
Abstract
The new histone deacylases inhibitors (HDACi) were synthesized in the class of 5-membered cyclic hydroxamic acids (5-CHA), showing medium size CHA as a new Zn-binding group. New reaction sequence was proposed for the synthesis of 5-membered alkylidene-cyclic-hydroxamic acids starting from butyrolactone. Compound 10c showed low µM activity on HeLa cell extracts. From these results, cyclic hydroxamic acids will be further investigated to find more potent compounds.
Introduction
One of research interest in “epi-drugs” is restoration of epigenetic regulation balance in histone acetylation/deacetylation processes. The acetylation state of lysine residues is controlled by two counteracting responsible enzymes – histone acetyl transferases (HATs) and histone deacylases (HDACs). HDACs catalyse the removal of acetyl groups from the ɛ-amino groups of the lysine side chainsCitation1,Citation2, leading to differentiation, growth arrest and apoptosis of malignant cells, representing a new strategy in cancer therapy. Moreover, HDAC inhibitors (HDACi) have anti-angiogenic, anti-invasive and immuno-modulatory activities in vitro and in vivoCitation3. Clinical trial data demonstrated that HDACi are efficacious as single agents and will be most broadly used in the clinics in combination with other anticancer agentsCitation3,Citation4. In recent years many synthetic hydroxamic acids as HDACi were developed for anticancer therapy. Thus, Vorinostat (SAHA) was the first unselective HDACi licenced by the FDA in October 2006 for the treatment of advanced forms of cutaneous T-cell lymphoma (CTCL)Citation5 and now continued to test in many other clinical trialsCitation6. There are many clinical trials for HDAC inhibitors and epigenetic drugs reviewed with the interpretation of data related to epigenetic measurementsCitation7. Due to the short half life and low oral bioavailability of hydroxamic acids very limited amount of new active compounds went through the clinical studies.
Almost all of the ∼300 Zn-dependant proteases are now known to be connected with tetrahedral Zn atom in the enzyme active centerCitation2. Despite a large number of publications in this field, discovery of new successful zinc binding groups (ZBGs) is very limitedCitation6,Citation7. There are only some ZBGs known for HDACi: bidentate ligands derived from hydroxamic acids, monoacylated phenylenediamines and monodentate ligands – carboxylic acids. The play for medicinal chemists in the development of new inhibitors for Zn-dependant enzymes is around the existing ZBGs. In spite of toxic side effects the main part of HDACi have a hydroxamic acid moiety as the binding group to Zn2+ ion in the active center of enzyme.
Only some recent publications were devoted to the development of the novel ZBGs for HDAC inhibitors. Among them are pyridine-2,6-dicarboxylic acidsCitation8, cysteinesCitation9, thiolsCitation10 and hydrazidesCitation11. Some of the compounds with these new ZBGs bind to HDACs with nM binding capacity. During the last decade, Cohen's groupCitation12–15 has investigated several new ZBGs for Zn-dependant enzymes, particularly matrix metalloproteinases. Among a wide variety of cyclic ketones and lactones they proposed some cyclic hydroxamic and thiohydroxamic acidsCitation13–15.
In the course of our interest in new ZBGs for HDACi and CAi, we were aimed to find new ZBGs and use the closest structural analog of the most exploited hydroxamic acids – medium size cyclic hydroxamic acids. These new structural units for HDACi will be broadened by enlarging the ring size and functionalizing the molecules. With this purpose we have analyzed all the synthetic routes to small and medium sizeCitation16 and mono-macrocyclicCitation17 hydroxamic acids. Current investigation is devoted to 5-membered cyclic hydroxamic acids (5-CHA), synthesis of the simplest representatives and evaluation of their potential as HDAC inhibitors.
Materials and methods
Chemistry
Commercial grade reagents and anhydrous solvents were used as received from suppliers and no attempts were made to purify or to dry these components further. All non-aqueous reactions were performed under dry conditions in the atmosphere of argon unless otherwise specified. Thin layer chromatography (TLC) was performed using silica gel 60 F254 (Merck, Darmstadt, Germany) plates. Visualization of TLC plates was made with short wave (254 nm) UV light. Flash column chromatography was carried out using silica gel 0.035–0.070 mm (Merck). 1H and 13C NMR spectra were obtained on NMR Spectrometers – Varian 200 Mercury (200 MHz) (Ex Varian, Agilent Technologies, Santa Clara, CA) and Varian Mercury plus 400 (400 MHz) (Ex Varian, Agilent Technologies) and are reported in ppm δ values, using tetramethylsilane as an internal reference. Microanalyses were performed using a Carlo Erba – 106 (Milan, Italy) elemental analyzer. Melting points were determined in capillary tubes using a Opti Melt automated melting point system (Stanford Research Systems, Sunnyvale, CA). IR spectra were recorded on Shimadzu IR Prestige 21 (Kyoto, Japan) IR spectrometer.
Trans-cinnamaldehyde, 2-quinolinecarbaldehyde and 3-formyl indole are commercialy available. General procedure for aldehyde chain elongationCitation18 was employed. 9H-Carbazole-2-carbaldehydeCitation19, 9-methyl-9H-carbazole-2-carbaldehydeCitation20 (8h), 3-formyl-indole-1-carboxylic acid tert-butyl esterCitation21 (8a), 3-formyl-N-phenyl-benzenesulfonamideCitation22 (8i), were synthesized according to literature procedures.
1-Benzyloxy-3-bromopyrrolidin-2-one (5a)
A solution of 2,4-dibromobutyroyl chlorideCitation23 (2; 1.32 g; 4.95 mmol) in dry DCM (30 mL) was added at −20 °C to a solution of O-benzylhydroxylamine (3a; 0.58 mL; 5.0 mmol) and triethylamine (0.70 mL; 5.0 mmol) in dry DCM (20 mL). After 30 min, the reaction mixture was extracted with water (20 mL) and the organic phase was dried over Na2SO4. Evaporation of the solvent afforded a colorless oil. Amberlite IRA-400 (in OH− form) was added to a vigorously stirred mixture of this oil in DCM (20 mL) and 1.5 g of 50% NaOH solution. After 4 h at room temperature the mixture was filtered, filtrate washed with water (2 × 5 mL) and the organic phase dried over Na2SO4. Evaporation of the solvent gave colorless oil, which was purified by flash chromatography on silica gel (eluent 20% EtOAc in PE) to afford the title compound as a white solid. Yield 800 mg (60%). 1H-NMR (CDCl3, 200 MHz, δ, ppm): 2.1–2.3 (m, 1H), 2.41–2.63 (m, 1H), 3.13–3.30 (m, 1H), 3.30–3.45 (m, 1H), 4.30–4.40 (m, 1H), 5.03 (s, 2H), 7.18–7.25 (m, 5H). MS (ESI), m/z: 270/272 (M + H)+, calcd for C11H12BrNO2: 270.13.
3-Bromo-1-(tetrahydro-pyran-2-yloxy)pyrrolidin-2-one (5b)
A solution of 2,4-dibromobutanoic acid chloride (2; 2.46 g; 9.3 mmol) in dry DCM (30 mL) at −20 °C was added to a solution of tetrahydropyranylhydroxylamine (3b; 1.11 g; 9.5 mmol)Citation24 and triethylamine (1.33 mL; 9.5 mmol) in dry DCM (30 mL). After 30 min stirring the reaction mixture was extracted with water (20 mL) and the organic phase was dried over Na2SO4. Evaporation of the solvent afforded a colorless oil. Amberlite IRA-400 (4.0 g; in OH− form) was added to a vigorously stirred solution of this oil in a mixture of DCM (50 mL) and 3.30 g of 50% NaOH solution. After 1 h at room temperature the mixture was filtered, the filtrate washed with water (2 × 10 mL), the organic phase separated and dried over Na2SO4. Evaporation of the solvent gave colorless oil, which was purified by flash chromatography on silica gel (eluent 20–30% EtOAc in PE) to afford the title compound as a white solid. Yield 1.43 g (58%). 1H-NMR (CDCl3, 400 MHz, δ, ppm): 1.58–1.72 (m, 3H), 1.75–1.85 (m, 3H), 2.30–2.40 (m, 1H), 3.58–3.72 (m, 2H), 3.75–3.90 (m, 1H), 4.00–4.15 (m, 1H), 4.37–4.43 (m, 1H), 5.17–5.22 (m, 1H). MS (ESI), m/z: 264/266 (M + H)+, calcd for C9H14NO3: 264.12.
General Method A
(N-Benzyloxy-2-oxopyrrolid-3-yl)-triphenyl phosphonium bromide (7a)
A mixture of 1-benzyloxy-3-bromopyrrolidin-one (5a; 540 mg, 2.0 mmol) and triphenylphosphine (6; 524 mg, 5.0 mmol) in dry THF (2 mL) was stirred at 60 °C for 30 h. After cooling to room temperature, the solid was filtered off, washed with THF and dried to afford 645 mg (60%) of the Wittig salt (7a). 1H-NMR (DMSO-D6, 200 MHz, δ, ppm): 2.10–2.42 (m, 2H), 3.15–3.40 (m, 1H, overlapped with H2O), 3.42–3.62 (m, 1H), 4.62 (d, J = 2.6 Hz, 2H), 5.50–5.74 (m, 1H), 7.30–7.40 (m, 5H), 7.70–7.95 (m, 15H). MS (ESI), m/z: 452 (M+), calcd for C28H28 PNO2: 452.53.
[(N-Tetrahydro-pyran-2-yloxy)-2-oxopyrrolid-3-yl]-triphenylphosphonium bromide (7b)
This compound was obtained from 3-bromo-1-(tetrahydropyran-yloxy)pyrrolidin-2-one (5b) and triphenyl phosphine (6) according to the General Method A as a white solid. Yield 35%. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 1.41–1.61 (m, 4H), 1.73–1.78 (m, 2H), 2.21–2.40 (m, 2H), 3.23–3.40 (m, 1H, overlapped with H2O), 3.41–3.56 (m, 1H), 3.56–3.63 (m, 2H), 3.75–3.93 (m, 1H), 5.49–5.64 (m, 1H), 7.72–7.94 (m, 15H). MS (ESI), m/z: 446.5 (M+), calcd for C27H29PNO3: 446.52.
General Method B
(2E,4E)-5-Phenyl-penta-2,4-dienal (8c)
Solution of dioxolane (14a) (541 mg, 2.67 mmol) in a mixture of THF (7 mL) and 10% HCl in water (7 mL) was stirred at room temperature 2 h and analyzed by TLC (solvent system 5% EtOAc in PE). The mixture was diluted with water (50 mL), and extracted with ether (3 × 30 mL). The organic layer was washed with brine, dried over Na2SO4, and evaporated. The residual oil was purified by flash chromatography on silica gel (eluent 5% EtOAc in PE) to afford the title compound as yellow oil. Yield 420 mg (100%). 1H-NMR (CDCl3, 400 MHz, δ, ppm): 6.25 (dd, J = 15.1 and 8.0 Hz, 1H), 6.98–7.01 (m, 1H), 7.00 (s, 1H), 7.22–7.29 (m, overlapped with CHCl3, 1H), 7.32–7.39 (m, 3H), 7.47–7.50 (m, 2H). GC/MS: 158.1 (M+), calcd for C11H10O: 158.20.
(2E,4E,6E)-7-Phenyl-hepta-2,4,6-trienal (8d)
This compound was obtained from dioxolane (14b) according to the General Method B as an orange substance. Yield 91%; 1H-NMR (CDCl3, 400 MHz, δ, ppm): 6.20 (dd, J = 15.2 and 7.8 Hz, 1H), 6.57 (dd, J = 14.0 and 11.1 Hz, 1H), 6.78–6.95 (m, 3H), 7.19 (dd, J = 15.2 and 11.1 Hz, 1H), 7.27–7.32 (m, 1H), 7.33–7.38 (m, 2H), 7.43–7.47 (m, 2H), 9.59 (d, J = 7.8 Hz, 1H). GC/MS: 184.1 (M+), calcd for C13H12O: 184.24.
(E)-3-Quinolin-3-yl-propenal (8e)
This compound was obtained from dioxolane (14c) according to the General Method B as a yellowish substance. Yield 75%. 1H-NMR (CDCl3, 400 MHz, δ, ppm): 6.94 (dd, J = 16.2 and 7.5 Hz, 1H), 7.60–7.65 (m, 1H), 7.65 (d, J = 16.2 Hz, 1H), 7.78–7.83 (m, 1H), 7.89 (d, J = 8.4 Hz, 1H), 8.14 (d, J = 8.4 Hz, 1H), 8.32 (d, J = 2.2 Hz, 1H), 9.12 (d, J = 2.2 Hz, 1H), 9.79 (d, J = 7.4 Hz, 1H). GC/MS: 183.1(M+), calcd for C12H9NO: 183.22.
(2E,4E)-5-Quinolin-3-yl-penta-2,4-dienal (8f)
This compound was obtained from dioxolane (14d) according to the General Method B as a yellowish substance. Yield 78%. 1H-NMR (CDCl3, 400 MHz, δ, ppm): 6.36 (dd, J = 15.0 and 7.8 Hz, 1H), 7.19 (dd, J = 19.1 and 10.1 Hz, 2H), 7.32 (dd, J = 15.0 and 9.9 Hz, 1H), 7.55–7.61 (m, 1H), 7.72 (m, 1H), 7.84 (dd, J = 8.2 and 1.6 Hz, 1H), 8.10 (d, J = 8.6 Hz, 1H), 8.22 (d, J = 2.1 Hz, 1H), 9.07 (d, J = 2.1 Hz, 1H), 9.67 (d, J = 7.8 Hz, 1H). GC/MS: 209.1 (M+). MS (ESI), m/z: 210.2 (M + H+), calcd for C14H11NO: 209.25.
(E)-3-(9-Methyl-9H-carbazol-2-yl)-propenal (8g)
This compound was obtained from dioxolane (14e) according to the General Method B as a yellow substance. Yield 85%. 1H-NMR (CDCl3, 400 MHz, δ, ppm): 6.86 (dd, J = 15.8 and 7.8 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.47 (d, J = 8.2 Hz, 1H), 7.50–7.56 (m, 1H), 7.59 (s, 1H), 7.68 (d, J = 15.8 Hz, 1H), 8.09–8.13 (m, 2H), 9.76 (d, J = 7.8 Hz, 1H). GC/MS: 235.1 (M+). MS (ESI), m/z: 236.1 (M + H)+, calcd for C16H13NO: 235.29.
General Method C
3-[1-Benzyloxy-2-oxo-pyrrolidin-(3E)-ylidenemethyl]-indole-1-carboxylic acid tert-butyl ester (9a)
To a solution of phosphonium salt (7a) (166 mg, 0.31 mmol) and aldehyde (8a) (84 mg, 0.34 mmol) in abs. EtOH (6 mL) was added triethylamine (131 µL, 0.93 mmol). Reaction mixture was heated at 85 °C under argon atmosphere for 4 h with vigorosly stirring, then evaporated under reduced pressure and residual substance purified by flash chromatography on silica gel (eluent 20–25% EtOAc in PE) to afford the title compound as a white substance. Yield 89 mg (67%). 1H-NMR (CDCl3, 400 MHz, δ, ppm): 1.67 (s, 9H), 2.85 (dt, J = 6.4 and 2.7 Hz, 2H), 3.41 (t, J = 6.4 Hz, 2H), 5.10 (s, 2H), 7.28–7.40 (m, 5H), 7.45–7.49 (m, 2H), 7.62 (t, J = 2.8 Hz, 1H), 7.64 (s, 1H), 7.78 (d, J = 2.8 Hz, 1H), 8.08 (d, J = 8.2 Hz, 1H). MS (ESI), m/z: 419.5 (M + H)+, calcd for C25H26N2O4: 418.50.
3-[2-Oxo-1-(tetrahydro-pyran-2-yloxy)-pyrrolidin-(3E)-ylidenemethyl]-indole-1-carboxylic acid tert-butyl ester (9b)
This compound was obtained from phosphonium salt (7b) and aldehyde (8a) according to the General Method C as a white substance. Yield 44%. 1H-NMR (CDCl3, 400 MHz, δ, ppm): 1.60–1.69 (m, 3H), 1.70 (s, 9H), 1.81–1.97 (m, 3H), 3.00 (dt, J = 6.5 and 2.5 Hz, 2H), 3.64–3.71 (m, 1H), 3.77 (q, J = 7.0 Hz, 1H), 3.86–3.93 (m, 1H), 4.13–4.21 (m, 1H), 5.28 (s, 2H), 7.29–7.41 (m, 2H), 7.63 (t, J = 2.8 Hz, 1H), 7.71 (s, 1H), 7.79 (d, J = 7.5 Hz, 1H), 8.10 (d, J = 8.1 Hz, 1H). MS (ESI), m/z: 413.60 (M + H)+, calcd for C23H28N2O5: 412.49.
1-Benzyloxy-3[(E)-3-phenyl-prop-2-en-(E)-ylidene]-pyrrolidin-2-one (9c)
This compound was obtained from phosphonium salt (7a) and cinnamaldehyde (8b) according to the General Method C as a yellowish substance. Yield 84%. 1H-NMR (CDCl3, 400 MHz, δ, ppm): 2.76 (td, J = 6.7 and 2.6 Hz, 2H), 3.34 (t, J = 6.7 Hz, 2H), 5.06 (s, 2H), 6.77 (dd, J = 15.6 and 10.6 Hz, 1H), 6.89 (d, J = 15.6 Hz, 1H), 7.10 (dt, J = 10.6 and 2.6 Hz, 1H), 7.25–7.29 (m, 1H), 7.30–7.39 (m, 5H), 7.41–7.47 (m, 4H). MS (ESI), m/z: 306.4 (M + H)+, calcd for C20H19NO2: 305.50.
3-[(E)-3-Phenyl-prop-2-en-(E)-ylidene]-1-(tetrahydro-pyran-2-yloxy)-pyrrolidin-2-one (9d)
This compound was obtained from phosphonium salt (7b) and cinnamaldehyde (8b) according to the General Method C as a yellowish substance. Yield 49%. 1H-NMR (CDCl3, 400 MHz, δ, ppm): 1.58–1.73 (m, 3H), 1.78–1.95 (m, 3H), 2.90 (dd, J = 6.1 and 2.6 Hz, 1H), 2.92 (dd, J = 7.3 and 2.6 Hz, 1H), 3.62–3.73 (m, 2H), 3.78–3.85 (m, 1H), 4.10–4.18 (m, 1H), 5.23 (t, J = 3.0 Hz, 1H), 6.84 (dd, J = 21.6 and 15.2 Hz, 1H), 6.85 (s, 1H), 7.09–7.14 (m, 1H), 7.26–7.31 (m, 1H), 7.32–7.37 (m, 2H), 7.44–7.48 (m, 2H). MS (ESI), m/z: 300.5 (M + H)+, calcd for C18H21NO3: 299.39.
3-[(2E,4E)-5-Phenyl-penta-2,4-dien(E)-ylidene]-1-(tetrahydropyran-2-yloxy)-pyrrolidin-2-one (9e)
This compound was obtained from phosphonium salt (7b) and aldehyde (8c) according to the General Method C as a yellowish substance. Yield 28%. 1H-NMR (CDCl3, 400 MHz, δ, ppm): 1.56–1.71 (m, 3H), 1.76–1.92 (m, 3H), 2.82 (dd, J = 6.3 and 2.4 Hz, 1H), 2.84 (dd, J = 7.0 and 2.4 Hz, 1H), 3.60–3.69 (m, 2H), 3.74–3.81 (m, 1H), 4.07–4.16 (m, 1H), 5.20 (t, J = 3.0 Hz, 1H), 6.37 (dd, J = 14.8 and 11.9 Hz, 1H), 6.66 (t, J = 11.8 Hz, 1H), 6.67 (d, J = 15.6 Hz, 1H), 6.88 (dd, J = 15.6 and 10.6 Hz, 1H), 7.02 (td, J = 11.8 and 2.4 Hz, 1H), 7.20–7.27 (m, overlapped with CHCl3, 1H), 7.28–7.34 (m, 2H), 7.38–7.43 (m, 2H). MS (ESI), m/z: 326.6 (M + H)+, calcd for C20H23NO3: 325.41.
3-[(2E,4E,6E)-7-Phenyl-hepta-2,4,6-trien-(E)-ylidene]-1-(tetrahydro-pyran-2-yloxy)-pyrrolidin-2-one (9f)
This compound was obtained from phosphonium salt (7b) and aldehyde (8d) according to the General Method C as a yellow substance. Yield 23%. 1H-NMR (CDCl3, 400 MHz, δ, ppm): 1.58–1.74 (m, 3H), 1.76–1.94 (m, 3H), 2.83 (dd, J = 6.0 and 2.4 Hz, 1H), 2.85 (dd, J = 7.0 and 2.4 Hz, 1H), 3.61–3.70 (m, 2H), 3.75–3.82 (m, 1H), 4.09–4.17 (m, 1H), 5.21 (t, J = 3.0 Hz, 1H), 6.32 (dd, J = 14.6 and 11.8 Hz, 1H), 6.44 (dd, J = 14.6 and 11.0 Hz, 1H), 6.55 (dd, J = 14.6 and 10.4 Hz, 1H), 6.61 (dd, J = 14.6 and 11.0 Hz, 1H), 6.64 (d, J = 15.6 Hz, 1H), 6.85 (dd, J = 15.6 and 10.4 Hz, 1H), 7.02 (td, J = 11.8 and 2.6 Hz, 1H), 7.21–7.26 (m, 1H), 7.29–7.35 (m, 2H), 7.39–7.44 (m, 2H). MS (ESI), m/z: 352.6 (M + H)+, calcd for C22H25NO3: 351.45.
3-[(E)-3-Ouinolin-3-yl-prop-2-en-(E)-ylidene]-1-(tetrahydro-pyran-2-yloxy)-pyrrolidin-2-one (9g)
This compound was obtained from phosphonium salt (7b) and aldehyde (8e) according to the General Method C as a yellow substance. Yield 38%. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 1.50–1.63 (m, 3H), 1.65–1.76 (m, 3H), 2.99 (dt, J = 6.6 and 1.9 Hz, 2H), 3.53–3.59 (m, 1H), 3.62–3.76 (m, 2H), 4.04–4.12 (m, 1H), 5.16 (unresolved t, J = 2.3 Hz, 1H), 6.97 (td, J = 11.4 and 2.5 Hz, 1H), 7.16 (d, J = 15.6 Hz, 1H), 7.37 (dd, J = 15.6 and 11.4 Hz, 1H), 7.60–7.63 (m, 1H), 7.70–7.75 (m, 2H), 7.96 (dd, J = 8.5 and 1.2 Hz, 1H), 8.00 (d, J = 8.5 Hz, 1H), 8.49 (d, J = 2.0 Hz, 1H), 9.18 (d, J = 2.0 Hz, 1H). MS (ESI), m/z: 351.1 (M + H)+, calcd for C21H22N2O3: 350.42.
3-[(2E,4E)-5-Ouinolin-3-yl-penta-2,4-dien-(E)-ylidene]-1-tetrahydro-pyran-2-yloxy)-pyrrolidin-2-one (9h)
This compound was obtained from phosphonium salt (7b) and aldehyde (8f) according to the General Method C as a yellow substance. Yield 30%. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 1.49–1.62 (m, 3H), 1.62 (m, 3H), 2.87 (dt, J = 6.4 and 2.1 Hz, 1H), 3.51–3.58 (m, 1H), 3.59–3.72 (m, 2H), 4.00–4.10 (m, 1H), 5.14 (unres t, J = 2.3 Hz, 1H), 6.63 (dd, J = 14.7 and 11.7 Hz, 1H), 6.84–6.92 (m, 2H), 6.96 (d, J = 15.7 Hz, 1H), 7.41 (dd, J = 15.7 and 11.1 Hz, 1H), 7.51–7.54 (m, 2H), 7.69–7.74 (m, 1H), 7.91–8.00 (m, 2H), 8.40 (d, J = 2.0 Hz, 1H), 9.12 (d, J = 2.0 Hz, 1H). MS (ESI), m/z: 377.1 (M + H)+, calcd for C23H24N2O3: 376.46.
3-[(E)-3-(9-Methyl-9H-carbazol-2-yl)-prop-2-en-(E)-ylidene]-1-tetrahydro-pyran-2-yloxy)-pyrrolidin-2-one (9i)
This compound was obtained from phosphonium salt (7b) and aldehyde (8g) according to the General Method D as a yellowish substance. Yield 24%. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 1.50–1.64 (m, 3H), 1.64–1.77 (m, 3H), 2.97 (dd, J = 6.7 and 2.2 Hz, 1H), 2.99 (dd, J = 7.1 and 2.2 Hz, 1H), 3.53–3.60 (m, 1H), 3.62–3.75 (m, 2H), 3.90 (s, 3H), 4.03–4.13 (m, 1H), 5.15 (unres. t, J = 2.2 Hz, 1H), 6.97 (dt, J = 10.3 and 2.2 Hz, 1H), 7.12–7.25 (m, 3H), 7.43–7.48 (m, 2H), 7.58 (d, J = 8.3 Hz, 1H), 7.82 (s, 1H), 8.12 (d, J = 3.2 Hz, 1H), 8.14 (d, J = 2.4 Hz, 1H). MS (ESI), m/z: 403.2 (M + H)+, calcd for C25H26N2O3: 402.5
3-[1-(9-Methyl-9H-carbazol-2-yl)-meth-(E)-ylidene]-1-tetrahydro-pyran-2-yloxy)-pyrrolidin-2-one (9j)
This compound was obtained from phosphonium salt (7b) and aldehyde (8h) according to the General Method C as a white substance. Yield 45%. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 1.52–1.64 (m, 3H), 1.69–1.79 (m, 3H), 3.22 (dd, J = 7.2 and 2.4 Hz, 1H), 3.24 (dd, J = 6.3 and 2.4 Hz, 1H), 3.55–3.61 (m, 1H), 3.66–3.80 (m, 2H), 4.07–4.15 (m, 1H), 5.20 (unres. t, J = 2.5 Hz, 1H), 7.19–7.24 (m, 1H), 7.38–7.41 (m, 1H), 7.60 (d, J = 8.2 Hz, 1H), 7.76 (s, 1H), 8.17 (d, J = 7.8 Hz, 1H), 8.19 (d, J = 8.2 Hz, 1H). MS (ESI), m/z: 377.1 (M + H)+, calcd for C23H24N2O3: 376.5
3-[2-Oxo-1-(tetrahydro-pyran-2-yloxy)-pyrrolidin-(3E)-ylidenemethyl]-N-phenyl-benzenesulfonamide (9k)
This compound was obtained from phosphonium salt (7b) and aldehyde (8i) according to the General Method C as a white substance. Yield 32%. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 1.51–1.64 (m, 3H), 1.65–1.77 (m, 3H), 2.90–2.96 (m, 2H), 3.52–3.59 (m, 1H), 3.60–3.68 (m, 1H), 3.69–3.76 (m, 1H), 4.04–4.12 (m, 1H), 5.18 (s, 1H), 7.02 (t, J = 7.2 Hz, 1H), 7.10 (dd, J = 8.6 and 1.1 Hz, 2H), 7.15 (t, J = 2.7 Hz, 1H), 7.23 (t, J = 7.8 Hz, 2H), 7.60 (t, J = 7.8 Hz, 1H), 7.71 (d, J = 8.1 Hz 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.81 (s, 1H), 10.32 (s, 1H). MS (ESI), m/z: 429.2 (M + H)+, calcd for C22H24N2O5S: 428.5.
1-Hydroxy-3-[1-(1H-indol-3-yl)-meth-(E)-ylidene]-pyrrolidin-2-one (10a)
To a solution of the protected cyclic hydroxamic acid (9a) (56 mg, 0.125 mmol) in 5 mL dry DCM under argon atmosphere at 0 °C was added to 2.5 mL trifluoracetic acid. The mixture was stirred at room temperature 2 h and analyzed by TLC in solvent system. Then 10% EtOH in DCM (to ascertain that reaction had ended) was evaporated under reduced pressure and triturated with absolute ether. The precipitate formed was filtered off and dried in vacuo. The title compound was obtained as a slightly brown substance. Yield 23.4 mg (82%); m.p.: 245–250 °C. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 2.90 (dt, J = 6.4 and 2.4 Hz, 2H), 3.59 (t, J = 6.4 Hz, 2H), 7.11 (t, J = 7.5 Hz, 1H), 7.17 (t, J = 7.5 Hz, 1H), 7.40 (unres. t, J = 2.4 Hz, 1H), 7.43 (d, J = 8.0 Hz, 1H), 7.59 (d, J = 2.4 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 9.80 (s, 1H), 11.66 (s, 1H). MS (ESI), m/z: 229 (M + H)+, calcd for C13H12N2O2: 228.25. Elemental analysis: Found, %: C 61.86, H 4.76, N 10.14. Calcd for C13 H12 N2O2: C 62.03, H 6.27, N 9.91
General Method E
1-Hydroxy-3-[(E)-3-phenyl-prop-2-en-(E)-ylidene]-pyrro-lidin-2-one (10b)
Protected hydroxamic acid (9d) (89 mg, 0.297 mmol) was dissolved in a mixture of 2.0 mL THF, 4.0 mL AcOH and 1.0 mL water and heated at 60 °C for 5 h. The process was monitored by TLC in solvent system 5% EtOH in DCM. Reaction mixture was evaporated under reduced pressure, and the residual product purified by flash chromatography on silica gel (eluent 5% EtOH in DCM), to afford the title substance. Yield 50 mg (78%); m.p.: 190–195 °C. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 2.90 (dt, J = 6.2 and 2.2 Hz, 2H); 3.55 (t, J = 6.2 Hz, 2H); 6.82 (dt, J = 11.2 and 2.3 Hz, 1H); 6.91 (d, J = 15.6 Hz, 1H); 7.03 (dd, J = 15.6 and 11.2 Hz, 1H); 7.28 (t, J = 7.2 Hz, 1H); 7.36 (t, J = 7.4 Hz, 2H); 7.56 (d, J = 7.4 Hz, 2H), 9.95 (s, 1H). MS (ESI), m/z: 216 (M + H)+, calcd for C13H13NO2: 215.25. Elemental analysis. Found, %: C 70.12, H 6.32, N 6.14. Calcd for C13H13NO2: C 70.25, H 5.90, N 6.30.
1-Hydroxy-3-[(2E,4E)-5-phenyl-penta-2,4-dien-(E)-ylidene]-pyrrolidin-2-one (10c)
This compound was obtained from protected hydroxamic acid (9e) according to the General Method D. Yield (82%); m.p.: 222–226 °C. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 2.81 (dt, J = 6.5 and 2.4 Hz, 2H), 3.53 (t, J = 6.5 Hz, 2H), 6.53 (dd, J = 14.5 and 11.8 Hz, 1H), 6.70–6.80 (m, 3H), 7.09 (dd, J = 15.6 and 10.8 Hz, 1H), 7.22–7.28 (m, 1H), 7.34 (t, J = 7.6 Hz, 2H), 7.50 (d, J = 7.6 Hz, 2H), 9.95 (br s, 1H). MS (ESI), m/z: 242 (M + H)+, calcd for C15H15NO2: 241.29. Elemental analysis. Found, %: C 73.44, H 6.00, N 5.66. Calcd for C15H15NO2: C 73.83, H 6.28, N 5.66.
1-Hydroxy-3-[(2E,4E,6E)-7-phenyl-hepta-2,4,6-trien-(E)-ylidene]-pyrrolidin-2-one (10d)
This compound was obtained from protected hydroxamic acid (9f) according to the General Method D. Yield 69%; m.p.: 209–214 °C. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 2.80 (dt, J = 6.5 and 2.3 Hz, 2H); 3.52 (t, J = 6.5 Hz, 2H); 6.45 (dd, J = 14.4 and 12.0 Hz, 1H); 6.51–6.62 (m, 2H); 6.63–6.78 (m, 3H); 7.02 (dd, J = 15.5 and 9.8 Hz, 1H); 7.24 (t, J = 7.2 Hz, 1H); 7.34 (t, J = 7.7 Hz, 2H); 7.49 (d, J = 7.4 Hz, 2H), 9.94 (br s, 1H). MS (ESI), m/z: 268 (M + H)+, calcd for C17H17NO2: 267.33. Elemental analysis. Found, %: C 70.87, H 6.41, N 4.80. Calcd for C17H17NO2: C 70.82, H 5.94, N 4.86.
1-Hydroxy-3-[(E)-3-quinolin-3-yl-prop-2-en-(E)-ylidene]-pyrrolidin-2-one (10e)
This compound was obtained from protected hydroxamic acid (9g) according to the General Method D. Yield (69%); m.p.: 234–237 °C. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 2.94 (dt, J = 6.2 and 2.3 Hz, 2H), 3.55 (t, J = 6.2 Hz, 2H), 6.84–6.90 (m, 1H), 7.08 (d, J = 15.7 Hz, 1H), 7.33 (dd, J = 15.7 and 11.3 Hz, 1H), 7.55–7.60 (m, 1H), 7.68–7.73 (m, 1H), 7.91–7.99 (m, 2H), 8.45 (d, J = 1.8 Hz, 1H), 9.15 (d, J = 2.3 Hz, 1H), 9.99 (br s, 1H). MS (ESI), m/z: 267 (M + H)+, calcd for C16H14N2O2: 266.30. Elemental analysis: Found, %: C 70.87, H 4.11, N 10.21. Calcd for C16H14N2O2: C 70.94, H 5.21, N 10.34.
1-Hydroxy-3-[(2E,4E)-5-quinolin-3-yl-penta-2,4-dien-(E)-ylidene]-pyrrolidin-2-one (10f)
This compound was obtained from protected hydroxamic acid (9h) according to the General Method D. Yield 63%; m.p.: 244–245 °C. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 2.82 (dt, J = 5.7 and 2.3 Hz, 2H), 3.52 (t, J = 6.3 Hz, 2H), 6.53–6.64 (m, 1H), 6.75–6.95 (m, 1H), 7.38 (dd, J = 15.7 and 10.7 Hz, 1H), 7.53–7.60 (m, 1H), 7.65–7.72 (m, 1H), 7.86–7.98 (m, 2H), 8.36 (d, J = 1.7 Hz, 1H), 9.09 (d, J = 2.2 Hz, 1H), 9.96 (br s, 1H). MS (ESI), m/z: 293 (M + H)+, calcd for C18H16N2O2: 292.34. Elemental analysis. Found, %: C 72.86, H 4.45, N 9.27. Calcd for C18H16N2O2: C 72.86, H 4.43, N 9.44.
1-Hydroxy-3-[(E)-3-(9-methyl-9H-carbazol-2-yl)- prop-2-en-(E)-ylidene]-pyrrolidin-2-one (10g)
This compound was obtained from protected hydroxamic acid (9i) according to the General Method D. Yield 92%; m.p.: 268–270 °C. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 2.89–2.99 (m, 2H), 3.55 (t, J = 6.3 Hz, 2H), 3.87 (s, 1H), 6.86 (dt, J = 10.9 and 2.6 Hz, 1H), 7.03–7.21 (m, 3H), 7.43 (t, J = 8.3 Hz, 2H), 7.55 (d, J = 8.3 Hz, 1H), 7.77 (s, 1H), 8.07–8.12 (m, 2H), 9.92 (s, 1H). MS (ESI), m/z: 319 (M + H)+, calcd for C20H18N2O2: 318.38. Elemental analysis. Found, %: C 74.95, H 4.41, N 8.56. Calcd for C20H18N2O2: C 74.98, H 5.66, N 8.74.
1-Hydroxy-3-[(E)-3-(9-methyl-9H-carbazol-2-yl)-meth-(E)-ylidene]-pyrrolidin-2-one (10h)
This compound was obtained from protected hydroxamic acid (9j) according to the General Method E. Yield 71%; m.p.: 258–262 °C. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 3.19 (dt, J = 6.3 and 2.6 Hz, 2H), 3.60 (t, J = 6.3 Hz, 2H), 3.87 (s, 3H), 7.18 (t, J = 7.8 Hz, 1H), 7.30 (t, J = 2.7 Hz, 1H), 7.35 (unres. dd, J = 8.2 and 1.0 Hz, 1H), 7.43–7.48 (m, 1H), 7.57 (d, J = 8.2 Hz, 1H), 7.71 (s, 1H), 8.12–8.17 (m, 2H), 9.97 (br s, 1H). MS (ESI), m/z: 293 (M + H)+, calcd for C18H16N2O2: 292.34. Elemental analysis. Found, %: C 73.44, H 4.40, N 9.35. Calcd for C18H16N2O2: C 73.40, H 5.47, N 9.51.
3-[1-Hydroxy-2-oxo-pyrrolidin-(3E)-ylidenemethyl]-N-phenyl-benzenesulfonamide (10i)
This compound was obtained from protected hydroxamic acid (9k) according to the General Method E. Yield 67%; m.p. 192–195 °C. 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 2.90 (dt, J = 6.0 and 2.1 Hz, 2H), 3.57 (t, J = 6.0 Hz, 2H), 7.00–7.05 (m, 1H), 6.51–6.62 (m, 1H), 7.06–7.12 (m, 3H), 7.19–7.26 (m, 2H), 7.59 (t, J = 7.8 Hz, 1H), 7.67–7.76 (m, 2H), 7.79 (s, 1H), 10.11 (br s, 1H), 10.32 (br s, 1H). MS (ESI), m/z: 345 (M + H)+, 386 (M + MeCN)+, calcd for C17H16N2O2S: 344.39. Elemental analysis. Found, %: C 58.65, H 4.80, N 7.76. Calcd for C17H16N2O2S: C 58.78, H 4.64, N 8.06.
3-(2,3-Dihydro-1H-indol-3-ylmethyl)-1-hydroxy-pyrrolidin-2-one (11a)
To a solution of the protected hydroxamic acid (9a) (130 mg, 0.31 mmol) in 50 mL of EtOH was added to 300 mg 10% Pd/C and mixture stirred at room temperature with hydrogen bubbling. Reaction mixture was analyzed in solvent system 10% EtOAc in DCM. After the reaction was complete, the mixture was filtered through celite and the filtrate was evaporated. Residual oil was dissolved in dry DCM (5 mL) at 0 °C and 2.5 mL of trifluoroacetic acid added. Reaction was monitored by TLC, solvent system 5% EtOH in DCM. After disappearance of the starting substance spot, the mixture was evaporated to dryness and the residual product purified by flash chromatography (eluent 5–10% EtOH in DCM) to afford the title compound as the TFA salt. Treating with anionite IRA-400 in Cl form the substance was turned to hydrogen chloride. Yield of a hygroscopic foam was 33.7 mg (40%). 1H-NMR (DMSO-D6, 400 MHz, δ, ppm): 1.43–1.87 (m, 3H), 2.09–2.24 (m, 1H), 2.30–2.46 (m, 1H), 3.15–3.24 (m, 1H), 3.30–3.48 (m, 3H), 3.61–3.70 (m, 1H), 6.78–6.87 (m, 2H), 7.04–7.11 (m, 1H), 7.15–7.21 (m, 1H), 9.56 (br s, 1H). MS (ESI), m/z: 233.1 (M + H)+ calcd for C13H16N2O2: 232.3. Elemental analysis. Found, %: C 56.90, H 6.55, N 9.36. Calcd for C13H16N2O2.HCl: C 56.98, H 6.25, N 10.22.
1-Hydroxy-3-phenethyl-pyrrolidin-2-one (11b)
To a solution of protected hydroxamic acid (9c) (45.7 mg, 0.15 mmol) in 40 mL of EtOH was added 200 mg 10% Pd/C and the mixture stirred with hydrogen bubbling. Reaction mixture was analyzed in solvent systems 10% EtOAc in DCM and 3% MeOH in DCM. After disappearance of the starting spot the mixture was filtered through celite, filtrate evaporated to dryness and the was residue dried in vacuo. Yield 20.0 mg (60%) of an off-white powder; m.p. 113–115 °C. 1H-NMR (CDCl3, 400 MHz, δ, ppm): 1.35–1.47 (m, 1H), 1.59–1.72 (m, 3H), 1.78–1.88 (m, 1H), 2.11–2.21 (m, 1H), 2.38–2.48 (m, 1H), 2.55–2.69 (m, 2H), 3.51–3.60 (m, 2H), 7.13–7.21 (m, 3H), 7.24–7.30 (m, overlapped with CHCl3, 2H), 9.0–10.5 (br s, 1H). MS (ESI), m/z: 220 (M + H)+, calcd for C13H17NO2: 219.3. Elemental analysis. Found, %: C 70.03, H 7.79, N 6.12. Calcd for C13H17NO2: C 70.24, H 7.71, N 6.30.
General Method B
[(1E,3E)-4-Phenyl-buta-1,3-dienyl]-1,3-dioxolane (14a)
To a mixture of cinnamaldehyde (1.03 g, 8.16 mmol) and (1,3-dioxolan-yl)-tributyl-phosphonium bromideCitation18,Citation25 (13) in dry DMF (20 mL) at 90 °C under argon atmosphere 1M NaOEt solution in abs EtOH was added (12.24 mL, 12.24 mmol). Reaction mixture was heated for 16 h at the same temperature, then cooled and diluted with water (200 mL), and extracted with EtOAc (3 × 50 mL). The organic layer was washed with brine, dried over Na2SO4 and evaporated. Residual substance was purified by flash chromatography on silica gel (eluent 5–10% EtOAc in PE) to afford 14a as a yellow substance. Yield 555 mg (34%). 1H-NMR (CDCl3, 400 MHz, δ, ppm): 3.87–4.07 (m, 4H), 5.34 (d, J = 6.0 Hz, 1H), 5.75 (dd, J = 15.2 and 6.0 Hz, 1H), 6.55 (dd, J = 15.2 and 10.6 Hz, 1H), 6.62 (d, J = 15.6 Hz, 1H), 7.20–7.26 (m, 1H, overlapped with CHCl3), 7.27–7.34 (m, 2H), 7.36–7.41 (m, 2H). GC/MS: 202.1 (M+), calcd for C13 H14O2: 202.26.
[(1E,3E,5E)-6-Phenyl-hexa-1,3,5-trienyl]-1,3-dioxolane (14b)
This compound was obtained from aldehyde (8c) according to the General Method B as a yellowish substance. Yield 29%. 1H-NMR (CDCl3, 400 MHz, δ, ppm): 3.86–4.06 (m, 4H), 5.32 (d, J = 6.1 Hz, 1H), 5.68 (dd, J = 15.1 and 6.1 Hz, 1H), 6.34 (dd, J = 14.5 and 10.3 Hz, 1H), 6.40–6.52 (m, 2H), 6.58 (d, J = 15.8 Hz, 1H), 6.80 (dd, J = 15.8 and 10.6 Hz, 1H), 7.21 (t, J = 7.1 Hz, 1H), 7.27–7.33 (m, 2H), 7.35–7.41 (m, 2H), GC/MS: 228 (M+), calcd For C15H16O: 228.3.
3-[(E)-2-1,3-Dioxolan-2-yl-vinyl]-quinoline (14c)
This compound was obtained from 2-quinolinealdehyde according to the General Method B as a white substance. Yield 29%. 1H-NMR (CDCl3, 400 MHz, δ, ppm): 3.94–4.04 (m, 2H), 4.04–4.11 (m, 2H), 5.49 (d, J = 5.8 Hz, 1H), 6.39 (dd, J = 16.2 and 5.8 Hz, 1H), 6.93 (d, J = 16.2 Hz, 1H), 7.50–7.55 (m, 1H), 7.65–7.70 (m, 1H), 7.79 (dd, J = 8.2 and 1.2 Hz, 1H), 8.06 (d, J = 8.7 Hz, 1H), 8.08 (d, J = 2.3 Hz, 1H), 9.00 (d, J = 2.3 Hz, 1H). GC/MS: 227.1 (M+), calcd for C14H13NO2: 227.3.
3-[(1E,3E)-4-1,3-Dioxolan-2-yl-buta-1,3-dienyl]-quinoline (14d)
This compound was obtained from aldehyde (8e) as a yellowish substance according to the General Method B. Yield 34%. 1H-NMR (CDCl3, 400 MHz, δ, ppm): 3.91–4.00 (m, 2H), 4.00–4.10 (m, 2H), 5.39 (d, J = 5.9 Hz, 1H), 5.88 (dd, J = 15.3 and 5.9 Hz, 1H), 6.63 (dd, J = 15.3 and 10.6 Hz, 1H), 7.51–7.56 (m, 1H), 7.65–7.70 (m, 1H), 7.79 (dd, J = 8.2 and 1.1 Hz, 1H), 8.05–8.09 (m, 2H), 9.00 (d, J = 2.1 Hz, 1H). GC/MS: 253.2(M+), calcd for C16H15NO2: 253.3.
2-[(E)-2-1,3-Dioxolan-2-yl-vinyl]-9-methyl-9H-carbazole (14e)
This compound was obtained from 9-methyl-9H-carbazole-2-carbaldehyde (8h) according to the General Method B as a white substance. Yield 39%. 1H-NMR (CDCl3, 400 MHz, δ, ppm): 3.85 (s, 3H), 3.97–4.05 (m, 2H), 4.05–4.13 (m, 2H), 5.50 (d, J = 6.0 Hz, 1H), 6.29 (dd, J = 16.1 and 6.3 Hz, 1H), 6.99 (d, J = 16.1 Hz, 1H), 7.20–7.25 (m, 1H), 7.33 (dd, J = 8.1 and 1.5 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.44 (s, 1H), 7.45–7.50 (m, 1H), 8.03 (d, J = 8.1 Hz, 1H), 8.06 (dt, J = 7.7 and 1.0 Hz, 1H). GC/MS: 279.1(M+). MS (ESI), m/z: 280.2 (M + H)+, calcd for C18H17NO2: 279.3.
HDAC activity evaluation
HDAC activity in HeLa nuclear extracts
5-Membered cyclic hydroxamic acids 10a–d and 11a were assayed for the inhibitory activities against HDAC using BioVision “HDAC inhibitor drug screening kit” according to the manufacturer’s protocol. Briefly, chemical compounds were dissolved into DMSO in concentration 10 mM and diluted to 2× the desired test concentration with double distilled water (ddH2O). 50 µL of diluted compounds as well as positive control without inhibitors, were added to the wells of 96-well plate. For negative control, 48 µL of ddH2O and 2 µL of Trichostatin A (TSA) were mixed in the well. For each well 50 µL of reaction mixture containing 10 µL 10X HDAC Assay Buffer was also added, 2 µL HeLa nuclear extract, 5 µL HDAC fluorescent substrate Boc-Lys(Ac)-AMC and 33 µL ddH2O. Plate was incubated for 30 min at 37 °C. The reaction was stopped by adding 10 µL of Lysine Developer and mixed well. Plate was incubated for 30 min at 37 °C. The relative fluorescent units (RFU) were measured in fluorimeter Tecan (Infinite M1000) with Ex./Em. = 350 nm/460 nm. Set of the RFUs and positive control with 100% HDAC activity, was used to calculate the relative HDAC inhibitory activity of chemical compounds as follow: activity remain with compounds = (RFU of compound/RFU of Positive Control) × 100%.
IC50 calculation
Detection of the IC50 (HDACi) values was revealed after measurement of RFU for at least five concentrations of compounds in a range from 1 to 40 µM. Calculation of IC50 was done with GraphPad Prism 5.03 software (GraphPad Software, La Jolla, CA) on the basis of detected RFUs. All data are the mean of three independent experiments.
Results and discussion
Chemistry
Synthesis of compounds 10a–d and 11a is described in Scheme 1. Starting materials - N-protected 3-bromopyrrolidin-2-onesCitation24 were prepared according to the literature procedure.
Scheme 1. Synthesis of 5-CHA 10 and 11.
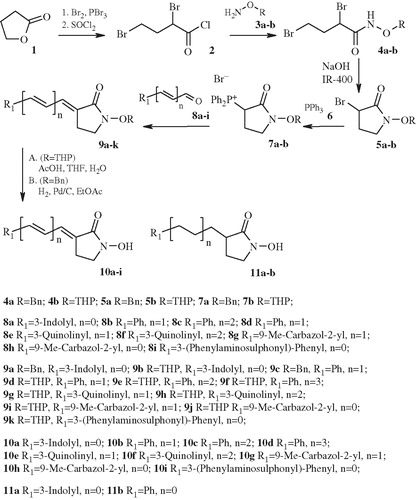
2,4-Dibromobutyryl chloride (2) was prepared from butyrolactone (1) in a two-step process and coupled with O-protected hydroxylamines 3a–b to give O-protected derivatives 4a–b. They were cyclized to cyclic hydroxamic acid (5a–b) with NaOH in the presence of strong anionite IRA-400 in OH− form. Introduction of the side chain was started in the two-step process from bromides 5a–b. Exchange of bromide functionality was performed with triphenylphosphine (6) affording phosphonium bromides 7a–b. Side chain elongation was performed from appropriate aldehydes (12) and tributyl phosphonium bromide (13) prepared after literature precedent depicted in Scheme 2Citation18.
Scheme 2. Unsaturated aldehyde 8c–g synthesis.
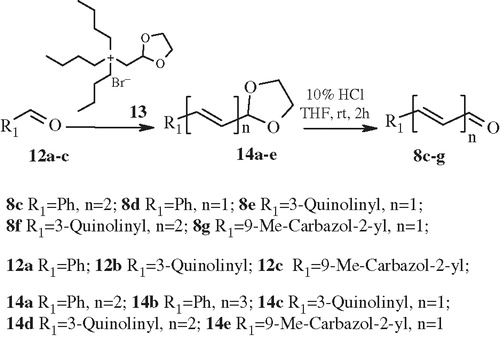
Acetals 14a–e prepared by this methodology were deprotected to give the target aldehydes 8c–g. Further reaction of them with earlier prepared phosphonium bromides 7a–b afforded protected cyclic hydroxamic acids 9a–k. Deprotection of the O-THP protecting group with AcOH in THF-H2O mixture yielded title compounds 10a–i. O-Benzyl group in compounds 9 was cleaved in Pd catalyzed reduction conditions and gave cyclic hydroxamic acids 11a–b.
Biological activity
HDAC activity with cyclic hydroxamic acids
Cyclic hydroxamic acids 10a–d and 11a were tested on BioVision HDAC Inhibitor Drug Screening Kit. Inhibition of histone deacetylases (HDACs) has been implicated to modulate transcription and to induce apoptosis or differentiation in cancer cells. We have used HDAC Inhibitor Drug Screening Kit, which provides a fast, fluorescence-based method that eliminates radioactivity, extractions or chromatography, as used in traditional assays. The new procedure requires only two easy steps, both performed on the same microtiter plate. First, tested substances are mixed with HeLa Nuclear Extract and HDAC fluorometric substrate, which comprises an acetylated lysine side chain. Deacetylation of the substrate sensitizes the substrate, so that, in the second step, treatment with the Lysine Developer produces a fluorophore. The fluorophore was analyzed using a fluorometer.
Screening of cyclic hydroxamic acids 10a–d and 11a for HDAC activity modulation has been done and IC50 were calculated ().
Table 1. IC50 values (µM) for the HDAC inhibitory activity of 5-CHA.
A comparison between IC50 values of HDAC inhibition revealed, the compound 10c was found to be the most efficient HDAC inhibitor (IC50 = 7.4 µM) between tested compounds, while other cyclic hydroxamic acids such as 10b (IC50 = 33.5 µM) and 10d (IC50 = 40 µM) were less potent in the same assay. The tendency in the increase of HDAC inhibition activity for classical hydroxamic acids with linkers reaching the higher activity with a distance of 7–9 bonds between ZBG and aromatic substituent was partially observed also in cyclic hydroxamic acids. However, the best binding was observed for compound 10c (7 bonds beytween Ph group and carbonyl in CHA moiety). At the same time distances in 5 bonds and 9 bonds, correspondingly for compounds 10b and 10d showed weaker activity. The compound 10a did not inhibit HDAC activity, while 11a inhibited about 10% of HDAC activity only in the range from 5 to 40 µM in a concentration independent manner. These data showed the necessity of the spacer in the HDACi molecules.
Thus, inhibition of HDAC activity was showed for the first time for the compounds from the class of cyclic hydroxamic acids 10b–d. Moreover, the lengths of the spacer between cyclic hydroxamic acid function and aromatic phenyl ring is important for the development of new zinc binding groups (ZBGs) for histone deacetylases inhibition.
Conclusions
In conclusion, the present work reports on the search of new ZBG for the development of HDAC inhibitors. We have designed and synthesized series of 5-CHA as HDAC inhibitors. The new derivatives have been tested in nuclear extracts from HeLa cell cultures using tert-butoxycarbonyl-lysine (acetylated)-4-amino-7-methylcoumarin (Boc-Lys(Ac)-AMC) as the substrate. Some of the reported derivatives showed low μM activity and they are good examples for starting fragment-based structure design.
Thus, we have developed – cyclic hydroxamic acids (CHA) as a new ZBG for HDAC inhibition. Although synthesized compounds are not as effective as known hydroxamic acids, their some structure-activity profile shows some kind of selectivity, and make these derivatives interesting for more detailed SAR studies. Further investigations on design and synthesis of other CHAs and their development as metallo-enzyme inhibitors are under the way.
Declaration of interest
The authors declare that there are no conflicts of interest.
This study was supported by the Latvian National Research Programme 2010–2013. “BIOMEDICINE” and COST Action TD0905 “Epigenetics: from Bench to Bedside”.
References
- Sippl W, Jung M. Epigenetic targets in drug discovery. In: Methods and principles in medicinal chemistry. Vol. 42. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co., KGaA; 2009
- Supuran CT, Winum J-Y, eds. Drug design of zinc-enzyme inhibitors. Hoboken: John Wiley & Sons; 2009
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006;5:769–84
- Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res 2007;5:981–9
- Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol 2007;25:84–90
- Mai A, Altucci L. Epi-drugs to fight cancer: from chemistry to cancer treatment, the road ahead. Int J Biochem Cell Biol 2009;41:199–213
- Martinet N, Bertrand P. Interpreting clinical assays for histone deacetylase inhibitors. Cancer Manage Res 2011;3:117–41
- Valente S, Conte M, Tardugno M, et al. Developing novel non-hydroxamate histone deacetylase inhibitors: the chelidamic warhead. Med Chem Comm 2012;3:298–304
- Butler KV, He R, McLaughlin K, et al. Stereoselective HDAC inhibition from cysteine-derived zinc-binding groups. Chem Med Chem 2009;4:1292–301
- Cole KE, Dowling DP, Boone MA, et al. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J Am Chem Soc 2011;33:12474–7
- Suzuki T, Nagano Y, Matsuura A, et al. Novel histone deacetylase inhibitors: design, synthesis, enzyme inhibition, and binding mode study of SAHA-based non-hydroxamates. Bioorg Med Chem Lett 2003;13:4321–6
- Day JA, Cohen SM. Investigating the selectivity of metalloenzyme inhibitors. J Med Chem 2013;56:7997–8007
- Jacobsen JA, Fullagar JL, Miller MT, Cohen SM. Identifying chelators for metalloprotein inhibitors using a fragment-based approach. J Med Chem 2011;54:591–602
- Jacobsen FE, Breece RM, Myers WK, et al. Model complexes of cobalt-substituted matrix metalloproteinases: tools for inhibitor design. Inorg Chem 2006;45:7306–15
- Puerta DT, Lewis JA, Cohen SM. New beginnings for matrix metalloprotei-nase inhibitors: identification of high-affinity zinc-binding groups. J Am Chem Soc 2004;126:8388–9
- Trapencieris P, Strazdina J, Bertrand P. Synthesis of small and medium size monocyclic hydroxamic acids. Chem Heterocycl Comp (Engl. Ed.) 2012;48:833–55
- Borovika D, Bertrand P, Trapencieris P. Synthesis of macro-monocyclic hydroxamic acids. Chem Heterocycl Comp (Rus. Ed.) 2013;49:1685–705
- Spangler CW, McCoy RK. Preparation of conjugated aromatic polyenals by Wittig oxopropenylation. Synth Comm 1988;18:51–9
- Freeman AW, Urvoy M, Criswell ME. Triphenylphosphine-mediated reductive cyclization of 2-nitrobiphenyls: a practical and convenient synthesis of carbazoles. J Org Chem 2005;70:5014–19
- Molina P, Fresneda PM, Almendros P. Fused carbazoles by tandem aza Wittig/electrocyclic ring closure. Preparation of 6H-pyrido[4,3-b] carbazole, 11H-pyrido[4,3-a]carbazole and 11H-pyrido[3,4-a]carbazole derivatives. Tetrahedron 1993;49:1223–36
- Camp JE, Craig D, Funai K, White AJP. Decarboxylative Claisen rearrangement reactions: synthesis and reactivity of alkylidene-substituted indolines. Org Biomol Chem 2011;9:7904–12
- Finn PW, Bandara M, Butcher C, et al. Novel sulfonamide derivatives as inhibitors of histone deacetylase. Helv Chim Acta 2005;88:1630–57
- Ikuta H, Shirota H, Kobayashi S, et al. Synthesis and antiinflammatory activities of 3-(3,5-di-tert-butyl-4-hydroxybenzylidene)pyrrolidin-2-ones. J Med Chem 1987;30:1995–8
- Heinze-Krauss I, Angehrn P, et al. Synthesis and structure-activity relationship of (lactamylvinyl)cephalosporins exhibiting activity against staphylococci, pneumococci, and enterococci. J Med Chem 1996;39:1864–71
- Li Y, Josowicz M, Tolbert LM. Diferrocenyl molecular wires. The role of heteroatom linkers. J Am Chem Soc 2010;132:10374–82