Gagne D, Pons M, Philibert D. RU 38486: a potent antiglucocorticoid in vitro and in vivo. J Steroid Biochem 1985;23:247–51
- Lebeau M-C, Baulieu EE. Steroid antagonists and receptor-associated proteins. Hum Reprod 1994;9:437–44
- Cadepond PF, Ulmann MDPA, Baulieu MDPEE. RU486 (MIFEPRISTONE): mechanisms of action and clinical uses. Annu Rev Med 1997;48:129–56
- Gravanis A, Schaison G, George M, et al. Endometrial and pituitary responses to the steroidal antiprogestin RU 486in postmenopausal women. J Clin Endocrinol Metab 1985;60:156–63
- Attardi BJ, Burgenson J, Hild SA, Reel JR. In vitro antiprogestational/antiglucocorticoid activity and progestin and glucocorticoid receptor binding of the putative metabolites and synthetic derivatives of CDB-2914, CDB-4124, and mifepristone. J Steroid Biochem Mol Biol 2004;88:277–88
- Attardi BJ, Burgenson J, Hild SA, et al. CDB-4124 and its putative monodemethylated metabolite, CDB-4453, are potent antiprogestins with reduced antiglucocorticoid activity: in vitro comparison to mifepristone and CDB-2914. Mol Cell Endocrinol 2002;188:111–23
- Melis GB, Piras B, Marotto MF, et al. Pharmacokinetic evaluation of ulipristal acetate for uterine leiomyoma treatment. Expert Opin Drug Metab Toxicol 2012;8:901–8
- Donnez J, Tatarchuk TF, Bouchard P, et al. Ulipristal acetate versus placebo for fibroid treatment before surgery. N Engl J Med 2012;366:409–20
- Lusher SJ, Raaijmakers HCA, Vu-Pham D, et al. X-ray structures of progesterone receptor ligand binding domain in its agonist state reveal differing mechanisms for mixed profiles of 11β-substituted steroids. J Biol Chem 2012;287:20333–43
- Han SJ, Tsai SY, Tsai M-J, O’Malley BW. Distinct temporal and spatial activities of RU486 on progesterone receptor function in reproductive organs of ovariectomized mice. Endocrinology 2007;148:2471–86
- Meyer ME, Pornon A, Ji J, et al. Agonistic and antagonistic activities of RU486 on the functions of the human progesterone receptor. EMBO J 1990;9:3923–32
- Robertson JFR, Willsher PC, Winterbottom L, et al. Onapristone, a progesterone receptor antagonist, as first-line therapy in primary breast cancer. Eur J Cancer 1999;35:214–18
- Chen W, Ohara N, Wang J, et al. A novel selective progesterone receptor modulator asoprisnil (J867) inhibits proliferation and induces apoptosis in cultured human uterine leiomyoma cells in the absence of comparable effects on myometrial cells. J Clin Endocrinol Metab 2006;91:1296–304
- Afhüppe W, Beekman JM, Otto C, et al. In vitro characterization of ZK 230211 – A type III progesterone receptor antagonist with enhanced antiproliferative properties. J Steroid Biochem Mol Biol 2010;119:45–55
- Jonat W, Bachelot T, Ruhstaller T, et al. Randomized phase II study of lonaprisan as second-line therapy for progesterone receptor-positive breast cancer. Ann Oncol 2013;24:2543–8
- Klijn JGM, Setyono-Han B, Foekens JA. Progesterone antagonists and progesterone receptor modulators in the treatment of breast cancer. Steroids 2000;65:825–30
- Yuan H, Upadhyay G, Lu J, et al. The chemopreventive effect of mifepristone on mammary tumorigenesis is associated with an anti-invasive and anti-inflammatory gene signature. Cancer Prev Res 2012;5:754–64
- Chwalisz K, Perez MC, DeManno D, et al. Selective progesterone receptor modulator development and use in the treatment of leiomyomata and endometriosis. Endocr Rev 2005;26:423–38
- Williams ARW, Critchley HOD, Osei J, et al. The effects of the selective progesterone receptor modulator asoprisnil on the morphology of uterine tissues after 3 months treatment in patients with symptomatic uterine leiomyomata. Hum Reprod 2007;22:1696–704
- Pasqualini JR. Progestins in the menopause in healthy women and breast cancer patients. Special issue: progestogens and the climacteric female. Maturitas 2009;62:343–8
- Klein-Hitpass L, Cato ACB, Henderson D, Ryffel GU. Two types of antiprogestins identified by their differential action in transcriptionally active extracts from T47D cells. Nucleic Acids Res 1991;19:1227–34
- Trussell J. Update on and correction to the cost effectiveness of contraceptives in the United States. Contraception 2012;85:218
- Dahmoun M, Boman K, Cajander S, Bäckström T. Intratumoral effects of medroxy-progesterone on proliferation, apoptosis, and sex steroid receptors in endometrioid endometrial adenocarcinoma. Gynecol Oncol 2004;92:116–26
- Katsuki Y, Shibutani Y, Aoki D, Nozawa S. Dienogest, a novel synthetic steroid, overcomes hormone-dependent cancer in a different manner than progestins. Cancer 1997;79:169–76
- Nakamura M, Katsuki Y, Shibutani Y, Oikawa T. Dienogest, a synthetic steroid, suppresses both embryonic and tumor-cell-induced angiogenesis. Eur J Pharmacol 1999;386:33–40
- Oettel M, Carol W, Elger W, et al. A 19-norprogestin without 17α-ethinyl group II: dienogest from a pharmacodynamic point of view. Drugs Today 1995;31:517–36
- Hapgood JP, Africander D, Louw R, et al. Potency of progestogens used in hormonal therapy: toward understanding differential actions. J Steroid Biochem Mol Biol. [Epub ahead of print]. doi: 10.1016/j.jsbmb.2013.08.001
- Escande A, Servant N, Rabenoelina F, et al. Regulation of activities of steroid hormone receptors by tibolone and its primary metabolites. J Steroid Biochem Mol Biol 2009;116:8–14
- Rižner TL, Brožič P, Doucette C, et al. Selectivity and potency of the retroprogesterone dydrogesterone in vitro. Steroids 2011;76:607–15
- Loose DS, Stancel GM. Estrogens and progestins. In: Brunton LL, Lazo JS, Parker KL, eds. Goodman & Gilman's the pharmacological basis of therapeutics. 11th ed. New York: McGraw-Hill; 2006:1541–71
- Wiebe J, Zhang G, Welch I, Cadieux-Pitre H-A. Progesterone metabolites regulate induction, growth, and suppression of estrogen- and progesterone receptor-negative human breast cell tumors. Breast Cancer Res 2013;15:R38
- Cabeza M, García-Lorenzana M, García M, et al. New-D-homoandrost-4,6-diene derivatives as potent progesterone receptor antagonist. Steroids 2010;75:101–8
- Garrido M, Cabeza M, Cortés F, et al. Cytotoxic effect of novel dehydroepiandrosterone derivatives on different cancer cell lines. Eur J Med Chem 2013;68:301–11
- Chabbert-Buffet N, Meduri G, Bouchard P, Spitz IM. Selective progesterone receptor modulators and progesterone antagonists: mechanisms of action and clinical applications. Hum Reprod Update 2005;11:293–307
- DeManno D, Elger W, Garg R, et al. Asoprisnil (J867): a selective progesterone receptor modulator for gynecological therapy. Steroids 2003;68:1019–32
- Madauss KP, Grygielko ET, Deng S-J, et al. A structural and in vitro characterization of asoprisnil: a selective progesterone receptor modulator. Mol Endocrinol 2007;21:1066–81
- Busia L, Faus H, Hoffmann J, Haendler B. The antiprogestin Lonaprisan inhibits breast cancer cell proliferation by inducing p21 expression. Mol Cell Endocrinol 2011;333:37–46
- Van Look PFA, von Hertzen H. Clinical uses of antiprogestogens. Hum Reprod Update 1995;1:19–34