
Abstract
A novel proton transfer compound (HClABT)+(HDPC.H2DPC)− (1) and its Fe(III), Co(II), Ni(II) and two different Cu(II) complexes (2–6) have been prepared and characterized by spectroscopic techniques. Additionally, single crystal X-ray diffraction techniques were applied to all complexes. All compounds, including acetazolamide (AAZ) as the control compound, were also evaluated for their in vitro inhibition effects on human hCA I and hCA II for their hydratase and esterase activities. Although there is no inhibition for hydratase activities, all compounds have inhibited the esterase activities of hCA I and II. The comparison of the inhibition studies of 1–6 to parent compounds, ClABT and H2DPC, indicates that 1–6 have superior inhibitory effects. The inhibition effects of 2–6 are also compared to the inhibitory properties of the simple metal complexes of ClABT and H2DPC, revealing an improved transfection profile. Data have been analysed by using a one-way analysis of variance for multiple comparisons.
Introduction
2,6-Pyridinedicarboxylic acid (or dipicolinic acid, H2DPC) is commonly used as an acid in proton transfer systemsCitation1. Continuing the path to synthesize proton transfer compounds, our group has focused on forming ion pairs between H2DPC and various organic bases, such as 2-aminobenzothiazoleCitation2, 2-amino-6-methylbenzothiazoleCitation3, 2-amino-6-methoxybenzothiazoleCitation4, 2-amino-4-methylpyridineCitation5, 2-hydroxyethylpiperazineCitation6. The interest of metal complexes for H2DPC and its deprotonated forms, HDPC− and DPC2−, stems from their interesting structural features with various coordination modesCitation1, stabilization of unusual oxidation statesCitation7 and insulin-mimetic effectsCitation8,Citation9.
Benzothiazoles and their simple metal complexes are well known for their biological activities, such as antitumor agents, antimicrobial, antifungal, analgesics, anti-inflammatory, anti HIV, carbonic anhydrase inhibition and local anesthesticCitation2–4,Citation10–15. In recent studies, the mixed ligand metal complexes between 2-amino-6-chlorobenzothiazole and other ligands, such as R3E (R = alkyl or phenyl, E = Sn or Pb)Citation16 and palmitic acidCitation17, have been reported in literature. The mixed ligand metal complexes of these compounds have shown better biological activities than the simple onesCitation4,Citation18,Citation19. In order to prepare the mixed ligand complexes, two types of ligands, generally acids and bases, are brought together before coordination to the metal ionCitation1,Citation6,Citation20,Citation21.
In this study, a novel proton transfer compound (HClABT)+(HDPC.H2DPC)− (1), and its Fe(III), Co(II), Ni(II) and two different Cu(II) complexes, (HClABT)[Fe(DPC)2] ċ 5H2O (2), (HClABT)2[Co(DPC)2] ċ H2O (3), (HClABT)2[Ni(DPC)2] ċ H2O (4), [Cu(ClABT)(DPC)(H2O)]2 (5) and (HClABT)2[Cu(DPC)2] ċ H2O (6), have been prepared and characterized by elemental, spectral (1H-NMR, IR and UV-Vis.) and thermal analyses, as well as using magnetic measurement and molar conductivity techniques. Single crystal X-ray analyses of the complexes (2–6) were also reported.
Furthermore, we have investigated the potential use of these compounds as new inhibitors of human carbonic anhydrase (hCA I and hCA II) isoenzymes in the treatment of glaucoma, which is a group of diseases characterized by the gradual loss of visual field due to an elevation in intraocular pressure (IOP)Citation22,Citation23. Carbonic anhydrases (CAs, EC 4.2.1.1) are ubiquitous metalloenzymes to catalyze interconversion of carbon dioxide and water to bicarbonate and proton and are encoded by five unrelated gene families: α-, β-, γ-, δ- and ζ-CAs. There are sixteen α-CA isoforms or CA-related proteins identified in mammalsCitation24.
Experimental section
General methods and materials
All chemicals used were analytical reagents and were commercially purchased from Aldrich. FeSO4 ċ 7H2O, Co(CH3COO)2 ċ 4H2O, Ni(CH3COO)2 ċ 4H2O, Cu(CH3COO)2 ċ H2O, 2-amino-6-chlorobenzothiazole and pyridine-2,6-dicarboxylic acid were used as received. Elemental analyses for C, H, N and S were performed on Elementar Vario III EL (Hanau, Germany) and Fe, Co, Ni and Cu were detected with Perkin Elmer Optima 4300 DV ICP-OES (Perkin Elmer Inc., Wellesley, MA). 1H-NMR spectra were recorded with Bruker DPX FT NMR (500 MHz) spectrometer (Karlsruhe, Germany) (SiMe4 as internal standard and 85% H3PO4 as an external standard). FT-IR spectra were recorded in the 4000–400 cm−1 region with Bruker Optics, Vertex 70 FT-IR spectrometer using ATR techniques (Ettlingen, Germany). Thermal analyses were performed on SII Exstar 6000 TG/DTA 6300 model (Shimadzu Co, Kyoto, Japan) using platinum crucible with 10 mg sample. Measurements were taken in the static air within a 30–900 °C temperature range. The UV–Vis spectra were obtained for aqueous solutions of the compounds (10−3 M) with a SHIMADZU UV-2550 spectrometer (Shimadzu Co, Kyoto, Japan) in the range of 200–900 nm. Magnetic susceptibility measurements at room temperature were performed using a Sherwood Scientific Magway MSB MK1 model (Sherwood Scientific Ltd, Cambridge, UK) magnetic balance by the Gouy method using Hg[Co(SCN)4] as calibrant. The molar conductances of the compounds were determined in water:ethanol (1:1) and in DMSO (10−3 M) at room temperature using a WTW Cond 315i/SET Model conductivity meter (Weilheim, Germany).
Synthesis of (HClABT)+(HDPC.H2DPC)− (1) and metal complexes (2–6)
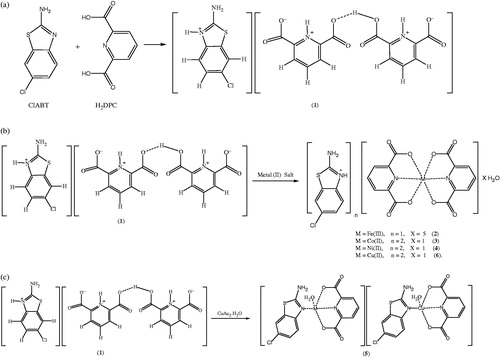
A solution of ClABT (0.923 g, 5 mmol) in 25 mL ethanol was added to the solution of H2DPC (0.836 g, 5 mmol) in 25 mL ethanol. The mixture was refluxed for 3 hours and then was cooled to room temperature. The reaction mixture was kept at room temperature for three hours to give white solid of 1 (1.232 g, 95 % yield).
A solution of 1 mmol metal(II) salt [0.278 g FeSO4 ċ 7H2O or 0.249 g Co(CH3COO)2 ċ 4H2O or 0.248 g Ni(CH3COO)2 ċ 4H2O or 0.199 g Cu(CH3COO)2 ċ H2O] in water (10 mL) was added dropwise to the solution of 1 (0.519 g, 1 mmol) in water:ethanol (1:1) (20 mL) with stirring at room temperature for two hours. The reaction mixture was kept at room temperature for two weeks to give yellow crystalline solid for 2 (0.397 g, 60% yield), brown crystalline solid for 3 (0.584 g, 75% yield), green crystalline solid for 4 (0.583 g, 75% yield) and dark green crystalline solid for 5 (0.347 g, 40% yield). Compound 6 was obtained from the mother liquor of 5 after one week as turquoise crystalline solid (0.313 g, 40% yield) (). The single crystals of all complexes suitable for X-ray diffraction were separated and washed with EtOH:water (1:1).
Figure 1. Syntheses of compounds 1–6 (a for 1 and b for 2, 3, 4, 6 and c for 5).
Anal Calcd. for 1 (C21H15ClN4O8S): C, 48.61%; H, 2.91%; N, 10.80%; S, 6.18%. Found: C, 48.60%; H, 2.90%; N, 10.78%; S, 6.20%; for 2 (C21H22ClN4O13SFe): C, 38.11%; H, 3.35%; N, 8.47%; S, 4.85%; Fe, 8.44. Found: C, 38.10%; H, 3.30%; N, 8.45%; S, 4.80%; Fe, 8.40%; for 3 (C28H20Cl2N6O9S2Co): C, 43.20%; H, 2.59%; N, 10.80%; S, 8.24%; Co, 7.57%. Found: C, 43.18%; H, 2.50%; N, 10.81%; S, 8.23%; Co, 7.75%; for 4 (C28H20Cl2N6O9S2Ni): C, 43.21%; H, 2.59%; N, 10.80%; S, 8.24%; Ni, 7.54%. Found: C, 43.19%; H, 2.50%; N, 10.79%; S, 8.23%; Ni, 7.72%; for 5 (C28H20Cl2N6O10S2Cu2): C, 38.99%; H, 2.34%; N, 9.74%; S, 7.43%; Cu, 14.73%. Found: C, 38.98%; H, 2.30%; N, 9.73%; S, 7.45%; Cu, 15.38%; for 6 (C28H20Cl2N6O9S2Cu): C, 42.95%; H, 2.57%; N, 10.73%; S, 8.19%; Cu, 8.11%. Found: C, 42.90%; H, 2.50%; N, 10.75%; S, 8.15%; Cu, 8.20%.
In addition, simple metal complexes of ClABT (FeClABT, CoClABT, NiClABT and CuClABT) and of H2DPC (FeDPC, CoDPC, NiDPC and CuDPC) were synthesized in order to compare the inhibition studies with the complex compounds of the proton transfer salt.
X-ray data collection and structure refinement
The crystal and instrumental parameters used in the unit-cell determination and data collection are summarized in Table S1 for compounds 2–6. Crystallographic data of 2–6 were recorded on a Bruker Kappa APEX II CCD area-dedector X-ray diffractometer using graphite monochromatized with MoKα radiation (ë = 0.71073 Å), using ω-2θ scan mode. The empirical absorption corrections were applied by multi-scan via Bruker, SADABS softwareCitation25. The structures were solved by the direct methods and subsequently completed by difference Fourier recycling. All non-hydrogen atoms were refined anisotropically using the full-matrix least-squares techniques on F2. The SHELXS-97 and SHELXL-97Citation26 programs were used for all the calculations. The H atoms were placed in idealized positions and constrained to ride on their parent atoms with distances in the range of N–H = 0.86–88 Å, C–H = 0.93–0.95 Å and with Uiso(H) = 1.2Ueq(C,N). Hydrogen atoms of water molecules were located from difference Fourier maps and refined with isotropic thermal parameters with a distance of O–H = 0.73(4)–0.94(5) Å and with Uiso(H) = 1.2Ueq(O). The drawings of molecules were accomplished with the help of ORTEP-3 for WindowsCitation27.
Purification of isoenzymes hCA-I and II from human erythrocytes
In order to purify hCA I and II isoenzymes, first, human blood was centrifuged at 1500 rpm for 20 min, and, after the removal of the plasma, the erythrocytes were washed with an isotonic solution (0.9% NaCl). After that, the erythrocytes were lysed with 1.5 volume of ice-cold water. The lysate was centrifuged at 20 000 rpm for 30 min to remove cell membranes and non-lysed cells. The pH of the supernatant was adjusted to 8.7 with tris and was then loaded onto an affinity column containing Sepharose-4B-l-tyrosine-p-aminobenzene sulfonamide as the binding group. After extensive washing with 25 mM tris-HCl/22 mM Na2SO4 (pH 8.7), the hCA I and II isoenzymes were eluted with 1.0 M NaCl/25 mM Na2HPO4 (pH 6.3) and 0.1 M CH3COONa/0.5 M NaClO4 (pH 5.6)Citation28,Citation29. The amount of purified protein was estimated by the Bradford methodCitation30 and SDS–PAGE was carried out to determine whether the elute contained the enzymeCitation31.
Hydratase and esterase activity assay
On hydration of CO2Citation32, CO2-hydratase activity as an enzyme unit (EU) was calculated by using the equation ((t0 − tc)/tc), where t0 and tc are the times for pH change of the nonenzymatic and the enzymatic reactions, respectively.
Carbonic anhydrase esterase activity was assayed by following the change in the absorbance at 348 nm of 4-nitrophenylacetate (NPA) to 4-nitrophenylate ion over a period of 3 min at 25 °C using a spectrophotometer (SHIMADZU UV 1700 PharmaSpec) according to the method described in the literatureCitation33,Citation34. The enzymatic reaction, in a total volume of 3.0 mL, contained 1.4 mL of 0.05 M tris-SO4 buffer (pH 7.4), 1 mL of 3 mM 4-nitrophenylacetate, 0.5 mL H2O and 0.1 mL enzyme solution. A reference measurement was obtained by preparing the same cuvette without enzyme solution.
IC50 values (the concentration of inhibitor producing a 50% inhibition of CA activity) have been obtained as in vitro for free ligands (ClABT and H2DPC) and their simple metal complexes, and the synthesized compounds 1–6, and acetazolamide (AAZ) as the control compound for their hydratase and esterase activities.
Statistical analysis
All the presented data were confirmed with three independent experiments and were expressed as the mean ± standard deviation (SD). Data were analyzed by using a one-way analysis of variance for multiple comparisons (SPSS 13.0, SPSS Inc., Chicago, IL). p < 0.0001 was considered to be statistically significant.
Results and discussion
Crystal structures of 2–6
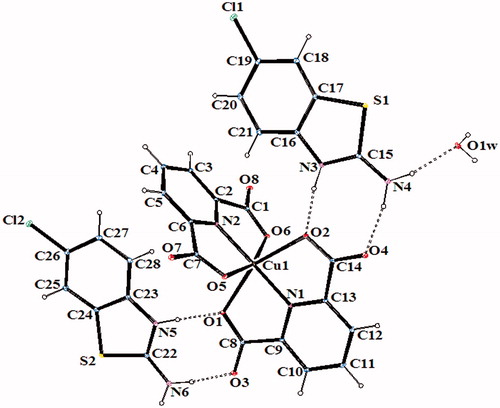
The molecular structures of 2–6, with the atom labeling of symmetric units, are shown in , respectively. The details of the crystal structure solutions are summarized in Table S1 and the selected bond lengths and angles are listed in Table S2.
Figure 2. An ORTEP drawing of asymmetric unit of 2 with the atom-numbering scheme. Displacement ellipsoids are drawn at the 40% probability level.
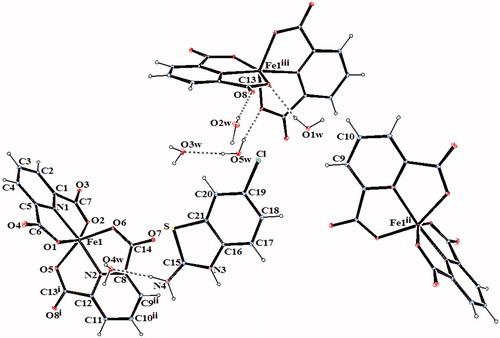
Figure 3. An ORTEP drawing of asymmetric unit of 3 with the atom-numbering scheme. Displacement ellipsoids are drawn at the 40% probability level.
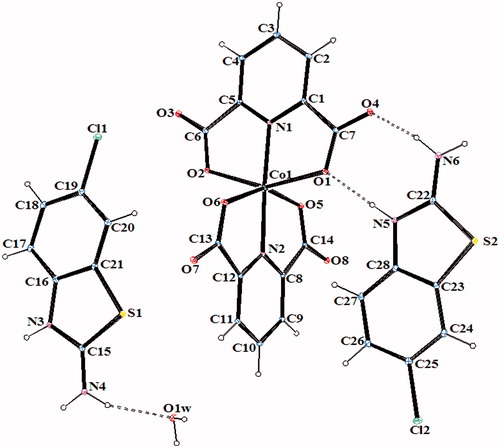
Figure 4. An ORTEP drawing of asymmetric unit of 4 with the atom-numbering scheme. Displacement ellipsoids are drawn at the 40% probability level.
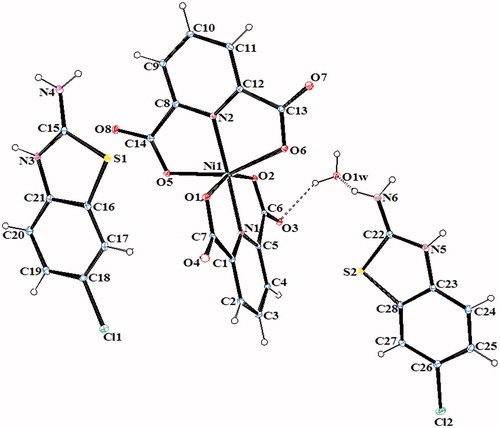
Figure 5. An ORTEP drawing of asymmetric unit of 5 with the atom-numbering scheme. Displacement ellipsoids are drawn at the 40% probability level.
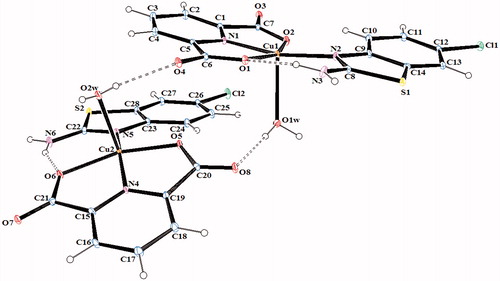
Figure 6. An ORTEP drawing of asymmetric unit of 6 with the atom-numbering scheme. Displacement ellipsoids are drawn at the 40% probability level.
The complex 2, (HClABT)[Fe(DPC)2] ċ 5H2O, crystallizes in the monoclinic P2(1)/n space group. Structure of 2 consists of one HMeABT+ cation, one [Fe(DPC)2]− anion and five uncoordinated water molecules. The complex 5, [Cu(DPC)(ClABT)(H2O)]2, crystallizes in the monoclinic P2(1)/c space group and the asymmetric unit consists of two independent and similar molecules. Structure of 5 contains two DPC2− ions, two ClABT molecules, two Cu(II) ions and two coordinated water molecules. All the other complexes, (HClABT)2[M(DPC)2] ċ H2O [M = Co(II) for 3, Ni(II) for 4 and Cu(II) for 6], crystallize in the triclinic P 1 space group. Structures of 3, 4 and 6 consist of one [M(DPC)2]2− anion and two counter HClABT+ cations and one uncoordinated water molecule.
In complexes 2, 3, 4 and 6, the metal ion coordinates to four oxygen atoms and two nitrogen atoms of two dipicolinate ions resulted with a distorted octahedral conformation. Both carboxylate oxygen atoms from DPC2− occupy the trans-apical positions of the metal, and define low trans-angle value around metal ion for all complexes within the range of 151.56(5)–158.85(5)° for O-M-O which reveal a rather rigid structure of such tri-dentate ligands (Table S2). In the structures of 2, 3, 4 and 6, the N-M-N trans-angles are much closer to 180° [174.76(7)–177.08(7)°] and the dihedral angles defined by the mean planes of two DPC2− ligands are 88.36(9)° for 2, 89.11(11)° for 3, 89.30(6)° for 4 and 89.45(10)° for 6, showing that they fall almost perpendicular. The M–N and M–O bond distances lie within expected range of 1.920(15)–2.0588(15) Å and 1.9832(13)–2.3156(13) Å, respectively (Table S2). In all essential details, the geometries of the molecules regarding bond lengths and angles of the compounds are in good agreement with the values observed in similar Fe(III)Citation2,Citation3,Citation35, Co(II)Citation3,Citation36, Ni(II)Citation3,Citation36,Citation37 and Cu(II)Citation3,Citation5,Citation37 complexes.
The complex 5 consists of two independent and different cationic Cu2+ sites (). Cu1 and Cu2 atoms in complex 5 are coordinated by two N atoms, one from DPC (N1 for Cu1 and N4 for Cu2) and one from ClABT ring (N2 for Cu1 and N5 for Cu2), two carboxylate O atoms from DPC (O1 and O2 for Cu1 and O5 and O7 for Cu2) and one water molecule (O1w for Cu1 and O2w for Cu2) to give distorted square pyramidal structures. In complex 5, the coordination arrangement of the Cu(II) ions are slightly distorted from regular square pyramidal since the values of the structural index τ are 0.16 for Cu1 and 0.05 for Cu2Citation38. The Cu–O bond distances [Cu1–O1 = 2.055(2), Cu1–O2 = 1.994(2), Cu2–O5 = 2.000(2), and Cu2–O6 = 2.057(2), Å], observed from the carboxylate coordination to the center metal ion, are well consistent with those observed in the literature dataCitation2,Citation4,Citation39. Two N atoms from two different ligands, DPC and ClABT, occupy equatorial positions with Cu–N distances of 1.932(2) Å (Cu1–N1), 2.008(3) Å (Cu1–N2), 1.936(2) Å (Cu2–N4) and 2.015(3) Å (Cu2–N5), and the bond distances are similar to those found in other related Cu(II) complexesCitation2,Citation4,Citation39. Square pyramidal coordination arrangements around Cu ions are also characterized by O–Cu–O, N–Cu–N, O–Cu–N, O–Cu–Ow and N–Cu–Ow angles (Table S2)Citation2,Citation4,Citation39. The dihedral angles between the planes of the DPC2− and ClABT are 5.42(5)° and 21.23(11)°, respectively. The Cu1 and Cu2 atoms lie 0.087(11) and 0.0223(11) Å out of the planes, respectively. The Cu–Owater bond distances, arising from coordination of the water molecule to the central metal ion, are 2.296(2) Å for Cu1–O1w and 2.259(2) Å for Cu2–O2w, which are much longer than the Cu–O (carboxylate) bond distances.
In all complexes, hydrogen bonds between the carboxylate group and water molecules play important roles in stabilizing the crystal structures. The ranges of the D–H…A angles and those of the H…A and D…A distances indicate the presence of strong and weak hydrogen bondings in the structures 2–6 (Table S3).
1H NMR studies of (HClABT)+(HDPC.H2DPC)− (1)
The 1H-NMR spectrum of compound 1 was obtained in d6-DMSO at room temperature using TMS as internal Standard (Figure S1). The 1H signals were assigned on the basis of chemical shifts, multiplicities, intensities of the signals and coupling constants. Table S4 lists complete 1H-NMR assignments for compound 1.
The H3, H4 and H5 protons of the (HClABT)+ ring are doublet with 1H intensity and they are observed at 7.30 ppm (H3, 3JH3−H4 = 8.50 Hz), 7.22 ppm (H4, 3JH4−H3 = 8.50, 4JH4−H5 = 2.00 Hz) and 7.75 ppm (H5, 4JH5−H4 = 2.00 Hz). The symmetrical H8 and H12 protons of the (HDPC)− rings are triplet with 2H intensity and they are observed at 8.18 ppm (3JH7−H6,9 or H12−H11,13 = 7.75 Hz). The H7, H9 and H11, H13 protons are also symmetrical and they are observed at 8.25 ppm as doublet with 4H intensity (3JH7,9−H8 or H11,13−H12 = 8.00 Hz). The hydrogen atoms bonded to N atoms, –NH2 (H1), =N+-H (H2), =N+-H (H6 and H14), and hydrogen atom (H10) on carboxylate in compound 1 were not observed in the 1H NMR spectrum. The room temperature 1H-NMR spectrum for compound 1 indicates clearly the formation of the proton transfer compound with 1:2 ratio of ClABT and H2DPC (Figure S1). The structure of 1 has been suggested as in on the basis of other experimental data, charge balance and a former study in literatureCitation40.
FT-IR measurements
The infrared spectral data of the starting compounds (ClABT and H2DPC) and compounds 1–6 are given in Table S5. In the high-frequency region, weak bands 3110–3058 cm−1 are attributed to the stretching vibrations of aromatic of C–H. There are also broad absorption bands at 3475–3303 cm−1 which are attributed to the ν(OH) vibrations of uncoordinated water molecules for compounds 2–4 and 6, and coordinated water molecules for compound 5. The relatively weak and broad bands at 2770–2527 cm−1 are attributed to the ν(N+–H) vibration for 1–4 and 641. These bands were not observed for compound 5 due to the deprotonation of the salt during the complex formation (). The absorption bands at 3453 and 3265 cm−1 of NH2 group of ClABT are slightly shifted from those found for 1 (3314 and 3200 cm−1), for 2 (3356 and 3244 cm−1), for 3 (3375 and 3300 cm−1), for 4 (3303 and 3224 cm−1), for 5 (3414 and 3263 cm−1) and for 6 (3312 and 3266 cm−1) due to the weak intermolecular interactions. The carboxylate groups exhibit strong carbonyl bands in the region 1700–1456 cm−1. These bands are reflected by IR spectrum of the asymmetric (νas) and symmetric (νs) stretching vibrations at 1701 and 1456 cm−1 for H2DPC, 1647 and 1469 cm−1 for 1, 1666 and 1470 cm−1 for 2, 1640 and 1463 cm−1 for 3, 1660 and 1462 cm−1 for 4, 1660 and 1453 cm−1 for 5 and 1655 and 1517 cm−1 for 6. The differences (Δν) between the asymmetric and symmetric stretches of the carboxylate groups of 2–6 are 196, 178, 177, 198 and 207, respectively, which suggests a monodentate binding of the carboxylate group to the metal ion in all complexesCitation42. The C–O vibrations data for all compounds are between 1382 and 1080 cm−1 as expected. The strong absorption bands at the region of 1635–1415 cm−1 are attributed to the ν(C=N) and ν(C=C) vibrations for all compounds. The ring wagging vibrations of the pyridine groups are also observed at 782–680 cm−1 region for compounds H2DPC and 1–6. The weak bands at 590–442 cm−1 and 658–422 cm−1 are from the M–N and M–O vibrations of compounds 2–6.
Thermal analyses of 1–6
Figures S2–S7 show the TG-DTG and DTA curves of compounds 1–6 and the thermal analyses results are given in Table S6. For compound 1, two stages are observed and the first endothermic stage corresponds to the loss of C15H12N4O8 unit and the second exothermic one is the decomposition of the residue of C6H3ClS unit.
For compounds 2, 3 and 4, three stages are observed. The first endothermic peak corresponds to the loss of five moles of water for 2 and one mole of water for 3 and 4. The second endothermic stage is consistent to the loss of one or two or two moles of (HClABT)+, respectively. Two moles of DPC2− are decomposed exothermically in the third stages for 2, 3 and 4. The final decomposition products are FeO, CoO and NiO and they are identified by IR spectroscopy.
For compounds 5 and 6, three stages are observed, and the first endothermic stage corresponds to the loss of two or one moles of water, respectively. The endothermic second stage is consistent to the loss of C12H6N2O2 or C8H10Cl2N4S2 units, respectively, and C16H10Cl2N4O6S2 or C20H10N2O8 units are also decomposed exothermically in the following stage. The final decomposition products are CuO for 5 and 6 and they are identified by IR spectroscopy.
UV/vis Spectrum, magnetic susceptibility and molar conductivity
The electronic spectra of compounds 1–6 and the free ligands ClABT and H2DPC were recorded in water/ethanol (1:1) and in DMSO solution with 1 × 10−3 M concentrations at room temperature (Table S7). Characteristic π–π* transitions are observed in the range of 232–288 nm (13000–34750 L mol−1 cm−1) in water/ethanol and 245–293 nm (15 600–49 660 L mol−1 cm−1) in DMSO for 1–6. The same ð–ð* transition profiles are also detected for the free ligands ClABT and H2DPC and there is no marked difference from those of either proton transfer compound or metal complexes in both solutions. The intensities of the absorption bands for all compounds in DMSO are, in general, higher than in water/ethanol. The bands for the d–d transitions in water/ethanol and in DMSO are observed at 786 (260 L mol−1 cm−1) and 781 nm (100 L mol−1 cm−1) for 3, 768 nm (240 L mol−1 cm−1) and 796 nm (270 L mol−1 cm−1) for 4, 766 nm (110 L mol−1 cm−1) and 769 nm (200 L mol−1 cm−1) for 5, 787 nm (410 L mol−1 cm−1) and 769 nm (490 L mol−1 cm−1) for 6, respectively. The d–d transition for complex 2 containing Fe(III) ion with d5 has not been observed.
The room temperature magnetic moment of the metal complexes are 5.86 for 2, 3.85 for 3, 2.80 for 4, 1.70 for 5 and 1.65 for 6 BM per metal ion, indicating the presence of five (d5), three (d7), two (d8), one (d9) and one (d9) unpaired electrons, respectively.
The molar conductivity data in water/ethanol and in DMSO are 42.8 and 43.5 Ù−1 cm2 mol−1 for 2, 87.2 and 49.5 Ù−1 cm2 mol−1 for 3, 81.6 and 43.3 Ù−1 cm2 mol−1 for 4, 2.4 and 0.5 Ù−1 cm2 mol−1 for 5 and 90.0 and 67.5 Ù−1 cm2 mol−1 for 6, respectively, indicating that the complexes 3, 4 and 6 are ionic with a 2:1 ratio, the complex 2 is ionic with a 1:1 ratio and the complex 5 is non-ionicCitation43.
In vitro inhibition studies
In the present study, human carbonic anhydrase isoforms (hCA I and hCA II) were purified from human erythrocytes using Sepharose 4B-l-Tyrosine-sulfanilamide affinity chromatography method. Then the synthesized compounds (1–6), ClABT, H2DPC and metal complexes of ClABT and H2DPC, as well as standard, clinically used CAI (AAZ) have been tested for the inhibition of two cytosolic isoforms hCA I and hCA II. The inhibition assays were performed by hydratase and esterase activity method under in vitro conditions. The following structure-activity relationships can be observed in the .
Table 1. Human carbonic anhydrase isozymes (hCA I and hCA II) inhibition data with newly synthesized compounds by an esterase assay with 4-nitrophenylacetate as substrate.
All of the synthesized compounds have not inhibited the hydratase activity of hCA I and hCA II isoforms. In addition, no inhibition effect of H2DPC and its metal complexes have been observed on esterase activities of hCA I and II. Other studied compounds act as moderate inhibitors against to esterase activities of hCA I and hCA II isoforms. All compounds, except H2DPC and its metal complexes have more powerful inhibition effect for hCA II than hCA I, but the differences of inhibition potentials are little for these isozymes. Overall differences between the lowest and the highest inhibition potentials are 6-fold (i.e. IC50 value of simple ligant, ClABT, for hCA I = 0.85 ± 0.02 mM, IC50 value of 5, for hCA II = 0.14 ± 0.003 mM). It has also been observed that the inhibition potentials of the metal complexes of ClABT are nearly 3-fold higher than ClABT for both hCA I and hCA II (i.e. IC50 values of CuClABT for hCA I and hCA II 0.29 ± 0.005 mM and 0.27 ± 0.003 mM, respectively, IC50 values of ClABT for hCA I and hCA II 0.85 ± 0.02 mM and 0.82 ± 0.006 mM, respectively).
While H2DPC does not show any inhibition effects on hCA I and hCA II, the proton transfer salt (1) has inhibition effect due to structural changes leading to impressive differences of activityCitation20,Citation21,Citation44. In addition, the salt has more effective inhibition effect than ClABT (IC50 value of 1 for hCA I = 0.76 ± 0.002 mM, for hCA II = 0.82 ± 0.006 mM, IC50 value of ClABT for hCA I = 0.85 ± 0.02 mM, for hCA II = 0.74 ± 0.007 mM). Similarly, the metal complexes of 1 (0.16–0.26 mM for hCA I and 0.14–0.24 mM for hCA II) have more potent effects than 1 (0.76 ± 0.002 mM for hCA I and 0.74 ± 0.007 mM for hCA II) and than all simple metal complexes ClABT (0.29–0.36 mM for hCA I and 0.27–0.30 mM for hCA II), revealing the structural changes increasing the inhibition effects. Hence, we conclude that metal ions facilitate the enzyme–inhibitor interactions. All inhibition values found in this study are in good agreement with those found in our similar previous studiesCitation2–4.
Conclusions
In the present work, Fe(III), Co(II), Ni(II) and Cu(II) ionic complexes and Cu(II) non-ionic complex (5) with (HClABT)+(HDPC.H2DPC)− (1) have been prepared for the first time. In complexes 2, 3, 4 and 6, the metal ion is coordinated by two pyridine-2,6-dicarboxylate anions to give a distorted octahedral conformation with one mole of counter ion (HClABT)+ for 2 and two moles of counter ions (HClABT)+ for 3 and 4 and 6. There are also hydrate water molecules in their crystal structures (five moles for 2, one mole for 3, and 4, and 6). The complex 5 consists of two independent and different cationic Cu2+ sites which are coordinated by one DPC2− anion and one ClABT ring and one water molecule to give distorted square pyramidal structures. For all complexes, intermolecular N–H…O and O–H…O hydrogen bonds and π–π stacking interactions seem to be effective in the stabilization of the crystal structure.
The novel compounds (1–6) possess significant inhibition effect on hCA I and on hCA II for esterase activity. These results suggest that further inhibition studies are worthwhile in order to obtain correlation in such compounds and the derivatives of such compounds should be subject to further inhibition in vivo tests. The order of the inhibition effects increasing through starting compounds to proton transfer salt and to the complexes of these compounds might occur due to the structural changes leading to an impressive difference of activity.
Supplementary data
CCDC 927071 (2), CCDC 927072 (3), CCDC 927073 (4), CCDC 927074 (5) and CCDC 927075 (6) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk (the Chambridge Crystallographic Data Centre, 12 Union Road, Chambridge CB2 1EZ, UK; fax +44 1123 336 033; or e-mail: [email protected]).
Declaration of interest
The authors acknowledge the support provided by Dumlupýnar University Research Fund (grant No. 2012/16).
Supplementary material available online
SUPPLEMENTAL
Download PDF (148.9 KB)Acknowledgements
The authors would like to thank the Medicinal Plants and Medicine Research Center of Anadolu University Eskişehir for allowing to use the X-ray facility.
References
- Aghabozorg H, Manteghi F, Sheshmani S. A brief review on structural concepts of novel supramolecular proton transfer compounds and their metal complexes. J Iran Chem Soc 2008;5:184–227
- İlkimen H, Yenikaya C, Sarı M, et al. Synthesis and characterization of a proton transfer salt between 2,6-pyridinedicarboxylic acid and 2-aminobenzothiazole, and its complexes and their inhibition studies on carbonic anhydrase isoenzymes. J Enzyme Inhib Med Chem 2013. [Epub ahead of print]. DOI:10.3109/14756366.2013.782299
- İlkimen H, Yenikaya C, Sarı M, et al. Synthesis and characterization of a proton transfer salt between dipicolinic acid and 2-amino-6-methylbenzothiazole and its complexes, and their inhibition studies on carbonic anhydrase isoenzyme. Polyhedron 2013;61:56–64
- İlkimen H, Yenikaya C, Sarı M, et al. Synthesis and characterization of some metal complexes of a proton transfer salt, and their inhibition studies on carbonic anhydrase isoenzymes and the evaluation of the results by statistical analysis. J Enzyme Inhib Med Chem 2013. [Epub ahead of print]. DOI: 10.3109/14756366.2013.843172
- Büyükkıdan N, Yenikaya C, Sarı M, et al. Synthesis, characterization and biological evaluation of novel Cu(II) complexes with proton transfer salt of 2,6-pyridinedicarboxylic acid and 2-amino-4-methylpyridine. J Coord Chem 2011;64:3353–5
- Büyükkıdan N, Yenikaya C, İlkimen H, et al. Synthesis, characterization and antimicrobial activity of a novel proton salt and its Cu(II) complex. Russian J Coord Chem 2013;39:24–31
- Hoof DL, Tisley DG, Walton RA. Studies on metal carboxylates. Part III. Pyridine-2,6-dicarboxylates of the lanthanides. Synthesis and spectral studies and the X-ray photoelectron spectra of several pyridine carboxylate complexes. J Chem Soc Dalton Trans 1973;2:200–4
- Yang L, Crans DC, Miller SM, et al. Cobalt(II) and Cobalt(III) dipicolinate complexes: solid state, solution, and in vivo insulin-like properties. Inorg Chem 2002;41:4859–71
- Buglyó P, Crans DC, Nagy EM, et al. Aqueous chemistry of the vanadiumııı (VIII) and the VIII−dipicolinate systems and a comparison of the effect of three oxidation states of vanadium compounds on Diabetic Hyperglycemia in rats. Inorg Chem 2005;44:5416–27
- Prakash A, Adhikari D. Application of Schiff bases and their metal complexes – a review. Int J Chem Tech Res 2011;3:1891–6
- Briganti F, Tilli S, Mincione G, et al. Carbonic anhydrase inhibitors. Metal complexes of 5-(2-chlorophenyl)-1,3,4-thiadiazole-2-sulfonamide with topical intraocular pressure lowering properties: The influence of metal ions upon the pharmacological activity. J Enzyme Inhib Med Chem 2000;15:185–200
- He XF, Vogels CM, Decken A, Westcott SA. Pyridyl benzimidazole, benzoxazole, and benzothiazole platinum complexes. Polyhedron 2004;23:155–60
- Mortimer CG, Wells G, Crochard JP, et al. Antitumor benzothiazoles. 2-(3,4-dimethoxyphenyl)-5-fluorobenzothiazole (GW 610, NSC 721648), a simple fluorinated 2-arylbenzothiazole, shows potent and selectve inhibitory activity against lung, colon and breast cancer cell lines. J Med Chem 2006;49:179–85
- Paramashivappa R, Phani KP, Rao PVS, Rao S. Replacement of the ureas moiety by benzothiazolesulfonamide provided inhibitors of HIV-1 protease with improved potency and antiviral activities. Bioorg Med Chem Lett 2003;13:657–60
- Vara-Prasad JVN, Panapoulous A, Rubin JR. Thiocyanation of alkylanilines. A simple and efficient synthesis of thiosulfonates containing 2-aminobenzothiazole. Tetrahedron Lett 2000;41:4065–8
- Ibrahim AMA, Etaiw SEDH. Paramagnetic charge transfer complexes: new molecular composites via intercalation of thiazole and benzothiazole derivatives within the cavities of the 3D host polymers [(Me3E)3Fe(CN)6]∞: E = Sn or Pb. Polyhedron 1997;16:1585–94
- Mathur N. Studies of solute-solvent interactions and applications of green and blue complexes of copper (II) palmitate with 2-aminobenzothiazoles. J Curr Chem Pharm Sci 2011;1:37–51
- Neelakantan MA, Mariappan SS, Dharmaraja J, Muthukumaran K. pH metric, spectroscopic and thermodynamic study of complexation behavior of 2-aminobenzothiazole with Ni (II) in presence of amino acids. Acta Chimica Slov 2010;57:198–205
- Shukla SN, Gaur P, Kaur H, Prasad M, et al. Synthesis, spectroscopic characterization and antibacterial sensitivity of some chloro dimethylsulfoxide/tetramethylenesulfoxide ruthenium(II) and ruthenium(III) complexes with 2-aminobenzothiazole. J Coord Chem 2008;61:441–9
- Yenikaya C, Sarı M, İlkimen H, et al. Synthesis, structural and antiglaucoma activity studies of a novel amino salicylato salt and its Cu(II) complex. Polyhedron 2011;30:535–41
- Yenikaya C, Sarı M, Bülbül M, et al. Synthesis, characterization and antiglaucoma activity of a novel proton transfer compound and a mixed-ligand Zn(II) complex. Bioorganic Med Chem 2010;18:930–8
- Schuman JS. Antiglaucoma medications: a review of safety and tolerability issues related to their use. Clin Ther 2000;22:167–208
- Scozzafava A, Banciu MD, Popescu A, Supuran CT. Carbonic anhydrase inhibitors: Inhibition of isozymes I, II and IV by sulfamide and sulfamic acid derivatives. J Enzyme Inhib Med Chem 2000;15:443–553
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;2:168–81
- Bruker, SADABS. Madison: Bruker AXS Inc.; 2005
- Sheldrick GM. SHELXS97 and SHEXL97. Program for crystal structure solution and refinement. Germany: University of Gottingen; 1997
- Farrugia LJ. ORTEP-3 for Windows – a version of ORTEP-III with a Graphical User Interface (GUI). J Appl Cryst 1997;30:565
- Arslan O, Nalbantoğlu B, Demir N, et al. A new method for the purification of carbonic anhydrase isozymes by affinity chromatography. Trop J Med Sci 1996;26:163–6
- Rickli EE, Ghazanfar SA, Gibbson BH, Edsall JT. Carbonic anhydrases from human erythrocytes: Preparation and properties of two enzymes. J Biol Chem 1964;239:1065–8
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of Bacteriophage T4. Nature 1970;227:680–5
- Wilbur KM, Anderson NG. Electrometric and colorimetric determination of carbonic anhydrase. J Biol Chem 1948;176:147–54
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–9
- Innocenti A, Scozzafava A, Parkkila S, et al. Investigations of the esterase, phosphatase, and sulfatase activities of the cytosolic mammalian carbonic anhydrase isoforms I, II, and XIII with 4-nitrophenyl esters as substrates. Bioorg Med Chem Lett 2008;18: 2267–71
- Tabatabaee M, Abbasi F, Kukovec BM, Nasirizadeh, N. Preparation and structural, spectroscopic, thermal, and electrochemical characterizations of iron(III) compounds containing dipicolinate and 2-aminopyrimidine or acridine. J Coord Chem 2011;64:1718–28
- Borah MJ, Singh RKB, Sinha UB, et al. Synthesis and crystal structure determination of dimeric Co(II) and Ni(II) with pyridine 2,6-dicarboxylic acid. J Chem Cryst 2012;42:67–75
- Zhang GC, Chen SP, Yang Q, Gao SL. Thermochemistry on K2[M(DPA)2]·7H2O(s) (M = Cu and Ni, H2DPA = pyridine-2,6-dicarboxylic acid). Thermochimica Acta 2011;518:66–71
- Addison AW, Rao TN. Synthesis, structure, and spectroscopic prorerties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane] copper(II) perchlorate. Chem Soc J Dalton Trans 1984;1:1349–55
- Yenikaya C, Poyraz M, Sarı M, et al. Synthesis, characterization and biological evaluation of a novel Cu(II) complex with the mixed ligands 2,6-pyridinedicarboxylicacid and 2-aminopyridine. Polyhedron 2009;28:3526–32
- Aghabozorg H, Soleimannejad J, Sharif MA, et al. Crystal structure of a proton-transfer compound between 2,6-pyridinedicarboxylic acid and N, N′-diethyl-2-amino-6-methyl-4-pyrimidinol. Anal Sci 2005;21:73–4
- Cook D. Vibrational spectra of pyridinium salts. Canadian J Chem 1961;39:2009–24
- Nakamoto K. Infrared and Raman spectra of inorganic and coordination compounds. 5th ed. New York: Wiley-Interscience; 1997
- Geary WJ. The use of conductivity measurements in organic solvents for the characterisation of coordination compounds. Coord Chem Rev 1971;7:81–121
- Yenikaya C, Sarı M, Bülbül M, et al. Synthesis and characterization of two novel proton transfer compounds and their inhibition studies on carbonic anhydrase isoenzymes. J Enzyme Inhib Med Chem 2011;26:104–14