
Abstract
As a part of trials to target ROS1 kinase with potential inhibitors, a novel series of pyrimidin-4-yl-ethanol and ethanone derivatives (4a–f, 5a–f, 6a–f and 7a–f) have been designed based on previously discovered lead compounds KIST301072 and KIST301080, and synthesized on 4–5 steps according to compounds. The structures of the newly synthesized compounds have been confirmed on 1H-NMR, 13C-NMR and IR. Most of the tested compounds showed ROS1 kinase inhibitory activity in micromolar range.
Introduction
Protein kinases are defined by their ability to catalyze the transfer of the terminal phosphate of ATP to substrates that usually contain a serine, threonine or tyrosine residueCitation1. All protein kinases, thus far characterized with regard to substrate specificity, fall within one of three classes, tyrosine kinases, serine/threonine kinases and dual specificity protein kinasesCitation2. Tyrosine kinases are further divided into two subgroups; receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinasesCitation3. The protein kinase family plays an important role in regulating various cellular processes such as cell proliferation, cell death, cell cycle progression, differentiation and cell survival. Therefore, it is not surprising that the deregulation of many kinases is usually directly linked to cancer developmentCitation4. Hence, protein kinase has become the most intensively pursued anticancer drug targetCitation5,Citation6. It is also worthy to mention that, since protein kinases are quite selective in their actions, they represent a wide spectrum of specific and exploitable targets for drugsCitation7. Currently, more than 14 small molecule inhibitors have become available on the market for cancer treatments since the first kinase inhibitor (imatinib) was approved by FDA in 2001 for chronic myeloid leukaemiaCitation8.
There are 58 RTKs in humans that belong to 20 distinct families, and ROS1 kinase is one of themCitation9,Citation10. The ectopic expression, as well as the production of variable mutant forms of ROS1 kinase, has been reported in a number of cancers, such as glioblastoma multiforme, cholangiocarcinoma, gastric adenocarcinoma and non-small cell lung cancer, suggesting a role for ROS1 kinase in deriving such tumorsCitation11. Hence, the targeting of the tyrosine kinase ROS1 could be a useful strategy for treatment of such tumors.
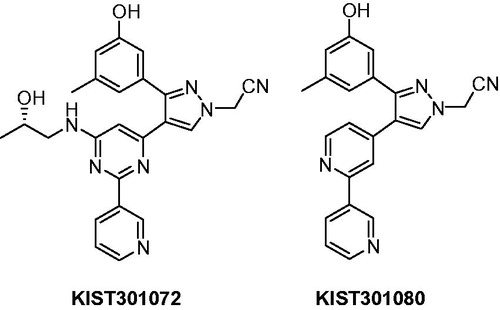
In aspiration of interest in developing novel ROS1 inhibitors, we recently detailed the discovery of KIST301072 as the first selective inhibitor for ROS1 RTK on 45 kinases with an IC50 of 199 nMCitation12, followed by the development of more simplified and equipotent derivative KIST301080 with an IC50 value of 209 nMCitation13. These two compounds represented new promising leads for the development of more potent and more selective ROS1 kinase inhibitors, and following this discovery, it was important to explore the new chemical scaffold in order to have a clear picture about the structure–activity relationship (SAR) of the new group of ROS1 kinase inhibitors ().
Figure 1. Structure of the lead compounds KIST301072 and KIST301080.
Therefore, as a continuation of our study, a further SAR study was conducted to explore the effect of changing the central pyrazole ring into an open form. Furthermore, considering the importance of polar side chains, which act as water-solubilizing groups to interact with water molecules within the kinase enzymesCitation14,Citation15, efforts to improve the potency of this scaffold was also made by exploring more water-solubilizing groups.
Experimental
Chemistry
General
NMR spectra were recorded with a Bruker NMR spectrometer (Billerica, MA), operating at 400 MHz for 1H NMR and 100 MHz for 13C NMR. The multiplicities were abbreviated as s: singlet, d: doublet, t: triplet, m: multiplet and q: quartet. The coupling constants J are recorded in hertz (Hz), and the relative shift values of peak are recorded by ppm unit using tetramethylsilane as standard material. Melting points were determined on a SRS OPTIMELT (Sunnyvale, CA). FT-IR spectra were obtained on Perkin Elmer 16E PC FT-IR Spectrometer (Waltham, MA). Thin-layer chromatography was performed using precoated plates (0.25 mm, Merck) of silica gel 60 F254 (230 ∼ 400 mesh) for monitoring all reactions and under ultraviolet irradiation (254 nm). Column chromatography separations are performed using silica gel (230 ∼ 400 mesh, Merck). All the commercially available reagent chemicals were obtained from Aldrich, TCI, Wako Pure Chemical, Acros and Dae-Jung Chemicals, and generally used without further purification.
2-(2,6-Dichloropyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl)ethanone (2)
To a solution of methyl 3-methoxy-5-methylbenzoate (1) (5.0 g, 27.7 mmol) and 2,4-dichloro-6-methylpyrimidine (5.4 g, 33.1 mmol) in 75 mL of dry THF was added a 1.0 M solution of lithium bis(trimethylsilyl)amide (LHMDS) in THF (55.6 mL, 55.6 mmol) at 0 °C dropwise using dropping funnel, and reaction mixture was stirred at room temperature for 12 h. The reaction was quenched with 1 N aqueous HCl solution for neutralization and then extracted with ethyl acetate (2 × 150 mL). The combined organic layers were washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography using the mixed solvent of hexane and dichloromethane (4:1, v/v) to give the title compound as a mixture of keto/enol tautomers (1:1.5). Yield: 7 g (81%) and mp 121–122 °C. IR (KBr) 3061, 2920, 1687, 1627, 1589, 1220, 1061, 832 and 678 cm−1. 1H NMR (CDCl3) δ 2.42 (s, 3H), 3.86 (s, 3H), 6.01 (s, 1H), 6.88 (s, 1H), 7.01 (s, 1H), 7.21 (s, 1H), 7.26 (s, 1H) and 13.54 (s, 1H). 13C NMR (CDCl3) δ 21.47, 21.60, 46.81, 55.39, 55.49, 93.09, 108.70, 110.03, 114.72, 118.12, 119.52, 120.81, 121.28, 122.00, 135.24, 136.99, 139.83, 140.23, 157.82, 159.84, 160.00, 160.48, 161.10, 162.55, 167.65, 168.11, 169.46 and 193.85.
General procedure for the synthesis of compounds 3a–f
A mixture of 2-(2,6-dichloropyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl)ethanone (2) (1.0 g, 3.23 mmol) and the appropriate amine in THF was heated to 80 °C in an oil bath for three hours. The reaction mixture was left to cool at room temperature and then concentrated in vacuo. The residue was partitioned between water and ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography using the proper mobile phase.
2-(2-Chloro-6-(3-hydroxypropylamino)pyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl)ethanone (3a)
Flash column chromatography was carried out using ethyl acetate-hexane (2:1, v/v) to give the title compound as a mixture of keto/enol tautomers (1:1.6). Yield: 550 mg (49%) and mp 105–106 °C. IR (KBr) 3274, 2918, 2843, 1636, 1590, 1461, 1224, 1193, 851 and 689 cm−1. 1H NMR (CDCl3) δ 1.90 (m, 2H), 2.40 (s, 3H), 3.59–3.65 (m, 4H), 3.87 (s, 3H), 5.86 (s, 1H), 6.33 (s, 1H), 6.83 (s, 1H), 7.16 (s, 1H), 7.23 (s, 1H) and 15.33 (s, 1H). 13C NMR (CDCl3) δ 21.46, 21.59, 32.60, 38.07, 55.36, 55.47, 58.85, 93.38, 105.64, 108.33, 110.10, 110.15, 117.33, 119.26, 120.94, 122.21, 136.36, 139.60, 140.03, 159.17, 159.75, 159.89, 160.40, 165.79 and 194.62.
2-(2-Chloro-6-(2-morpholinoethylamino)pyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl)ethanone (3b)
Flash column chromatography was carried out using ethyl acetate-hexane (2:1, v/v) to give the title compound as a mixture of keto/enol tautomers (1:2.3). Yield: 630 mg (48%) and mp 114–116 °C. IR (KBr) 3318, 2956, 1681, 1588, 1209, 1067, 854 and 683 cm−1. 1H NMR (CDCl3) δ 2.40 (s, 3H), 2.54 (m, 4H), 2.68 (m, 2H), 3.54 (m, 2H), 3.76 (m, 4H), 3.89 (s, 3H), 5.87 (s, 1H), 6.33 (s, 1H), 6.83 (s, 1H), 7.17 (s, 1H), 7.23 (s, 1H) and 15.03 (s, 1H). 13C NMR (CDCl3) δ 21.48, 21.58, 37.57, 37.69, 47.58, 53.24, 53.28, 55.34, 55.44, 66.69, 66.78, 93.51, 105.58, 108.22, 109.84, 110.33, 117.30, 119.19, 120.63, 122.23, 136.34, 137.58, 139.58, 139.89, 158.99, 159.78, 159.85, 160.67, 165.85, 166.19, 168.18 and 195.23.
2-(2-Chloro-6-((2-(piperidin-1-yl)ethyl)amino)pyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl)ethanone (3c)
Flash column chromatography was carried out using ethyl acetate-hexane (4:1, v/v) to give the title compound as a mixture of keto/enol tautomers (1:2.7). Yield: 250 mg (13 %) and mp 90–92 °C. IR (KBr) 3265, 2934, 1637, 1592, 1228, 1064, 820 and 691 cm−1. 1H NMR (CDCl3) δ 1.49 (m, 2H), 1.63 (m, 4H), 2.40 (s, 3H), 2.47 (m, 4H), 2.62 (m, 2H), 3.51 (m, 2H), 3.86 (s, 3H), 5.87 (s, 1H), 6.01 (s, 1H), 6.31 (s, 1H), 6.83 (s, 1H), 7.18 (s, 1H), 7.23 (s, 1H) and 15.17 (s, 1H). 13C NMR (CDCl3) δ 21.47, 21.57, 24.22, 24.31, 25.65, 25.77, 38.01, 38.10, 54.21, 54.28, 55.34, 55.43, 57.06, 93.43, 105.28, 109.56, 110.26, 117.29, 119.21, 120.69, 122.31, 136.51, 139.54, 159.76, 160.59, 165.69, 166.12, 168.41 and 193.46.
2-(2-Chloro-6-((2-(diethylamino)ethyl)amino)pyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl)ethanone (3d)
Flash column chromatography was carried out using ethyl acetate to give the title compound as a mixture of keto/enol tautomers (1:2.2). Yield: 350 mg (19%) and mp 65–67 °C. IR (KBr) 3275, 2964, 1637, 1225, 1063, 821 and 691 cm−1. 1H NMR (CDCl3) δ 1.07 (t, J = 7.2, 6H), 2.41 (s, 3H), 2.60–2.63 (m, 6H), 3.45 (m, 2H), 3.87 (s, 3H), 5.87 (s, 1H), 6.01 (s, 1H), 6.32 (s, 1H), 6.83 (s, 1H), 7.18 (s, 1H), 7.24 (s, 1H) and 15.22 (s, 1H). 13C NMR (CDCl3) δ 11.31, 11.43, 21.47, 21.58, 38.86, 46.39, 46.68, 51.26, 55.35, 55.44, 93.68, 108.15, 109.63, 117.31, 119.22, 122.31, 136.52, 139.55, 139.87, 159.76, 159.83, 160.61, 165.68, 166.12, 168.45 and 195.79.
2-(2-Chloro-6-(4-ethylpiperazin-1-yl)pyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl)ethanone (3e)
Flash column chromatography was carried out using ethyl acetate to give the title compound as a mixture of keto/enol tautomers (1:1.6). Yield: 512 mg (28%); sticky solid. IR (KBr) 2928, 2812, 1637, 1591, 1231, 1061, 821 and 691 cm−1. 1H NMR (CDCl3) δ 1.14 (t, J = 7.2, 3H), 2.39 (s, 3H), 2.44–2.58 (m, 6H), 3.84–3.86 (m, 7H), 5.32 (s, 1H), 5.89 (s, 1H), 6.28 (s, 1H), 6.82 (s, 1H), 7.17 (s, 1H), 7.23 (s, 1H) and 15.06 (s, 1H). 13C NMR (CDCl3) δ 11.89, 11.95, 21.45, 21.56, 43.69, 44.23, 47.93, 52.32, 52.40, 52.51, 53.42, 55.33, 55.41, 93.59, 105.11, 108.11, 108.79, 110.31, 117.33, 119.16, 120.59, 122.43, 136.34, 137.63, 139.55, 139.76, 158.48, 159.77, 159.81, 160.66, 161.18, 161.39, 165.57, 165.75, 168.02 and 195.41.
2-(2-Chloro-6-(4-(dimethylamino)piperidin-1-yl)pyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl) ethanone (3f)
Flash column chromatography was carried out using ethyl acetate-methanol (4:1, v/v) to give the title compound as a mixture of keto/enol tautomers (1:1.8). Yield: 345 mg (18%) and mp 73–75 °C. IR (KBr) 3388, 2927, 1682, 1637, 1570, 1231, 1061, 837 and 691 cm−1. 1H NMR (CDCl3) δ 1.42–1.57 (m, 2H), 1.87–1.91 (m, 2H), 2.26 (s, 6H), 2.31 (s, 3H), 2.39 (m, 1H), 2.92–2.97 (m, 2H), 3.78 (s, 3H), 4.10 (s, 1H), 4.47–4.49 (m, 2H), 5.77 (s, 1H), 6.15 (s, 1H), 6.43 (s, 1H), 6.73 (s, 1H), 7.10 (s, 1H), 7.16 (s, 1H) and 15.02 (s, 1H).
General procedure for the synthesis of compounds 4a–f
To a glass vial containing a magnetic stir bar was added a mixture of the starting compound 3a–f (1 mmol), 3-pyridineboronic acid (1.5 mmol), bis(triphenylphosphine)palladium(II) dichloride (0.05 mmol) and potassium carbonate (2 mmol) and the vial was purged with nitrogen gas for 10 min. Then, a solvent mixture of tetrahydrofuran/acetonitrile/water (8 mL/2:1:1) was injected, and the vial was once again bubbled with nitrogen for 10 min and then capped. The reaction mixture was stirred at 90 °C under nitrogen. After three hours, the reaction mixture was left to cool to ambient temperature. The reaction mixture was filtered through Celite. Then the filtrate was concentrated in vacuo, and the residue was purified by column chromatography using the proper mobile phase.
2-(6-((3-Hydroxypropyl)amino)-2-(pyridin-3-yl)pyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl)ethanone (4a)
Flash column chromatography was carried out using ethyl acetate–methanol (30:1, v/v) to give the title compound as a mixture of keto/enol tautomers (1:1.7). Yield: 137 mg (35%) and mp 99–100 °C.IR (KBr) 3302, 2929, 1673, 1587, 1216, 1064, 929 and 706 cm−1. 1H NMR (CDCl3) δ 1.88–1.92 (m, 2H), 2.38 (s, 3H), 3.64–3.71 (m, 4H), 3.82–3.85 (m, 2H), 3.87 (s, 3H), 5.97 (s, 1H), 6.04 (t, J = 6.4 Hz, 1H), 6.66 (s, 1H), 6.81 (s, 1H), 7.19 (s, 1H), 7.25 (s, 1H), 7.38–7.42 (m, 1H), 8.27 (dd, J = 1.6, 8.0 Hz, 1H), 8.67 (dd, J = 1.6, 5.2 Hz, 1H), 9.19 (d, J = 1.6 Hz, 1H) and 15.39 (s, 1H). 13C NMR (CDCl3) δ 21.46, 21.60, 32.20, 32.93, 37.81, 47.94, 55.37, 55.46, 59.07, 59.90, 93.67, 103.10, 106.77, 108.39, 110.25, 117.26, 119.35, 120.79, 122.31, 123.57, 123.63, 132.89, 134.41, 134.55, 137.12, 137.61, 139.54, 139.96, 148.32, 148.54, 151.12, 151.27, 158.59, 159.79, 159.89, 161.03, 163.14, 164.70 and 195.62.
2-(6-(2-Morpholinoethylamino)-1-(3-methoxy-5-methylphenyl)-2-(pyridin-3-yl)pyrimidin-4-yl)ethanone (4b)
Flash column chromatography was carried out using ethyl acetate-methanol (10:1, v/v) to give the title compound as a mixture of keto/enol tautomers (1:2.8). Yield: 317 mg (71%) and mp 72–74 °C. IR (KBr) 3259, 2926, 1680, 1588, 1216, 1065, 854 and 706 cm−1. 1H NMR (CDCl3) δ 2.36 (s, 3H), 2.50 (m, 4H), 2.65 (m, 2H), 3.59 (m, 2H), 3.74 (m, 4H), 3.82 (s, 3H), 5.88 (s, 1H), 5.99 (s, 1H), 6.70 (s, 1H), 6.80 (s, 1H), 7.20 (s, 1H), 7.39–7.40 (m, 1H), 8.29 (d, J = 8.0 Hz, 1H), 8.67 (d, J = 3.6 Hz, 1H), 9.21 (s, 1H) and 15.38 (s, 1H). 13C NMR (CDCl3) δ 21.48, 21.58, 22.96, 23.83, 28.79, 30.52, 37.70, 37.74, 38.73, 48.06, 53.37, 53.40, 55.33, 55.41, 57.33, 66.75, 66.80, 66.97, 93.71, 102.98, 106.41, 108.16, 110.51, 117.14, 119.22, 120.40, 122.30, 123.43, 123.46, 128.76, 130.34, 133.24, 134.34, 134.5, 137.23, 137.84, 139.48, 139.82, 148.44, 148.57, 151.06, 151.14, 159.77, 162.73, 165.47 and 196.09.
1-(3-Methoxy-5-methylphenyl)-2-(6-((2-(piperidin-1-yl)ethyl)amino)-2-(pyridin-3-yl)pyrimidin-4-yl)ethanone (4c)
Flash column chromatography was carried out using ethyl acetate-methanol (2:1, v/v) to give the title compound as a mixture of keto/enol tautomers (1:2.6). Yield: 100 mg (40%) and mp 57–58 °C. IR (KBr) 3293, 2932, 1719, 1633, 1588, 1216, 1063, 840 and 705 cm−1. 1H NMR (CDCl3) δ 1.50 (m, 2H), 1.64–1.68 (m, 4H), 2.39 (s, 3H), 2.52 (m, 4H), 2.64–2.70 (m, 2H), 3.64 (m, 2H), 3.87 (s, 3H), 6.02 (s, 1H), 6.72 (s, 1H), 6.82 (s, 1H), 6.96 (s, 1H), 7.22 (s, 1H), 7.29 (s, 1H), 7.37–7.50 (m, 1H), 8.30–8.34 (m, 1H), 8.70–8.71 (m, 1H), 9.23 (s, 1H) and 15.47 (s, 1H). 13C NMR (CDCl3) δ 21.47, 21.58, 24.11, 24.28, 25.49, 25.71, 29.69, 38.07, 38.13, 48.31, 54.35, 54.42, 55.34, 55.43, 57.09, 57.47, 93.59, 102.75, 106.32, 108.13, 110.33, 117.16, 11.05, 117.16, 119.26, 12.63, 122.38, 122.85, 123.46, 133.14, 133.34, 134.38, 134.50, 137.31, 137.72, 139.45, 139.84, 148.46, 148.58, 151.02, 151.11, 159.76, 159.82, 162.65, 164.47, 165.57 and 195.93.
2-(6-((2-(Diethylamino)ethyl)amino)-2-(pyridin-3-yl)pyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl) ethanone (4d)
Flash column chromatography was carried out using ethyl acetate-methanol (4:1, v/v) to give the title compound as a mixture of keto/enol tautomers (1:2.6). Yield: 130 mg (33%); sticky solid. IR (KBr) 3248, 2967, 1751, 1633, 1583, 1215, 1063, 838 and 705 cm−1. 1H NMR (CDCl3) δ 1.07 (m, 6H), 2.40 (s, 3H), 2.59–2.63 (m, 4H), 2.75–2.78 (m, 2H), 3.58–3.60 (m, 2H), 3.85 (s, 3H), 5.90 (s, 1H), 6.02 (s, 1H), 6.72 (s, 1H), 6.82 (s, 1H), 7.23 (s, 1H), 7.40 (m, 1H), 8.33 (m, 1H), 8.69 (m, 1H), 9.23 (s, 1H) and 15.94 (s, 1H). 13C NMR (CDCl3) δ 11.41, 21.47, 21.58, 38.94, 46.52, 46.78, 51.19, 51.59, 55.35, 55.43, 102.74, 106.24, 108.10, 110.30, 117.19, 119.28, 120.66, 122.41, 123.44, 134.39, 134.49, 139.46, 148.49, 148.61, 151.04, 151.11, 159.77 and 164.49.
2-(6-(4-Ethylpiperazin-1-yl)-2-(pyridin-3-yl)pyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl)ethanone (4e)
Flash column chromatography was carried out using ethyl acetate-methanol (4:1, v/v) to give the title compound as a mixture of keto/enol tautomers (1:1.6). Yield: 450 mg (90%) and mp 81–82 °C. IR (KBr) 2926, 2852, 1737, 1634, 1575, 1217, 1062, 841 and 705 cm−1. 1H NMR (CDCl3) δ 1.17 (t, J = 7.2, 3H), 2.28 (s, 3H), 2.35–2.43 (m, 6H), 3.74 (s, 3H), 3.84–3.86 (m, 4H), 5.91 (s, 1H), 6.58 (s, 1H), 6.69 (s, 1H), 7.11 (s, 1H), 7.17 (s, 1H), 7.25–7.27 (m, 1H), 8.18 (d, J = 8.0 Hz, 1H), 8.59 (s, 1H), 9.16 (s, 1H) and 15.41 (s, 1H). 13C NMR (CDCl3) δ 11.89, 11.96, 21.46, 21.57, 43.61, 44.17, 48.70, 52.38, 52.46, 52.53, 52.66, 53.44, 55.31, 55.38, 93.90, 102.31, 105.41, 108.03, 110.36, 117.18, 119.19, 120.54, 122.58, 123.37, 123.41, 133.24, 133.29, 134.31, 134.40, 137.04, 137.71, 139.47, 139.70, 148.52, 148.63, 151.08, 158.27, 159.76, 161.38, 161.83, 162.27, 164.63, 165.29, 169.12 and 196.06.
2-(6-(4-(Dimethylamino)piperidin-1-yl)-2-(pyridin-3-yl)pyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl) ethanone (4f)
Flash column chromatography was carried out using ethyl acetate-methanol-triethylamine (1:1: 0.01 v/v/v) to give the title compound as a mixture of keto/enol tautomers (1:2.3). Yield: 300 mg (83%) and mp 184–185 °C. IR (KBr) 3381, 2949, 1681, 1636, 1581, 1215, 1061, 843 and 707 cm−1. 1H NMR (CDCl3) δ 1.47–1.54 (m, 2H), 1.89–1.92 (m, 2H), 2.25 (s, 6H), 2.31 (s, 3H), 2.36–2.42 (m, 1H), 2.94–3.00 (m, 2H), 3.77 (s, 3H), 4.61–4.64 (m, 2H), 5.93 (s, 1H), 6.57 (s, 1H), 6.72 (s, 1H), 7.15 (s, 1H), 7.21 (s, 1H), 7.28–7.31 (m, 1H), 8.21 (dt, J = 2.0, 8.0 Hz, 1H), 8.61 (dd, J = 1.6, 4.8 Hz, 1H), 9.17 (d, J = 1.6 Hz, 1H) and 15.53 (s, 1H). 13C NMR (CDCl3) δ 21.40, 21.52, 28.08, 41.52, 41.60, 43.16, 43.62, 48.63, 55.22, 55.29, 62.08, 62.51, 93.69, 101.80, 104.94, 108.05, 110.38, 117.04, 119.14, 120.39, 122.52, 123.32, 133.22, 134.20, 134.32, 137.13, 137.71, 139.32, 139.59, 148.40, 148.51, 150.89, 157.77, 159.69, 161.12, 161.65, 162.12, 164.35, 165.25, 169.33 and 196.04.
General procedure for the synthesis of compounds 5a–f
To a solution of the starting compound 4a–f (0.3 mmol) in 5 mL of dry dichloromethane was added boron trifluoride–methylsulfide complex (3 mmol) dropwise. The resulting suspension was stirred at room temperature for 12 h. The mixture was concentrated, and the residue was partitioned between ethyl acetate and sodium bicarbonate solution. The layers were separated, and the aqueous layer was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by column chromatography using the proper mobile phase.
1-(3-Hydroxy-5-methylphenyl)-2-(6-((3-hydroxypropyl)amino)-2-(pyridin-3-yl)pyrimidin-4-yl)ethanone (5a)
Flash column chromatography was carried out using ethyl acetate–methanol (10:1, v/v) to give the title compound. Yield: 22 mg (19%) and mp 97–99 °C. IR (KBr) 3336, 2923, 1738, 1585, 1243, 1019 and 851 cm−1. 1H NMR (DMSO-d6) δ 1.82 (d, J = 6.4, 12.8 Hz, 2H), 2.35 (s, 3H), 3.56 (q, J = 6.0 Hz, 1H), 3.73 (q, J = 6.4 Hz, 1H), 4.69 (s, 1H), 6.61 (s, 1H), 6.79 (s, 1H), 7.14 (s, 1H), 7.21 (s, 1H), 7.65 (ddd, J = 0.8, 4.8, 8.0 Hz, 1H), 8.54 (dt, J = 1.6, 8.0 Hz, 1H), 8.80 (dd, J = 1.6, 4.8, 1H), 9.36 (d, J = 1.6 Hz, 1H) and 9.71 (s, 1H). 13C NMR (DMSO-d6) δ 21.51, 21.72, 31.91, 59.10, 93.79, 102.13, 110.62, 118.30, 119.84, 124.53, 131.46, 134.69, 135.38, 140.01, 149.05, 152.95, 157.37, 158.12, 163.21 and 163.49.
1-(3-Hydroxy-5-methylphenyl)-2-(6-((2-morpholinoethyl)amino)-2-(pyridin-3-yl)pyrimidin-4-yl) ethanone (5b)
Flash column chromatography was carried out using ethyl acetate-methanol (10:1, v/v) to give the title compound. Yield: 22 mg (17%) and mp 143–145 °C IR (KBr) 3428, 2955, 2924, 1723, 1248, 1099, 874 and 728 cm−1. 1H NMR (DMSO-d6) δ 2.33 (s, 3H), 2.50 (m, 4H), 2.65 (t, J = 6.4, 2H), 3.61 (t, J = 4.4, 4H), 3.75 (m, 2H), 6.61 (s, 1H), 6.80 (s, 1H), 7.14 (s, 1H), 7.22 (s, 1H), 7.48 (s, 1H), 7.56 (s, 1H), 7.65 (dd, J = 4.8, 8.0 Hz, 1H), 8.52 (dt, J = 2.0, 8.0 Hz, 1H), 8.79 (d, J = 3.6 Hz, 1H), 9.36 (s, 1H) and 9.69 (s, 1H).
1-(3-Hydroxy-5-methylphenyl)-2-(6-((2-(piperidin-1-yl)ethyl)amino)-2-(pyridin-3-yl)pyrimidin-4-yl)ethanone (5c)
Flash column chromatography was carried out using ethyl acetate-methanol (4:1, v/v) to give the title compound. Yield: 34 mg (26%) and mp 126–128 °C. IR (KBr) 3424, 2955, 2924 1723, 1593, 1248, 1099, 875 and 728 cm−1. 1H NMR (DMSO-d6) δ 1.43–1.46 (m, 2H), 1.53–1.58 (m, 4H), 2.33 (s, 3H), 2.45–2.48 (m, 4H), 2.61 (t, J = 6.4 Hz, 1H), 3.70–3.75 (m, 2H), 6.59 (s, 1H), 6.80 (s, 1H), 7.15 (s, 1H), 6.96 (s, 1H), 7.22 (s, 1H), 7.44 (s, 1H), 7.53–7.54 (m, 1H), 7.63 (ddd, J = 0.4, 4.4, 8.0 Hz, 1H), 8.52 (dt, J = 1.6, 8.0 Hz, 1H), 8.79 (dd, J = 1.6, 4.4 Hz, 1H), 9.35 (d, J = 1.6 Hz, 1H) and 9.56 (s, 1H). 13C NMR (DMSO-d6) δ 21.47, 24.47, 26.03, 54.16, 56.31, 93.78, 102.27, 110.76, 118.37, 119.89, 124.41, 131.53, 134.82, 135.31, 139.95, 149.05, 152.88, 154.93, 157.44, 158.15, 163.58 and 163.65.
2-(6-((2-(Diethylamino)ethyl)amino)-2-(pyridin-3-yl)pyrimidin-4-yl)-1-(3-hydroxy-5-methylphenyl) ethanone (5d)
Flash column chromatography was carried out using ethyl acetate-methanol (4:1, v/v) to give the title compound. Yield: 37 mg (29%) and mp 135–136 °C. IR (KBr) 3428, 2956, 2924, 1723, 1247, 1099, 874 and 758 cm−1. 1HNMR (DMSO-d6) δ 1.05 (t, J = 7.2 Hz, 6H), 2.33 (s, 3H), 2.65 (m, 4H), 2.79 (m, 2H), 3.73–3.75 (m, 2H), 6.59 (s, 1H), 6.81 (s, 1H), 7.15 (s, 1H), 7.22 (s, 1H), 7.44 (s, 1H), 7.52–7.54 (m, 1H), 7.63 (dd, J = 4.8, 8.0 Hz, 1H), 8.52 (dt, J = 1.6, 8.0 Hz, 1H), 8.80 (m, 1H), 9.36 (s, 1H) and 9.57 (s, 1H). 13C NMR (DMSO-d6) δ 12.16, 21.47, 47.09, 47.33, 50.80, 93.78, 102.39, 110.77, 118.37, 119.91, 124.40, 131.51, 134.80, 135.33, 139.95, 149.06, 152.90, 154.90, 157.49, 158.15 and 163.66.
2-(6-(4-Ethylpiperazin-1-yl)-2-(pyridin-3-yl)pyrimidin-4-yl)-1-(3-hydroxy-5-methylphenyl)ethanone (5e)
Flash column chromatography was carried out using ethyl acetate–methanol (4:1, v/v) to give the title compound. Yield: 100 mg (46%) and mp 149–150 °C. IR (KBr) 3419, 2955, 2924, 1723, 1567, 1246, 1099, 874 and 759 cm−1. 1H NMR (DMSO-d6) δ 1.17 (m, 3H), 2.30 (s, 3H), 2.35–2.44 (m, 4H), 3.79 (m, 4H), 6.24 (s, 1H), 6.72 (s, 1H), 7.19 (s, 1H), 7.20 (s, 1H), 7.30 (s, 1H), 7.54–7.59 (m, 1H), 8.44–8.73 (m, 1H), 8.70–8.73 (m, 1H), 9.27–9.29 (m, 1H) and 15.23 (s, 1H).
2-(6-(4-(Dimethylamino)piperidin-1-yl)-2-(pyridin-3-yl)pyrimidin-4-yl)-1-(3-hydroxy-5-methylphenyl) ethanone (5f)
Flash column chromatography was carried out using ethyl acetate–methanol–triethylamine (1:3:0.01, v/v/v) to give the title compound. Yield: 20 mg (16%) and mp 105–106 °C. IR (KBr) 3419, 2954, 2926, 1725, 1568, 1264, 813 and 756 cm−1. 1H NMR (CDCl3) δ 1.54–1.61 (m, 2H), 1.98–2.01 (m, 2H), 2.33–2.35 (m, 9H), 2.49–2.57 (m, 1H), 2.97–3.03 (m, 2H), 4.68–4.72 (m, 2H), 5.95 (s, 1H), 6.23 (s, 1H), 6.75 (s, 1H), 7.15 (s, 1H), 7.17 (s, 1H), 7.34–7.43 (m, 1H), 8.28–8.32 (m, 1H), 8.65–8.68 (m, 1H), 9.21–9.22 (m, 1H) and 15.57 (s, 1H).
General procedure for the synthesis of compounds 6a–f
Sodium borohydride was placed in round bottom flask, and the flask was closed with a rubber septum and purged with nitrogen gas for 10 min. Then 10 mL of ethanol was injected, and the flask was once again bubbled with nitrogen and placed in an ice-bath. Then the compound 4a–f dissolved in 10 mL ethanol was injected. The reaction mixture was stirred at room temperature under nitrogen for 12 h. The mixture was concentrated, and the residue was partitioned between ethyl acetate and water. The layers were separated, and the aqueous layer was extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate and concentrated in vacuo. The residue was purified by column chromatography using the proper mobile phase.
3-((6-(2-Hydroxy-2-(3-methoxy-5-methylphenyl)ethyl)-2-(pyridin-3-yl)pyrimidin-4-yl)amino)propan-1-ol (6a)
Flash column chromatography was carried out using dichloromethane–methanol (10:1, v/v) to give the title compound. Yield: 23 mg (58%); sticky solid. IR (KBr) 3314, 2922, 2851, 1584, 1556, 1065, 846 and 704 cm−1. 1H NMR (CD3OD) δ 1.86–1.93 (m, 2H), 2.31 (s, 3H), 2.96–3.07 (m, 2H), 3.61 (t, 2H), 3.73 (t, 2H), 3.77 (s, 3H), 5.10 (dd, J = 5.2, 8.4 Hz, 1H), 6.66 (s, 1H), 6.80 (s, 1H), 6.83 (s, 1H), 6.97 (s, 1H), 7.53–7.64 (m, 1H), 8.45 (d, J = 8.0 Hz, 1H), 8.63 (dd, J = 1.6, 4.8 Hz, 1H) and 9.16 (d, J = 1.6 Hz, 1H). 13C NMR (CD3OD) δ 20.23, 32.09, 37.85, 54.18, 59.23, 72.79, 106.01, 108.23, 113.29, 118.74, 123.88, 133.77, 135.13, 139.07, 145.53, 147.44, 150.01, 159.82, 162.67 and 169.59.
1-(3-Methoxy-5-methylphenyl)-2-(6-((2-morpholinoethyl)amino)-2-(pyridin-3-yl)pyrimidin-4-yl)ethanol (6b)
Flash column chromatography was carried out using ethyl acetate–methanol (3:1, v/v) to give the title compound. Yield: 29 mg (58%); sticky solid. IR (KBr) 3331, 2927, 2855, 1583, 1556, 1067, 844 and 706 cm−1. 1H NMR (CD3OD) δ 2.31 (s, 3H), 2.58 (m, 4H), 2.65 (t, J = 6.8 Hz, 2H), 2.96–3.08 (m, 2H), 3.65 (t, J = 6.8 Hz, 2H), 3.74 (m, 4H), 3.77 (s, 3H), 5.13 (q, J = 4.8, 1H), 6.53 (s, 1H), 6.79 (s, 1H), 6.82 (s, 1H), 7.53 (dd, J = 4.8, 7.6, 1H), 8.43 (d, J = 8.0 Hz, 1H), 8.63 (dd, J = 1.6, 4.8 Hz, 1H) and 9.21 (d, J = 1.6 Hz, 1H).13C NMR (CD3OD) δ 20.27, 37.53, 53.43, 54.19, 57.45, 66.35, 72.77, 106.22, 108.31, 113.25, 118.74, 123.87, 133.70, 135.08, 139.07, 145.59, 147.44, 150.04, 159.82, 161.69, 162.44 and 169.65.
1-(3-Methoxy-5-methylphenyl)-2-(6-((2-(piperidin-1-yl)ethyl)amino)-2-(pyridin-3-yl)pyrimidin-4-yl)ethanol (6c)
Flash column chromatography was carried out using dichloromethane–methanol (10:1 to 4:1, v/v) to give the title compound. Yield: 16 mg (32%) and mp 123–125 °C. IR (KBr) 3397, 2926, 2854, 1583, 1556, 1064, 847 and 706 cm−1. 1H NMR (CDCl3) δ 1.53 (m, 2H), 1.74 (m, 4H), 2.36 (s, 3H), 2.65 (m, 4H), 2.78 (t, J = 5.6 Hz, 1H), 3.02–3.05 (m, 2H), 3.72 (m, 2H), 3.83 (s, 3H), 5.13 (dd, J = 3.6, 8.4 Hz, 1H), 6.10 (s, 1H), 6.67 (s, 1H), 6.85 (s, 1H), 6.86 (s, 1H), 6.87 (s, 1H), 7.43 (ddd, J = 0.8, 4.8, 8.0 Hz, 1H), 8.33 (dt, J = 2.0, 8.0 Hz, 1H), 8.71 (dd, J = 1.6, 4.8 Hz, 1H) and 9.21 (s, 1H). 13C NMR (CDCl3) δ 21.59, 23.83, 25.12, 37.91, 54.41, 55.23, 57.29, 72.63, 106.24, 108.30, 113.91, 118.97, 123.54, 133.03, 134.56, 139.52, 145.08, 148.49, 151.24, 159.79 and 161.74.
2-(6-((2-(Diethylamino)ethyl)amino)-2-(pyridin-3-yl)pyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl) ethanol (6d)
Flash column chromatography was carried out using dichloromethane–methanol (6:1, v/v) to give the title compound. Yield: 12 mg (40%); sticky solid. IR (KBr) 3302, 2964, 2926, 1583, 1556, 846 and 705 cm−1. 1H NMR (CD3OD) δ 1.15 (t, J = 7.2 Hz, 6H), 2.31 (s, 3H), 2.75 (q, J = 7.2 Hz, 4H), 2.83 (t, J = 7.2 Hz, 2H), 2.97–3.10 (m, 1H), 3.65 (t, J = 7.2, 2H), 3.76 (s, 3H), 5.11 (dd, J = 5.2, 8.4 Hz, 1H), 6.66 (s, 1H), 6.79 (s, 1H), 6.82 (s, 1H), 7.01 (s, 1H), 7.56 (ddd, J = 0.8, 5.2, 8.4 Hz, 1H), 8.46 (d, J = 8.0 Hz, 1H), 8.64 (dd, J = 1.6, 4.8 Hz, 1H) and 9.19 (s, 1H). 13C NMR (CD3OD) δ 10.06, 20.23, 38.04, 51.31, 54.17, 72.81, 106.35, 108.27, 113.25, 118.72, 123.87, 133.76, 135.12, 139.07, 145.57, 147.45, 150.04, 159.83, 161.87, 162.54 and 169.68.
2-(6-(4-Ethylpiperazin-1-yl)-2-(pyridin-3-yl)pyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl)ethanol (6e)
Flash column chromatography was carried out using dichloromethane-methanol (20:1, v/v) to give the title compound. Yield: 52 mg (52%); sticky solid. IR (KBr) 3223, 2929, 2852, 1582, 1065, 846 and 705 cm−1. 1H NMR (CDCl3) δ 1.17 (t, J = 7.2 Hz, 3H), 2.30 (s, 3H), 2.50 (q, J = 7.2, 2H), 2.56 (t, J = 5.2 Hz, 4H), 2.99–3.11 (m, 2H), 3.76 (s, 3H), 3.94 (m, 4H), 5.13 (dd, J = 5.2, 8.4 Hz, 1H), 6.64 (s, 1H), 6.78 (s, 1H), 6.81 (s, 1H), 6.98 (s, 1H), 7.51 (ddd, J = 0.8, 4.8, 8.0 Hz, 1H), 8.42 (dt, J = 1.6, 8.0 Hz, 1H), 8.6 (dd, J = 1.6, 4.8 Hz, 1H) and 9.15 (d, J = 1.6 Hz, 1H). 13C NMR (CDCl3) δ 10.41, 20.30, 43.10, 52.04, 52.26, 54.20, 72.75, 105.95, 108.49, 113.22, 118.87, 123.86, 133.80, 135.07, 139.01, 145.63, 147.40, 149. 98, 159.79, 161.24, 161.61 and 169.43.
2-(6-(4-(Dimethylamino)piperidin-1-yl)-2-(pyridin-3-yl)pyrimidin-4-yl)-1-(3-methoxy-5-methylphenyl) ethanol (6f)
Flash column chromatography was carried out using dichloromethane–methanol (5:1, v/v) to give the title compound. Yield: 33 mg (44%) and mp 95–96 °C. IR (KBr) 3341, 2925, 2853, 1583, 1552, 1055, 1026, 842 and 706 cm−1. 1H NMR (CDCl3) δ 1.51–1.55 (m, 2H), 1.57–1.60 (m, 2H), 1.99–2.02 (m, 9H), 2.52–2.58 (m, 1H), 2.98–3.05 (m, 4H), 3.83 (s, 3H), 4.92 (m, 2H), 5.13 (dd, J = 4.0, 8.4 Hz, 1H), 6.67 (s, 1H), 6.81 (s, 1H), 6.85 (s, 1H), 6.88 (s, 1H), 7.41 (dd, J = 4.8, 8.0 Hz, 1H), 8.32 (dt, J = 1.6, 8.0 Hz, 1H), 8.71 (d, J = 4.0 Hz, 1H) and 9.23 (s, 1H). 13C NMR (CDCl3) δ 21.59, 27.98, 28.02, 41.46, 43.41, 45.77, 55.22, 62.67, 72.63, 105.13, 108.26, 113.91, 118.96, 123.47, 133.21, 134.46, 139.49, 145.09, 148.59, 151.22, 159.79, 161.02, 162.62 and 169.51.
General procedure for the synthesis of compounds 7a–f
Synthesis of compounds 7a–f could be achieved through reaction with compounds 5a–f by the same method for synthesis of compounds 6a–f.
3-(1-Hydroxy-2-(6-((3-hydroxypropyl)amino)-2-(pyridin-3-yl)pyrimidin-4-yl)ethyl)-5-methylphenol (7a)
Flash column chromatography was carried out using dichloromethane–methanol (10:1, v/v) to give the title compound. Yield: 7 mg (35%) and mp 112–114 °C. IR (KBr) 3323, 2970, 2929, 1585, 1558, 1048, 849 and 705 cm−1. 1H NMR (CD3OD) δ 1.90 (dd, J = 6.4, 12.8 Hz, 2H), 2.27 (s, 3H), 2.96–3.08 (m, 2H), 3.61 (t, J = 6.4, 2H), 3.72 (t, J = 6.4, 2H), 5.05 (dd, J = 4.8, 8.4 Hz, 1H), 6.55 (s, 1H), 6.69 (s, 1H), 6.72 (s, 1H), 6.98 (s, 1H), 7.56 (ddd, J = 0.8, 4.8, 8.0 Hz, 1H), 8.46 (d, J = 8.0 Hz, 1H), 8.64 (dd, J = 1.6, 4.8 Hz, 1H) and 9.18 (d, J = 1.6 Hz, 1H). 13C NMR (CD3OD) δ 20.12, 32.08, 37.83, 59.22, 72.80, 106.01, 109.68, 114.58, 117.69, 123.88, 133.82, 135.16, 139.01, 145.49, 147.45, 149.99, 157.04, 162.68 and 169.63.
3-(1-Hydroxy-2-(6-((2-morpholinoethyl)amino)-2-(pyridin-3-yl)pyrimidin-4-yl)ethyl)-5-methylphenol (7b)
Flash column chromatography was carried out using dichloromethane-methanol (6:1, v/v) to give the title compound. Yield: 10 mg (50%) and mp 68–69 °C. IR (KBr) 3323, 2919, 2848, 1583, 1557, 1068, 844 and 704 cm−1. 1H NMR (CD3OD) δ 2.27 (s, 3H), 2.64 (m, 4H), 2.72 (t, J = 6.8 Hz, 2H), 2.97–3.09 (m, 2H), 3.69 (t, J = 6.4 Hz, 2H), 3.76 (m, 4H), 5.08 (dd, J = 4.8, 8.4 Hz, 1H), 6.54 (s, 1H), 6.67 (s, 1H), 6.71 (s, 1H), 7.01 (s, 1H), 7.57 (ddd, J = 0.8, 4.8, 8.0 Hz, 1H), 8.47 (d, J = 8.0 Hz, 1H), 8.64 (dd, J = 1.6, 4.8 Hz, 1H) and 9.19 (d, J = 1.6 Hz, 1H).
3-(1-Hydroxy-2-(6-((2-(piperidin-1-yl)ethyl)amino)-2-(pyridin-3-yl)pyrimidin-4-yl)ethyl)-5-methylphenol (7c)
Flash column chromatography was carried out using dichloromethane-methanol (6:1, v/v) to give the title compound. Yield: 8 mg (53%) and mp 72–73 °C. IR (KBr) 3265, 2930, 2848, 1581, 1557, 847, 805 and 705 cm−1. 1H NMR (CD3OD) δ 1.54–1.56 (m, 2H), 1.64–1.1.71 (m, 4H), 2.27 (s, 3H), 2.64 (m, 4H), 2.71 (t, J = 6.8 Hz, 2H), 2.96–3.09 (m, 2H), 3.69 (t, J = 6.8 Hz, 2H), 5.04–5.07 (dd, J = 5.2, 2.8, 1H), 6.54 (s, 1H), 6.67 (s, 1H), 6.72 (s, 1H), 7.00 (s, 1H), 7.56 (ddd, J = 0.8, 4.8, 8.0 Hz, 1H), 8.47 (d, J = 8.0 Hz, 1H), 8.64 (dd, J = 1.2, 4.8 Hz, 1H) and 9.19 (s, 1H). 13C NMR (CD3OD) δ 20.13, 23.59, 24.97, 37.60, 54.23, 57.17, 72.71, 106.29, 109.72, 114.59, 115.36, 117.68, 121.39, 123.88, 133.78, 135.13, 138.99, 145.51, 147.45, 150.01, 157.05, 161.60, 162.46 and 169.71.
3-(2-(6-((2-(Diethylamino)ethyl)amino)-2-(pyridin-3-yl)pyrimidin-4-yl)-1-hydroxyethyl)-5-methylphenol (7d)
The combined organic layers were dried over sodium sulfate and concentrated in vacuo to give 20 mg (80%) of the target product; mp 62–63 °C. IR (KBr) 3272, 2925, 2853, 1584, 1557, 1066, 845 and 705 cm−1. 1HNMR (CD3OD) δ 1.16 (t, J = 7.2 Hz, 6H), 2.27 (s, 3H), 2.77 (q, 4H), 2.86 (t, J = 6.8 Hz, 3H), 2.95–3.09 (m, 1H), 3.66 (t, J = 7.2, 3H), 5.06 (dd, J = 4.8, 3.2 Hz, 1H), 5.52 (s, 1H), 6.55 (s, 1H), 6.68 (s, 1H), 6.871 (s, 1H), 7.01 (s,1H), 7.55 (dd, J = 5.2, 8.0 Hz, 1H), 8.46 (d, J = 8.0 Hz, 1H), 8.64 (dd, J = 1.6, 5.2 Hz, 1H) and 9.19 (s, 1H). 13C NMR (CD3OD) δ 9.96, 20.14, 29.30, 37.97, 51.30, 72.82, 106.39, 109.70, 114.60, 117.67, 123.87, 133.76, 135.13, 139.00, 145.54, 147.45, 150.02, 157.07, 161.79, 162.53 and 169.73.
3-(2-(6-(4-Ethylpiperazin-1-yl)-2-(pyridin-3-yl)pyrimidin-4-yl)-1-hydroxyethyl)-5-methylphenol (7e)
Flash column chromatography was carried out using dichloromethane–methanol (5:1, v/v). Yield: 50 mg (83%) and mp 92–93 °C. IR (KBr) 3349, 2920, 2852, 1583, 1554, 1026, 842 and 702 cm−1. 1H NMR (CD3OD) δ 1.18 (t, J = 7, 6 Hz, 3H), 2.26 (s, 3H), 2.48–2.64 (m, 6H), 2.99–3.09 (m, 2H), 3.95 (t, J = 5.2 Hz, 4H), 5.08 (dd, J = 5.2, 8 Hz, 1H), 6.54 (s, 1H), 6.78 (s, 1H), 6.68 (s, 1H), 6.71 (s, 1H), 6.98 (s, 1H), 7.52 (ddd, J = 0.8, 4.8, 8.0 Hz, 1H), 8.42 (dt, J = 1.6, 8.0 Hz,1H), 8.6 (dd, J = 1.6, 4.8 Hz, 1H) and 9.15 (d, J = 1.6 Hz, 1H). 13C NMR (CD3OD) δ 10.40, 20.18, 43.09, 52.03, 52.26, 72.80, 105.94, 109.83, 114.58, 117.82, 123.87, 133.85, 135.10, 138.95, 145.57, 147.39, 149.94, 157.03, 161.23, 161.62 and 169.45.
3-(2-(6-(4-(Dimethylamino)piperidin-1-yl)-2-(pyridin-3-yl)pyrimidin-4-yl)-1-hydroxyethyl)-5-methyl phenol (7f)
Flash column chromatography was carried out using dichloromethane–methanol (5:1, v/v). Yield: 47 mg (94%) and mp 153–154 °C. IR (KBr) 3208, 2924, 2857, 1583, 1553, 1051, 844 and 705 cm−1. 1H NMR (CDCl3) δ 1.39–1.48 (m, 2H), 1.96–1.99 (m, 2H), 2.26 (s, 3H), 2.33 (s, 6H), 2.52–2.58 (m, 1H), 2.87–3.10 (m, 4H), 5.00 (m, 2H), 5.07 (dd, J = 5.6, 8.0 Hz, 1H), 6.54 (s, 1H), 6.68 (s, 1H), 6.72 (s, 1H), 6.93 (s, 1H), 7.52 (ddd, J = 0.8, 4.8, 8.0 Hz, 1H), 8.42 (dt, J = 1.6, 8.0 Hz, 1H), 8.60 (dd, J = 1.6, 4.8 Hz, 1H) and 9.23 (d, J = 1.6 Hz, 1H). 13C NMR (CDCl3) δ 20.20, 27.67, 40.30, 42.95, 48.26, 62.66, 72.81, 105.42, 109.88, 114.59, 117.82, 123.86, 133.95, 135.08, 138.95, 145.55, 147.37, 149.90, 157.03, 161.26, 161.44 and 169.43.
Enzyme inhibition assay
Kinase assays was performed at Reaction Biology Corporation using the “HotSpot” assay platformCitation16. Briefly, specific kinase/substrate pairs along with required cofactors were prepared in reaction buffer; 20 mM HEPES (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/mL BSA, 0.1 mM Na3VO4, 2 mM DTT and 1% DMSO. Compounds were delivered into the reaction, followed ∼20 min later by addition of a mixture of ATP (Sigma) and 33P ATP (Perkin Elmer) to a final concentration of 10 μM. Reactions were carried out at 25 °C for 120 min, followed by spotting of the reactions onto P81 ion exchange filter paper (Whatman # 3698–915). Unbound phosphate was removed by extensive washing of filters in 0.1% phosphoric acid.
After subtraction of background derived from control reactions containing inactive enzyme, kinase activity data were expressed as the percent remaining kinase activity in test samples compared to vehicle (dimethyl sulfoxide) reactions. IC50 values and curve fits were obtained using Prism (GraphPad Software). Compounds were tested against ROS1 kinase in a 10-dose IC50 mode with threefold serial dilutions starting at 20 μM, using staurosporine as a positive control.
Docking studies
Docking studies were performed using Molecular Operating Environment (MOE version 2008.10; Chemical Computing Group Canada). The program operated under “Windows XP” operating system installed on an Intel Pentium IV PC with a 2.8 MHz processor and 512 RAM. Compounds to be studied were built using the builder interface of the MOE program and subjected to energy minimization tool using the included MOPAC 7.0. The produced model was subjected to Systematic Conformational Search where all items were set as default with RMS gradient of 0.01 kcal/mol and RMS distance of 0.1 A. The X-ray crystallographic structure of ROS1 kinase enzyme co-crystallized with crizotinib (PDB code 3zbf) was obtained from the Protein Data BankCitation17. The enzyme was prepared for docking studies where: (i) the ligand molecule with any existing solvent molecules were removed from the enzyme active site; (ii) hydrogen atoms were added to the structure with their standard geometry; and (iii) MOE Alpha Site Finder was used for the active sites search in the enzyme structure and dummy atoms were created from the obtained alpha spheres. The obtained ligand–enzyme complex model was then used in calculating the energy parameters using MMFF94x force field energy calculation and predicting the ligand–enzyme interactions at the active site.
Results and discussion
Chemistry
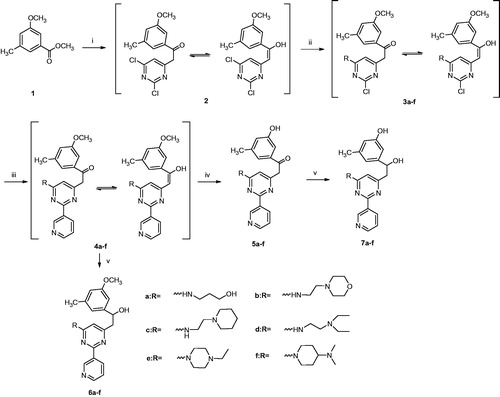
The target compounds were readily prepared according to Scheme 1. Briefly, the benzoate ester 1 underwent a nucleophilic attack at its carboxylic carbon by the activated methylene group of 2,4-dichloro-6-methylpyrimidine. The activation of this methyl group into an active methylene was achieved by dropwise addition of LHMDS in dry THF at room temperature. The resulted tautomeric α,β-unsaturated ketone 2 was then subjected to a nucleophilic substitution reaction with the amino group of the appropriate amine by heating at 80 °C in THF for 3 h to give compounds 3a–3f. The intermediates 3a–f was reacted with 3-pyridineboronic acid under palladium catalysis to afford the Suzuki coupling products 4a–f. Compounds 5a–f was obtained by demethylation of the methoxy group of the corresponding compounds 4a–f using 10 equivalent of boron trifluoride–dimethylsulfide complex in dichloromethane. The reduction of 4a–f and 5a–f with sodium borohydride in ethanol, afforded the alcohol derivatives 6a–f and 7a–f, respectively (Scheme 1).
Scheme 1. Synthesis of target compounds: (i) LHMDS, 2,4-dichloro-6-methylpyrimidine, THF, N2, rt, 24 h, 81%; (ii) THF, various amine, 80 °C, 3 h; (iii) 3-pyridineboronic acid, Pd(PPh3)2Cl2, K2CO3, N2, THF/CH3CN/H2O (2:1:1), 90 °C, 3 h; (iv) BF3 · S(CH3)2, dichloromethane, N2, rt, 12 h; and (v) NaBH4, ethanol, N2, 0 → rt, 12 h.
Evaluation of biological activity
In order to study the effect of structural modifications in the new group of compounds, all synthesized compounds were in vitro screened against ROS1 kinase enzyme. By referring to the IC50’s of the tested compounds as in , we can observe micromolar activity of most of them. But, unfortunately, it is obvious that the new compounds are much less active than the lead compound KIST301072. We think this is because of changing the central pyrazole ring into an open form. Keeping the rigid pyrazole ring linking the two aromatic moieties together seems essential for good ROS1 inhibitory activity. Changing the central pyrazole ring into an open form might lead to loss of proper orientation of the different groups in the active site of ROS1.
Table 1. The IC50 values of the target compounds against ROS1 kinase.
In addition, from all tested compounds, the compounds (4d, 5d, 6d and 7d) having N,N-diethyl amino ethylamine group as the polar side chain were the least active. This group seems to be not good water-solubilizing group in this compounds’ set.
Finally, reduction of ketone group into alcohol or demethylation of methoxy group did not have significant effect in ROS1 inhibitory activity.
Therefore, we think changing the central pyrazole ring into an open form is the major cause of decreasing ROS1 activity in this set of compounds. Other changes just have minor effect on activity.
Docking studies
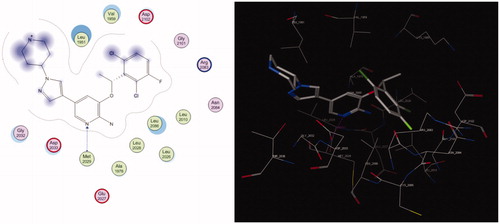
The 2.2 Å co-crystal structure of crizotinib with ROS1-kinase domain discloses how the inhibitor binds in the ATP-binding site. ROS1 binds crizotinib at the ATP-binding site in the cleft between the N-terminal and C-terminal domains of the kinase (). The G2032 residue sits at the solvent front in the distal end of the kinase hinge and creates a turn, putting the G2032 α-carbon in position to engage in a Van der Waals interaction with the pyrazole ring of crizotinibCitation17. The hinge interactions include a hydrogen bond between the pyridine of crizotinib and the backbone nitrogen of Met2029. Remarkably, this is the only direct polar interaction observed between ROS1 kinase domain and crizotinib.
Figure 2. Binding mode of crizotinib in ROS1 active pocket in 2D and 3D, respectively.
In order to find an explanation for the potency of the lead molecules and loss of potency of the designed analogues against ROS1 kinase, we applied docking simulation in the active site of ROS1 kinase using MOE.2008.10 software. Using the crystal structure ROS1 kinase enzyme co-crystallized with crizotinib (PDB code 3zbf), we found that KIST301072 exhibits similar binding behavior to that of crizotinib. The most stable docking pose of KIST301072 aligned with crizotinib in the binding pocket is shown in . KIST301072 occupies a comparable space in the binding pocket and overlays the bioactive conformation of crizotinib.
Figure 3. Overlay of crizotinib (green stick) and KIST301072 (element colored stick) bound to ROS1 active site.
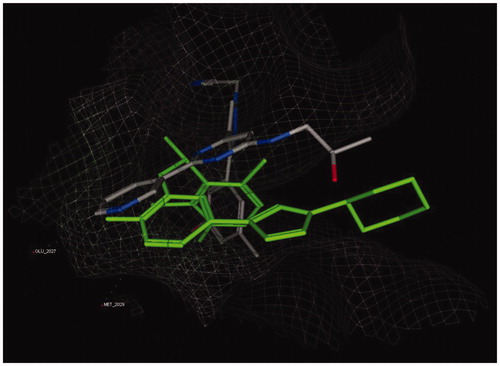
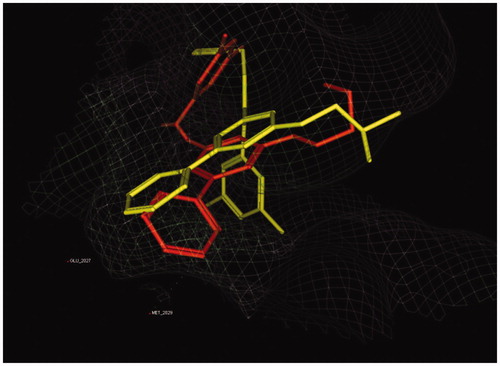
Then, compound 7a was selected as a representative from the synthesized analogues due to its similarity to KIST301072 except the absence of central pyrazole ring. Aligning the most stable docking pose of both compounds 7a and KIST301072 showed a similar orientation of both pyridine rings and solvent exposure moieties, but the lead compound occupied a different and more expanded space in ROS1 kinase active site (). Therefore, we think that the loss of potency in the designed analogues is due to their failure to occupy the same space and to have the same orientation as the lead compounds.
Figure 4. Overlay of KIST301072 (yellow stick) and compound 7a (orange stick) bound to ROS1 active site.
Conclusion
In conclusion, through synthesis and screening of this new set of compounds, we could explore an essential feature for ROS1 inhibitory activity, which is the rigidity found in diaryl pyrazole system. The flexible open form presented in this study could not achieve high ROS1 inhibitory activity of the lead. This may be due to lack of proper orientation in the active site of ROS1 kinase enzyme as it was supported by docking studies. Therefore, in future studies, we plan to preserve the rigid scaffold through replacing central pyrazole moiety with suitable isosteres.
Declaration of interest
The authors declare no conflict of interest.
The research was supported by Korea Institute of Science and Technology.
References
- Chen XW, Sun J, Zhou SF. Protein kinase inhibitors for clinical targeted cancer treatment. Clin Pharmacol Biopharmaceut 2013;2:1–2
- Dhanasekaran N, Reddy EP. Signaling by dual specificity kinases. Oncogene 1998;17:1447–55
- Paul MK, Mukhopadhyay AK. Tyrosine kinase – role and significance in cancer. Int J Med Sci 2004;1:101–15
- Giamas G, Man YL, Hirner H, et al. Kinases as targets in the treatment of solid tumors. Cell Signal 2010;22:984–1002
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009;9:28–39
- Cohen P. Protein kinases – the major drug targets of the twenty-first century? Nat Rev Drug Discov 2002;1:309–15
- Christoffersen T, Guren TK, Spindler KG, et al. Cancer therapy targeted at cellular signal transduction mechanisms: strategies, clinical results, and unresolved issues. Eur J Pharmacol 2009;625:6–22
- Tsai CJ, Nussinov R. The molecular basis of targeting protein kinases in cancer therapeutics. Sem Cancer Biol 2013;23:235–42
- Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene 2000;19:5548–57
- Blume-Jensen P, Hunter T. Oncogenic kinase signaling. Nature 2001;411:355–65
- El-Deeb IM, Yoo KH, Lee SH. ROS receptor tyrosine kinase: a new potential target for anticancer drugs. Med Res Rev 2011;31:794–818
- Park BS, El-Deeb IM, Yoo KH, et al. Design, synthesis and biological evaluation of new potent and highly selective ROS1-tyrosine kinase inhibitor. Bioorg Med Chem Lett 2009;19:4720–3
- El-Deeb IM, Park BS, Jung SJ, et al. Design, synthesis, screening, and molecular modeling study of a new series of ROS1 receptor tyrosine kinase inhibitors. Bioorg Med Chem Lett 2009;19:5622–6
- Berger D, Dutia M, Powell D, et al. Anilinoimidazo[4,5-g]quinoline-7-carbonitriles as Src Kinase Inhibitors. Bioorg Med Chem Lett 2002;12:2761–5
- Richard DJ, Verheijen JC, Curran K, et al. Incorporation of water-solubilizing groups in pyrazolopyrimidine mTOR inhibitors: discovery of highly potent and selective analogs with improved human microsomal stability. Bioorg Med Chem Lett 2009;10:6830–5
- Reaction Biology Corporation Web Site. Available from: www.reactionbiology.com
- Awad MM, Katayama R, McTigue M, et al. Acquired resistance to crizotinib from a mutation in CD74–ROS1. N Engl J Med 2013;368:2395–401