
Abstract
In this study, novel thiosemicarbazides and 1,3,4-oxadiazoles were synthesized and evaluated for their anticancer effects on human MCF-7 breast cancer cell lines. Among the synthesized derivatives studied, compound 2-(3-(4-chlorophenyl)-3-hydroxybutanoyl)-N-phenylhydrazinecarbothioamide 4c showed the highest cytotoxicity against MCF-7 breast cancer cells as it reduced cell viability to approximately 15% compared to approximately 25% in normal breast epithelial cells. Therefore, we focused on 4c for further investigations. Our data showed that 4c induced apoptosis in MCF-7 cells which was further confirmed by TUNEL assay. Western blotting analysis showed that compound 4c up-regulated the pro-survival proteins Bax, Bad and ERK1/2, while it down-regulated anti-apoptotic proteins Bcl-2, Akt and STAT-3. Additionally, 4c induced phosphorylation of SAPK/JNK in MCF-7 cells. Pretreatment of MCF-7 cells with 10 µM of JNK inhibitor significantly reduced 4c-induced apoptosis. Molecular docking results suggested that compound 4c showed a binding pattern close to the pattern observed in the structure of the lead fragment bound to JNK1. Collectively, the data of current study suggested that the thiosemicarbazide 4c might trigger apoptosis in human MCF-7 cells by targeting JNK signaling.
Introduction
Breast cancer is one of the most common malignancies and ranks the second leading cause of cancer death among women worldwide, with nearly 1.4 million new cases annuallyCitation1–4. Although significant progress has been made in breast cancer detection and treatment, survival tends to be poorer in economically developing countriesCitation5. In breast cancer, metastatic spread is responsible for virtually all of the deaths. About 6% of patients with newly diagnosed disease present with advanced or metastatic breast cancer (MBC) at the time of initial diagnosis, while about 40% of patients with early stage disease will subsequently develop metastasis. Despite the many advances in therapeutic regimens, MBC remains incurable, with an estimated 5-year overall survival rate of only 23%Citation6. Resistance to chemotherapy is also a major problem in the management of breast cancer, where many of the initially responsive tumors relapse and develop resistance to diverse chemotherapeutic agentsCitation7. Therefore, novel therapies and targets are still in demand for the management and treatment of various forms of breast cancer.
Apoptosis is a selective process of physiological cell deletion that plays an important role in the balance between cellular replication and deathCitation8. Uncontrolled cell cycle progression and deregulation of apoptosis are two characteristics of cancer. Therefore, inhibition of cell cycle progression and induction of apoptotic cancer cell death represent a key strategy for the prevention and treatment of cancer. Accordingly, many chemotherapeutic agents reportedly exert their antitumor effects by induction of apoptosis in various cancer cellsCitation9–15. c-Jun N-terminal kinases (JNKs) are members of a family of important signal transduction enzymes known as mitogen-activated protein kinases (MAPK) and can be activated by a variety of stress factorsCitation16. Some studies also indicated that JNK played a pivotal role in the induction of apoptosis in human breast cancer cells, and the cJun NH2-terminal kinase (JNK) signal transduction pathway has been implicated in mammary carcinogenesisCitation17.
Thiosemicarbazones constitute an interesting class of compounds with a wide spectrum of pharmacological applications. They have been evaluated as antiviralCitation18, antibacterialCitation19 and anticancer agentsCitation20,Citation21. Conjugated N-N-S tridentate ligand system of thiosemicarbazide (NH2-CS-NH-NH2) seems essential for anticancer activityCitation22. This structural assembly is found in compounds under clinical trials, such as the important chemotherapeutic anti-cancer agent, 3-aminopyridine-2-carboxaldehyde-thiosemicarbazone; Triapine A ()Citation23. Thus, such template could be seen as starting point for further optimization of novel anticancer agents. On the other hand, 1,3,4-oxadiazoles have received a great deal of attention owing to their wide variety of pharmacological activities especially as anticancerCitation24–28. Recently, some 3-morpholinomethyl-5-substituted-1,3,4-oxadiazole-2(3H)-thiones B () were found to possess potent cytotoxicity against human cancer cell linesCitation29. Moreover, 1,3,4-oxadiazoles are very good bioisosteres of amides and esters, which can contribute substantially in enhancing pharmacological activity by participating in hydrogen bonding interactions with the receptorsCitation30.
Figure 1. Chemical structures of triapine (A), reported antitumor 1,3,4-oxadiazoles (B) and the synthesized compounds (C and D).
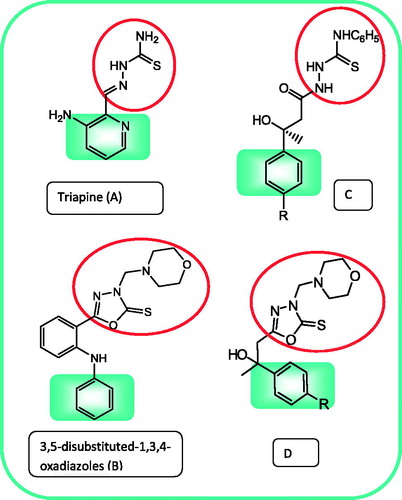
In view of the utility of thiosemicarbazide and 1,3,4-oxadiazole scaffolds for the discovery of novel antitumor agents, we thought of synthesizing a new set of thiosemicarbazides C and 1,3,4-oxadiazoles D () to be evaluated in our laboratory for their antitumor activity against breast cancer cells. The target compounds comprise some pharmacophoric elements () which influence the biological activity of the molecules, such as the aryl ring as hydrophobic binding site, the sulphur of thiosemicarbazide or oxadiazole as electron donor and the hydroxy group as hydrogen bonding domain. To understand the mechanism of action and the effects on cancer biology, we evaluated the effect of the most promising compound on cell cycle, apoptosis and expression of proteins related to cell cycle pathways.
Methods and materials
Chemistry
Melting points were determined in open glass capillaries on a Gallenkamp melting point apparatus and were uncorrected. The infrared (IR) spectra were recorded on Perkin-Elmer 1430 infrared spectrophotometer using the KBr plate technique. 1H-NMR spectra were determined either on a Bruker Avance spectrometer (300 MHz) at the microanalytical unit, Faculty of Science, Cairo University, or on Jeol (500 MHz) at the microanalytical unit, Faculty of Science, Alexandria University, using DMSO-d6 as a solvent and TMS as internal standard. The chemical shifts are given in δ ppm values (s, singlet; br s, broad singlet; d, doublet; dd, doublet of doublet; t, triplet; and m, multiplet). 13C-NMR spectra were determined on Jeol (300 MHz), Faculty of Science, Alexandria University, using TMS as internal standard. Mass spectra were run on a Finnigan mass spectrometer model SSQ/7000 (70 eV), Faculty of Science, Cairo University. Microanalyses were performed at the Microanalytical Unit, Faculty of Science, Cairo University, Egypt. The found values were within ± 0.4% of the theoretical values. Follow-up of the reactions and checking the homogeneity of the compounds were made by TLC on silica gel-protected glass plates, and the spots were detected by exposure to UV-lamp at λ 254.
General procedure for the synthesis of compounds 2a–c
The starting 3-substituted-3-hydroxy butanoic acid ethyl esters (2a–c) were prepared via Reformatsky reaction of ethyl bromoacetate with the appropriate ketones 1a–c following previously reported methodsCitation31–33.
General procedure for the synthesis of 3-(substituted) 3-hydroxy-butanoic acid hydrazides 3a–c
A mixture of 3-(substituted)-3-hydroxybutanoic acid ethyl esters 2a–c (0.01 mol) and hydrazine hydrate 98% (1.0 g, 0.97 mL, 0.02 mol) in absolute ethanol (20 mL) was heated under reflux for 3 h. The reaction mixture was concentrated, cooled and poured into ice-cold water. The separated solid was filtered, dried and crystallized from ethanol.
3-Hydroxy-3-phenylbutanoic acid hydrazide 3a
Yield: 78%, m.p.: 141–143 °C. [Reported: 81%, 140 °C]Citation34.
3-Hydroxy-3-p-tolylbutanoic acid hydrazide 3b
Yield: 73%, m.p.: 291–293 °C. IR (cm−1): 3455–3150 (OH, NH); 3062, 2978, 2930 (CH); 1640 (C=O); 1504, 1458 (C=C). 1H-NMR (δ ppm): 1.63 (s, 3H, CH3); 2.50 (s, 2H, CH2); 2.79 (s, 3H, CH3); 3.71 (br s, 2H, NH2, D2O exchangeable); 4.50 (s,1H, OH, D2O exchangeable); 7.28, 7.63 (2d, J = 8.1 Hz, each 2H, Ar–H); 8.44 (br s, 1H, NH, D2O exchangeable). Anal. Calcd for C11H16N2O2 (208.26): C, 63.44; H, 7.74; N, 13.45. Found: C, 63.11; H, 7.46; N, 13.21.
3-(4-Chlorophenyl)-3-hydroxybutanoic acid hydrazide 3c
Yield: 77%, m.p.: >300 °C. IR (cm−1): 3455–3150 (OH, NH); 3052, 2985, 2920 (CH); 1670 (C=O); 1510, 1492 (C=C). 1H-NMR (δ ppm): 1.58 (s, 3H, CH3); 2.48 (s, 2H, CH2); 3.73 (br s, 2H, NH2, D2O exchangeable); 4.70 (s, 1H, OH, D2O exchangeable); 7.38, 7.79 (2d, J = 8.2 Hz, each 2H, Ar–H); 8.44 (br s, 1H, –NH, D2O exchangeable). Anal. Calcd for C10H13ClN2O2 (228.68): C, 52.52; H, 5.73; N, 12.25. Found: C, 52.41; H, 5.71; N, 12.42.
General procedure for the synthesis of 3-(substituted)-3-hydroxybutanoic acid thiosemicarbazides 4a–c
A mixture of the corresponding hydrazides 3a–c (0.01 mol) and phenyl isothiocyanate (1.35 g, 1.2 mL, 0.01 mol) in ethanol (20 mL) was heated under reflux for 2–3 h. The reaction mixture was allowed to attain room temperature, and the formed solid was filtered, washed with ethanol and crystallized from ethanol.
1-(3-Hydroxy-3-phenylbutanoyl)-4-phenylthiosemicarbazide 4a
Yield: 87%, m.p.: 223–225 °C. IR (cm−1): 3455–3190 (OH, NH); 3057, 2893, 2838 (CH); 1680 (C=O); 1630 (C=N); 1503, 1456 (C=C); 1330 (C=S). 1H-NMR (δ ppm): 1.64 (s, 3H, CH3); 2.52 (br s, 2H, CH2); 5.71 (br s, 1H, OH, D2O exchangeable); 7.28–8.0 (m, 10H, Ar–H); 8.44, 9.5, 10.6 (3 br s, each 1H, 3NH, D2O exchangeable). 13C-NMR (DMSO-d6) δ (ppm): 29.46 (CH3); 48.23 (CH2CO); 75.92 (CH3–C–OH); 124.89 (phenyl C4); 126.01 (phenyl C4); 126.24 (phenyl C2,6); 126.58 (phenyl C2,6); 128.53 (phenyl C3,5); 129.17 (phenyl C3,5); 137.15 (phenyl C1); 149.98 (phenyl C1); 176.30 (C=O); 181.65 (C=S). MS (m/z, %): 329 (M+, 31.8), 78 (100). Anal. Calcd for C17H19N3O2S (329.42): C, 61.98; H, 5.81; N, 12.76; S, 9.73. Found: C, 61.57; H, 5.71; N, 12.67; S, 9.53.
1-(3-Hydroxy-3-(p-tolyl)butanoyl)-4-phenylthiosemicarbazide 4b
Yield: 82%, m.p.: 245–247 °C. IR (cm−1): 3440–3250 (OH, NH); 3063, 2964, 2931 (CH); 1677 (C=O); 1627 (C=N); 1598 (C=C); 1328 (C=S). 1H-NMR (δ ppm): 1.58 (s, 3H, CH3); 2.35 (s, 3H, CH3); 2.55 (s, 2H, CH2); 5.91 (br s, 1H, OH, D2O exchangeable); 7.28–7.66 (m, 5H, phenyl-H); 7.48, 7.98 (2d, J = 8.0 Hz, each 2H, tolyl-H); 9.11, 9.4, 10.5 (3 br s, each 1H, 3NH, D2O exchangeable). 13C-NMR (DMSO-d6) δ (ppm): 23.86 (p-tolyl CH3); 29.13 (CH3); 49.23 (CH2CO); 78.14 (CH3–C–OH); 124.89 (phenyl C4); 126.14 (p-tolyl C2,6); 126.98 (phenyl C2,6); 128.73 (p-tolyl C3,5); 129.29 (phenyl C3,5); 136.57 (p-tolyl C4); 138.10 (phenyl C1); 146.98 (p-tolyl C1); 178.30 (CH2CO); 181.65 (N–C=S). MS (m/z, %): 343 (M+, 13.6), 107 (100). Anal. Calcd for C18H21N3O2S (343.44): C, 62.95; H, 6.16; N, 12.23; S, 9.34. Found: C, 62.65; H, 5.95; N, 12.45; S, 9.22.
1-(3-(4-Chlorophenyl)-3-hydroxybutanoyl)-4-phenylthiosemicarbazide 4c
Yield: 89%, m.p.: 277–279 °C. IR (cm−1): 3445–3250 (OH, NH); 3057, 2893, 2838 (CH); 1680 (C=O); 1635 (C=N); 1595 (C=C); 1324 (C=S). 1H-NMR (δ ppm): 1.53 (s, 3H, CH3); 2.57(s, 2H, CH2); 5.96 (br s, 1H, OH); 7.34–7.58 (m, 5H, phenyl–H); 7.67, 7.89 (2d, J = 8.2 Hz, each 2H, chlorophenyl-H); 9.10, 9.55, 10.55 (3 br s, each 1H, 3 NH, D2O exchangeable). 13C-NMR (DMSO-d6) δ (ppm): 28.64 (CH3); 47.32 (CH2CO); 75.89 (CH3–C–OH); 124.97 (phenyl C4); 126.35 (phenyl C2,6); 126.58 (chlorophenyl C2,6); 128.78 (chlorophenyl C3,5); 129.12 (phenyl C3,5); 131.01 (chlorophenyl C4); 137.34 (phenyl C1); 149.65 (chlorophenyl C1); 175.32 (C=O); 179.56 (C=S). EI-MS (relative abundance %): 365 [M+ 2] (1.01), 363 [M+.] (2.88), 69 (100). Anal. Calcd for C17H18ClN3O2S (363.86): C, 56.12; H, 4.99; N, 11.55; S, 8.81. Found: C, 56.53; H, 4.68; N, 11.43; S, 8.64.
General procedure for the synthesis of 5-(substituted)-1,3,4-oxadiazole-2(3H)-thiones 5a–c
Carbon disulfide (2.28 g, 1.8 mL, 0.03 mol) was added dropwise over a period of 15 min to a suspension of the hydrazides 3a–c (0.01 mol) and potassium hydroxide (0.56 g, 0.01 mol) in ethanol (15 mL) at 0 °C. After complete addition, the reaction mixture was stirred at room temperature for 1 h followed by reflux until the evolution of H2S ceased. Excess solvent was then evaporated under reduced pressure, and the residue was dissolved in water and acidified with HCL to pH 5. The precipitate was filtered off, dried and crystallized from ethanol.
5-(2-Hydroxy-2-phenylpropyl)-1,3,4-oxadiazole-2(3H)-thione 5a
Yield: 86%, m.p.: 214–216 °C. IR (cm−1): 3465–3370 (OH, NH); 3105, 2963, 2910 (CH); 1616 (C=N); 1590 (C=C); 1349 (C=S). 1H-NMR (δ ppm): 1.55 (s, 3H, CH3); 2.56 (s, 2H, CH2); 5.72 (br s, 1H, OH, D2O exchangeable); 7.28–7.65 (m, 5H, Ar–H); 10.52 (br s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6) δ (ppm): 28.86 (CH3); 52.32 (CH2); 72.89 (CH3–C–OH); 126.17 (phenyl C4); 126.69 (phenyl C2,6); 128.58 (phenyl C3,5); 148.65 (phenyl C1); 157.32 (C=N); 179.64 (C=S). EI-MS m/z (relative abundance %): 237 [M+. +1] (3.45), 236 [M+.] (2.34), 55 (100). Anal. Calcd for C11H12N2O2S (236.29): C, 55.91; H, 5.12; N, 11.86; S, 13.57. Found: C, 56.12; H, 5.43; N, 11.67; S, 13.75.
5-(2-Hydroxy-2-p-tolylpropyl)-1,3,4-oxadiazole-2(3H)-thione 5b
Yield: 85%, m.p.: 223–225 °C. IR (cm−1): 3550–3235 (OH, NH); 1615 (C=N); 1595 (C=C); 1319 (C=S). 1H-NMR (δ ppm): 1.55 (s, 3H, CH3); 2.35 (s, 3H, CH3); 2.56 (s, 2H, CH2); 5.71 (br s, 1H, OH, D2O exchangeable); 7.30,7.72 (2d, J = 8.2 Hz,each 2H,Ar–H); 10.54 (br s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6) δ (ppm): 24.68 (p-tolyl CH3); 28.79 (CH3); 51.21 (CH2); 71.22(CH3–C–OH); 127.89 (phenyl C2,6); 128.97 (phenyl C3,5); 133.17 (phenyl C4); 146.65 (phenyl C1); 156.43 (C=N); 178.46 (C=S). MS (m/z, %): 250 (M+, 58.8), 119 (100). Anal. Calcd for C12H14N2O2S (250.32): C, 57.58; H, 5.64; N, 11.19; S, 12.81. Found: C, 57.85; H, 5.47; N, 11.34; S, 12.54.
5-(2-(4-Chlorophenyl)-2-hydroxypropyl)-1,3,4-oxadiazole-2(3H)-thione 5c
Yield: 72%, m.p.: 234–236 °C. IR (cm−1): 3550–3230 (OH, NH); 3099, 3052, 2917 (CH); 1617 (C=N); 1595 (C=C); 1327 (C=S). 1H-NMR (δ ppm): 1.57 (s, 3H, CH3); 2.62 (s, 2H, CH2); 5.74 (br s, 1H, OH, D2O exchangeable); 7.37, 7.77 (2d, J = 8.4 Hz, each 2H, Ar–H); 10.55 (br s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6) δ (ppm): 29.37 (CH3); 50.37 (CH2C=N); 72.38 (CH3-C); 128.32 (phenyl C2,6); 128.90 (phenyl C3,5); 129.50 (phenyl C4); 143.90 (phenyl C1); 150.00 (C=N); 172.20 (C=S). Anal. Calcd for C11H11ClN2O2S (270.74): C, 48.80; H, 4.10; N, 10.35; S, 11.84. Found: C, 48.58; H, 4.24; N, 10.68; S, 11.73.
General procedure for the synthesis of 5-(substituted)-3-(morpholinomethyl)-1,3,4-oxadiazole-2(3H)-thiones 6a–c
Formaline 40% (1.5 mL, 0.02 mol) was added to a stirred solution of 5-(substituted)-1,3,4-oxadiazole-2(3H)-thione (5a–c) (0.02 mol) in absolute ethanol (20 mL). An ethanolic solution (10 mL) of morpholine (1.74 g, 0.02 mol) was added dropwise to the reaction mixture and stirring was continued for 3 h at room temperature. The reaction mixture was then left over night in the refrigerator, and the obtained precipitate was filtered off, dried and crystallized from ethanol.
5-(2-Hydroxy-2-phenylpropyl)-3-(morpholinomethyl)-1,3,4-oxadiazole-2(3H)-thione 6a
Yield: 93%, m.p.: >300 °C. IR (cm−1): 3455 (OH); 3065, 2951 (CH); 1615 (C=N); 1325 (C=S). 1H-NMR (δ ppm): 1.59 (s, 3H, CH3); 2.4 (s, 2H, CH2); 2.97 (t, J = 6.9 Hz, 4H, morpholine C3,5–H2); 3.66 (t, J = 6.9 Hz, 4H, morpholine C2,6–H2); 4.72 (s, 2H, CH2); 5.71 (br s, 1H, OH, D2O exchangeable); 7.30–7.74 (m, 5H, Ar-H). 13C-NMR (DMSO-d6) δ (ppm): 27.96 (CH3); 50.43 (morpholine C3,5); 51.87 (CH2); 66.43 (N–CH2–N); 67.48 (morpholine C2,6); 71.98 (CH3–C–OH); 126.45 (phenyl C4); 126.78 (phenyl C2,6); 128.69 (phenyl C3,5); 147.97 (phenyl C1); 156.62 (C=N); 178.74 (C=S). EI-MS m/z (relative abundance %): 336 [M+. +1] (42.45), 335 [M+.] (43.34), 78 (100). Anal. Calcd for C16H21N3O3S (335.42): C, 57.29; H, 6.31; N, 12.53; Found: C, 57.01; H, 6.45; N, 12.34.
5-(2-Hydroxy-2-p-tolylpropyl)-3-(morpholinomethyl)-1,3,4-oxadiazole-2(3H)-thione 6b
Yield: 91%, m.p.: >300 °C. IR (cm−1): 3455–3240 (OH); 3056, 3006, 2956 (CH); 1616 (C=N); 1595 (C=C); 1338 (C=S). 1H-NMR (δ ppm): 1.62 (s, 3H, CH3); 2.32 (s, 3H, CH3); 2.49 (s, 2H, CH2); 3.00 (t, J = 6.9 Hz, 4H, morpholine C3,5–H2); 3.78 (t, J = 6.9 Hz, 4H, morpholine C2,6–H2); 4.99 (s, 2H, CH2); 5.79 (br s, 1H, OH, D2O exchangeable); 7.36, 7.68 (2d, J = 8.1 Hz, each 2H, Ar–H). 13C-NMR (DMSO-d6) δ (ppm): 24.68 (p-tolyl CH3); 27.86 (CH3); 50.45 (morpholine C3,5); 51.23 (CH2C=N); 66.25 (N–CH2–N); 66.98 (morpholine C2,6); 71.28 (CH3–C); 127.89 (phenyl C2,6); 128.97 (phenyl C3,5); 133.17 (phenyl C4); 146.65 (phenyl C1); 156.43 (C=N); 178.46 (C=S). EI-MS m/z (relative abundance %): 350 [M+. +1] (7.78), 349 [M+.] (12.14), 55 (100). Anal. Calcd for C17H23N3O3S (349.45): C, 58.43; H, 6.63; N, 12.02. Found: C, 58.21; H, 6.54; N, 11.97.
5-(2-(4-Chlorophenyl)-2-hydroxypropyl)-3-(morpholinomethyl)-1,3,4-oxadiazole-2(3H)-thione 6c
Yield: 94%, m.p.: >300 °C. IR (cm−1): 3555–3415 (OH); 3057, 2893, 2838 (CH); 1623 (C=N); 1337 (C=S). 1H-NMR (δ ppm): 1.57 (s, 3H, CH3); 2.42 (s, 2H, CH2); 3.02 (t, J = 6.9 Hz, 4H, morpholine C3,5–H2); 3.69 (t, J = 6.9 Hz, 4H, morpholine C2,6–H2); 4.88 (s, 2H, CH2); 5.78 (br s, 1H, OH, D2O exchangeable); 7.54, 7.92 (2d, J = 8.3 Hz, each 2H, Ar–H). 13C-NMR (DMSO-d6) δ (ppm): 29.37 (CH3); 50.43 (morpholine C3,5); 51.37 (CH2C=N); 66.21 (N–CH2–N); 66.84 (morpholine C2,6); 72.38 (CH3–C); 128.32 (phenyl C2,6); 128.90 (phenyl C3,5); 129.50 (phenyl C4); 148.90 (phenyl C1); 150.00 (C=N); 177.20 (C=S). EI-MS m/z (relative abundance %): 371(M+ 2) (4.5) 369 [M+.] (13.45), 109 (100). Anal. Calcd for C16H20ClN3O3S (369.87): C, 51.96; H, 5.45; N, 11.36; S, 8.67. Found: C, 52.12; H, 5.18; N, 11.09; S, 8.44.
Pharmacology
Cell culture and Compound treatment
MCF-7 breast cancer cells with wild-type p53 gene were purchased from ATCC (Manassas, VA). MCF-7 cells were maintained in Dulbecco’s modified essential media (DMEM) (Gibco, Carlsbad, CA) supplemented with 10% Fetal Bovine Serum, 100 Units/ml penicillin and 100 μg/ml streptomycin at 37 °C in a 5% CO2 atmosphere (Gibco, Carlsbad, CA). The synthesized compounds were prepared in 20 μM concentration and dissolved in suitable media.
Methyl tetrazolium (MTT) bromide mitochondrial activity assay
Cell viability was measured by methyl tetrazolium (MTT) (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay as described previouslyCitation35 (ATCC, Manassas, VA). Briefly, 4000–5000 cells/well in 100 µL of medium were seeded into 96-well plates. After 24 h incubation at 37 °C, the culture media were removed and replaced with fresh media containing the test compounds (100 μM). The cells were incubated for another 24 h, then 10 µL of 5 mg/ mL MTT reagent was added to each well and incubated for 4 h at 37 °C. After incubation, 100 µL of detergent reagent was added to each well to dissolve the formazan crystals. The absorbance was determined at 570 nm using Spectra Max Plus. Each assay was performed in triplicate, and the experiment was repeated three times and standard deviation was determined. To determine the IC50 of compound 4c, the assay was conducted using different concentrations of compound 4c, and the average 50% inhibitory concentration (IC50) was determined from the dose–response curve.
Enzyme linked immunosorbent apoptosis assay
Cells were seeded at a density of 2 × 104/well in a 96-well plate and incubated for 24 h. Media were changed to media containing compound 4c (20 μM). Cells were then incubated for 24 h. An ELISA assay was performed using Cell Death Detection ELISAPLUS kit (Roche-Applied Science, Indianapolis, IN) that measures histone release from fragmented DNA in apoptosing cells. Briefly, cells were lysed with 200-μL lysis buffer for 30 min at room temperature. The lysate was centrifuged at 200 g for 10 min. 150 μL of supernatant was collected, of which 20 μL was incubated with anti-histone biotin and anti-DNA peroxidase at room temperature for 2 h. After washing with incubation buffer three times, 100 μL of substrate solution (2,2′azino-di(3-ethylbenzthiazolin-sulphuric acid) was added to each well and incubated for 15–20 min at room temperature. The absorbance was measured using an ELISA reader (Spectra Max Plus) at 405 nm. The control group was cells treated with either UV or gamma radiation. Each assay was done in triplicate, and the standard deviation was determined.
TUNEL assay
For in situ detection of apoptotic cells, TUNEL (terminal-deoxynucleotidyl transferase meditated nick end labeling) assay was performed using DeadEnd™ fluorimetric tunnel system (Promega, Madison, WI). Cells were cultured on 4-chamber slides (VWR, Radnor, PA) at a density of 2 × 104cells/chamber. After treatment with 20 μM of compound 4c, cells were washed with phosphate-buffered saline (PBS) and fixed by incubation in 4% paraformaldehyde (PFA) for 20 min at 4 °C, then permeabilized with 0.05% triton X-100 for 5 min at 4 °C. The fixed cells were then incubated with digoxigenin-conjugated dUTP in terminal deoxynucleotide transferase recombinant (rTdT)-catalyzed reaction and nucleotide mixture for 60 min at 37 °C in a humidified atmosphere and then immersed in stop/wash buffer for 15 min at room temperature. Cells were then washed with PBS to remove unincorporated fluorescein-12-dUTP. After washing with PBS, cells were incubated in 1 μg/mL 2-(4-amidinophenyl)-6-indole carbamidine dihydrochloride (DAPI) and fluorescin isothiocyanate (FITC) solution for 15 min in dark (data not shown). Cells were observed with fluorescence microscopy (RT slider Spot, Diagnostic Instruments, Inc) and photographed at 100 × magnification.
Impact of compound 4c on cell cycle
Cells were seeded at a density of 3–5 × 105/10 cm plate and incubated for 24 h before treatment. Media were changed to media containing 20 μM of compound 4c; 24 h later, cells were harvested by trypsinization. The cells were washed with PBS and fixed with ice-cold 70% ethanol while vortexing. Finally, the cells were washed and resuspended in PBS containing 5 μg/mL RNase A (Sigma, St. Louis, MO) and 50 μg/mL propidium iodide (Sigma, St. Louis, MO) for analysis. Cell cycle analysis was performed using FACScan Flow Cytometer (Becton Dickson, East Rutherford, NJ) according to the manufacturer’s protocol. Windows multiple document interfaces (WinMDI) software was used to calculate the cell-cycle phase distribution from the resultant DNA histogram and expressed as a percentage of cells in the G0/G1 and G2/M phases. The apoptotic cells were identified on the DNA histogram as a subdiploid peak.
Determination of pro-apoptotic, anti-apoptotic proteins and protein kinases by Western blotting analysis
Total protein was extracted from cells treated with 20 µM of compound 4c and untreated cells using lysis buffer (10 mM Tris HCl, pH 7.5, 1 mM EDTA, 1% triton X-100, 150 mM NaCl, 1 mM dithiothretol, 10% glycerol, 0.2 mM phenylmethysulphonyl fluoride and protease inhibitors) for 30–50 min on ice. The extracts were centrifuged at 13,000 rpm for 15 min at 4 °C to remove cell debris. Folin Lowry (Pierce, Rockford, IL) protein assay was used to determine the protein concentration in the cell lysates. Proteins were resolved by electrophoresis on 8–10% sodium dodecyl sulphate–polyacrylamide gel loading equal amount of proteins per lane. The resolved proteins were transferred to polyvinylidene difluoride (PVDF) membrane and then probed with primary antibody against the protein of interest prepared in 5% milk/PBS-T. The membrane was washed using PBS with Tween 20 (PBS-T), and then appropriate secondary antibody conjugated to horseradish peroxidase (HRP) was used for visualization of the bands using ECL chemiluminescence kit (GE Healthcare Bio-Sciences, Pittsburgh, PA). Anti-Akt, anti-Bcl-2, anti-Bax, anti-Bad and anti-JNK were purchased from Santa Cruz, USA. MEK1 and SP600125 were purchased from Santa Cruz Biotechnology, Dallas, TX. Pixel density of the proteins studied was calculated using Image J, version 1.41o, NIH. The obtained values were first normalized to loading control β-actin, and folds increase was measured by normalizing to the control (0 h) value. At least two independent experiments were performed.
Docking study
Computer-assisted simulated docking experiments were carried out under an MMFF94X force field using Molecular Operating Environment (MOE Dock 2009) software, Chemical Computing Group, Montréal, QC.
Docking protocol
The coordinates from the X-ray crystal structure of JNK1 used in this simulation were obtained from the Protein Data Bank (PDB ID: 2H96), where the active site is bound to pyridine carboxamide molecule. The ligand molecules were constructed using the builder module and were energy minimized. The active site of JNK1 was generated using the MOE-Alpha Site Finder, and then ligands were docked within this active site using the MOE Dock. The lowest energy conformation was selected, and the ligand interactions (hydrogen bonding and hydrophobic interaction) with JNK1 were determined.
Results and discussion
Chemistry
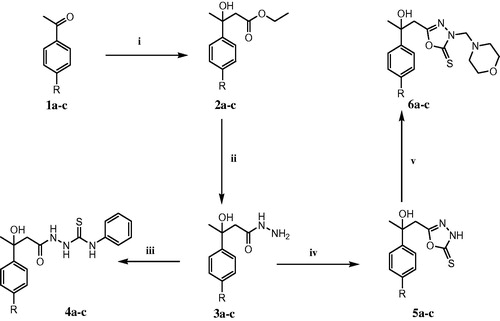
Synthetic strategies adopted for the synthesis of the intermediates and target compounds are depicted in Scheme 1. The starting compounds Ethyl 3-substituted-3-hydroxybutanoates 2a–c were prepared via Reformatsky reaction from the readily available acetophenones 1a–c with ethyl bromoacetate in the presence of zinc following previously published proceduresCitation31–33. Hydrazinolysis of the esters 2a–c afforded the corresponding key intermediate hydrazides 3a–c. 1H-NMR spectra of 3a–c lacked the triplet and quartet of the ethyl moiety and revealed the broad exchangeable protons of NH and NH2 characteristic for the hydrazide moiety at their expected chemical shifts. Refluxing equimolar amounts of the hydrazides 3a–c and phenyl isothiocyante in ethanol yielded the expected thiosemicarbazides 4a–c. 1H-NMR spectra of compounds 4a–c displayed the three amidic NH protons characteristic for thiosemicarbazides. The oxadiazole-2-thione derivatives 5a–c were prepared via cyclization of the hydrazides 3a–c with carbon disulphide in refluxing ethanol containing potassium hydroxide adopting a procedure reported for the synthesis of analogous compoundsCitation36. IR spectra of 5a–c lacked C=O absorption bands (characteristic for the hydrazide) and showed vibrational bands at 3370–3230 cm−1 (NH) and 1617–1615 cm−1 (C=N) which support the conversion of the hydrazides to the corresponding oxadiazole-2-thiones. Additionally, the presence of bands due to NH and C=S suggests the existence of compounds 5a–c in the thione form rather than the thiol form. 1H-NMR spectra of compounds 5a–c displayed a downfield broad singlet at around 10.5 ppm characteristic for the oxadiazoline CS–NH. Reaction of the latter compounds with formaldehyde and morpholine afforded the target compounds 6a–c in good yield. IR spectra of compounds 6a–c generally showed the characteristic bands corresponding to hydroxyl group and thione function. The 1H NMR spectra displayed an extra singlet corresponding to CH2 protons and triplets of the morpholino protons, in addition to other protons which were observed at their expected chemical shifts.
Scheme 1. Synthetic pathways for the target compounds. For 1–6: a, R = hydrogen; b, R = methyl; c, R = chloro. Reagents and conditions: (i) ethyl bromoacetate/Zn/benzene; (ii) NH2NH2.H2O/absolute ethanol/reflux; (iii) phenyl isothiocyanate/ethanol/reflux; (iv) CS2/KOH/reflux; (v) HCHO 40%/morpholine.
Pharmacology
Thiosemicarbazide derivatives reduced cell viability and induced apoptosis in MCF-7 breast cancer cells
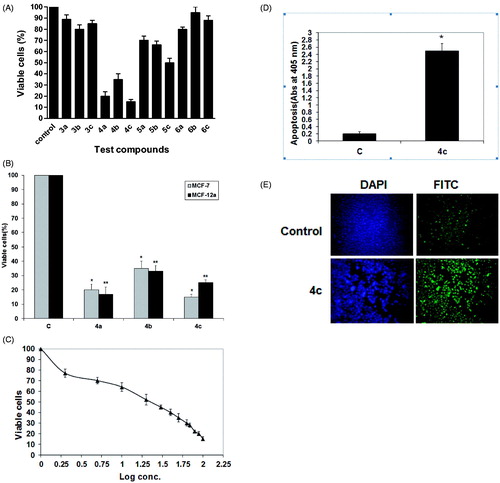
The synthesized compounds were assayed for their in vitro cytotoxicity on four breast cancer cell lines namely; MCF 7, MDA-MB-231, T47D and MDA-MB-157 cell lines. The obtained results showed that the MCF-7 cell line was the most sensitive towards some of the tested compounds (Supplementary Material). Among the synthesized compounds, the thiosemicarbazides 4a–c were the most potent derivatives in reducing viability in MCF-7 breast cancer cells (% viability = 15–35%) (). Accordingly, they were selected for further investigation in normal breast cells (MCF12a). The cell viability assay indicated that 4c reduced cell viability to approximately 15% in breast cancer cells compared to approximately 25% in normal breast epithelial cells (). Based on the fact that 4c had less cytotoxic effect on normal breast epithelial cells rather than the other compounds, we focused on 4c for further studies. The dose–response curve was constructed by plotting % of cell viability versus different concentrations of 4c ranging from 2 to 100 µM, and the IC50 for 4c was determined to be 20 µM (). To determine whether the inhibitory effects of 4c on MCF-7 cancer cell growth were correlated with apoptosis, we further investigated the effects of 4c on histone release from fragmented DNA in apoptosing cells using ELISA. The results revealed that addition of 20 µM 4c increased apoptosis in MCF-7 by approximately 12-fold increase compared to non-treated controls (). To ascertain induction of apoptosis after treating cells with 20 µM 4c, TUNEL assay was performed. TUNEL assay involves labeling of the 3′-hydroxyl DNA ends generated during DNA fragmentation by means of rTdT and labeled dUTP. The localized fluorescence of apoptotic cells (labeled-dUTP) in a blue background (DAPI) or green background (FITC) was visualized using fluorescence microscope. The assay results () revealed the presence of nuclear condensation and TUNEL-positive cells (stained cells).
Figure 2. Thiosemicarbazides reduced cell viability and induced apoptosis in human MCF-7 breast cancer cells. (A) MCF-7 cells were treated with 100 µM of test compounds for 24 h. Cells without test compounds were used as controls. *p < 0.05 as compared to the mock-treated controls using unpaired Student t test. The bars represent the mean ± SD (n = 3). (B) MCF-7 and MCF-12a were treated with 100 µM of compounds 4a–c for 24 h. Untreated cells were used as controls. Each data point was an average of results from three independent experiments performed in triplicate and presented as mean ± SD (n = 3). Significant differences between the control and 4c are indicated by *(***p < 0.05). (C) Dose–response curve for compound 4c. A dose–response curve for 4c was constructed, and IC50 was determined to be 20 µM. Each data point was an average of results from three independent experiments performed in triplicate and presented as mean ± SD. (D) MCF-7 cells were treated with 20 µM of compound 4c for 24 h. Untreated cells were used as control (C). ELISA assay was applied for apoptotic cell detection. The bars represent the mean ± SD (n = 3). Significant differences between the control and 4c are indicated by *(*p < 0.05). (E) TUNEL staining of MCF-7 cells after exposure to compound 4c at a concentration of 20 µM for 24 h. Stained cells represent TUNEL positive cells, and the control cells (C) act as control for the staining procedure.
Impact of compound 4c on cell cycle checkpoints and apoptotic signaling
Flow cytometry is regarded as one of the most powerful and specific methods for the integrated study of molecular and morphological events occurring during cell death and cell proliferationCitation37. Generally, anticancer drugs inhibit the proliferation of cancer cell either by induction of cell cycle arrest or by apoptosis, or by the combination of both these modes. However, by measuring the DNA content, it becomes possible to identify apoptotic cells; to recognize the cell cycle phase specificity and to quantitate apoptosis. Therefore, the effect of 4c on cell cycle checkpoints of MCF-7 cells was studied by flow cytometry in propidium iodide (PI) stained cells as shown in . After treating MCF-7 cells with 20 μM of 4c for 24 h, a significant decrease in the percent of G2 cells (25%) compared to control group (47%) was observed (); however, no significant difference was found in the percent of cells in the S phase. Moreover, the apoptotic cells increased to 23% after treatment with 4c compared to control (8%). Hence, it can be concluded that 4c inhibits the viability of MCF-7 cells, and the growth inhibition is associated with induction of apoptosis. A representative image of DNA histogram is illustrated in (supplementary material).
Figure 3. Impact of 4c on cell cycle and apoptotic signaling in MCF-7 cells. (A) Cells were treated as described under experimental. The % of cell cycle phases was determined in MCF-7 cells after treatment with 20 µM of 4c for 24 h. Each data point is the mean of three independent experiments and expressed as mean ± SD. Significant differences between the control and 4c are indicated by *(*p < 0.05, **p < 0.01). (B) A representative image of DNA histogram for control and treated cells with 20 µM of 4c for 24 h. (C) Cells were treated with 20 µM of compound 4c for 24 h. The cell lysates were collected, and the expression of Bcl-2, STAT-3, Akt, Bax, Bad and ERK1/2 was studied by Western blotting analysis using specific antibodies. β-actin was used as loading control. (C) Control (untreated cells).
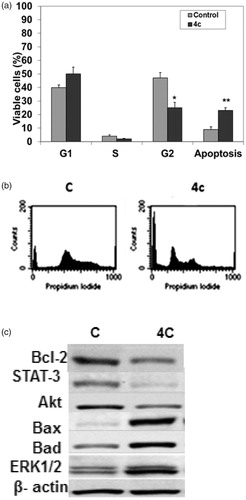
The mechanism of the pro-apoptotic effect of 4c was further investigated. Therefore, we monitored the levels of some mitochondrial proteins involved in apoptosis such as proapoptotic proteins; Bax, Bad, antiapoptotic proteins; Bcl-2 and protein kinases; ERK1/2, AKT and STAT 3. ERK1/2 is an important subfamily of mitogen-activated protein kinases that control a broad range of cellular activities and physiological processes involved in cell survival; however, recent evidence suggests that the activation of ERK1/2 also contributes to cell death in some cell types and organs under certain conditionsCitation38. Akt is a protein kinase involved in mitogenic signal transduction to the cell cycle control system and negatively controls apoptosisCitation39, whereas signal transducer and activator of transcription 3 (STAT3) is the most studied member of a family of STAT proteins that are involved in the regulation of angiogenesis, cell proliferation, differentiation and apoptosis. STAT3 is hyperactivated in many human tumors and represents an attractive therapeutic target for the treatment of various types of cancerCitation40. Our results from Western blot analysis showed that, in comparison to the non-treated control, treatment of MCF-7 cells with 4c increased proapoptotic proteins Bax, Bad and ERK1/2 levels by approximately 4-, 3- and 2.5-folds, respectively, in MCF-7 cells after 24 h. In contrast, the expression levels of anti-apoptotic proteins Bcl-2, STAT3 and Akt were reduced to approximately 3-, 2- and 1.5-fold, respectively (). These results suggest that 4c-induced apoptosis may be mediated through the down-regulation of anti-apoptotic proteins, Akt and STAT 3 together with upregulation of proapoptotic proteins and ERK1/2.
Effect of compound 4c on SAPK/JNK phosphorylation in MCF-7
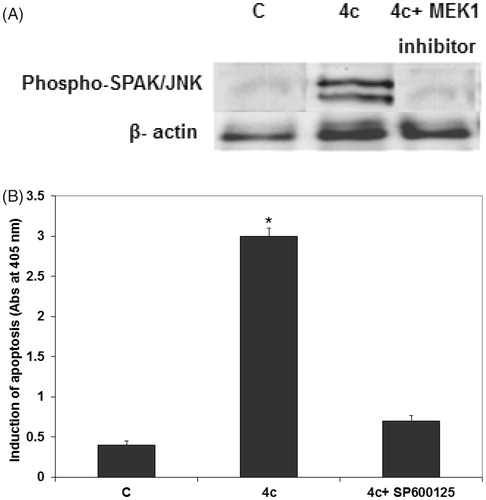
JNK exert tumor suppressive and oncogenic functions in a cell type-specific or context-specific mannerCitation41. Some studies have shown that JNK kinase may regulate the cancer cell cycle progression. Therefore, we proposed that 4c may be a promising chemical probe to help understand the role of JNK in cell cycle regulation. To test this hypothesis, we measured phosphorylated JNK levels using Western blotting analysis. Our data showed that the phosphorylation of SAPK/JNK on Thr 183 and Tyr 185 in MCF-7 cells significantly increased 24 h after 4c treatment (). Moreover, the phosphorylation of SAPK/JNK in MCF-7 was completely inhibited by 4c and MEK1 inhibitor (). In addition, pretreatment of MCF-7 cells with 10 µM of JNK inhibitor (SP600125) 1 h prior to 4c exposure significantly reduced 4c-induced cell apoptosis (). This suggests that SAPK/JNK phosphorylation activation possibly mediates the apoptosis induced by derivative 4c in MCF-7 cells.
Figure 4. 4c increased the expression levels of JNK in MCF-7 cells. (A) MCF-7 cells were treated with 20 µM of 4c alone and with 20 µM of 4c and 10 µM of MEK1 inhibitor. Total protein was extracted for Western blot analysis to determine SAPK/JNK phosphorylation under different treatment conditions. Non-treated or mock-treated cells were used as control (C) and β-actin was used as loading control. (B) MCF-7 cells were treated with 20 µM of 4c alone and with 20 µM 4c and 10 µM SP600125. Induction of apoptosis was determined at 405 nm and compared to control (untreated cells). Each data point is the mean of three independent experiments and expressed as mean ± SD.
Docking study
Docking study was carried out using the enzyme parameters obtained from the crystallographic structure of the complex between JNK1 with the co-crystallized pyridine carboxamide () (PDB ID: 2H96)Citation42. The docking simulation for the ligand was carried out using molecular operating environment (MOE) software supplied by the Chemical Computing Group, Inc., Montréal, QCCitation43. The X-ray crystal structure revealed that pyridine carboxamide inhibitor binds to the ATP binding site and makes several interactions with the hinge-binding region of the enzyme. The carbonyl group in the inhibitor is hydrogen-bonded to the Met 111 NH group. The pyridine ring interacts with the hydrophobic surface of the terminal methyl group of Met108 and the side chains of Ile 86, Leu168 and Val 158. The ethoxy group on the pyridine ring points to the ribose binding pocket, and the cyano group on the pyridine ring is in van der Waals contact with Lys55 and Met108. The phenyl ring in the molecule provides some hydrophobic interactions with Ile 32. The 5-methoxy group on the phenyl ring is in van der Waals contact with Ala 42.
Figure 5. (A) Binding mode of pyridine carboxamide inhibitor in the binding site of JNK1 using MOE software. (B) Binding mode of compound 4c docked and minimized in the JNK1 binding site using MOE software.
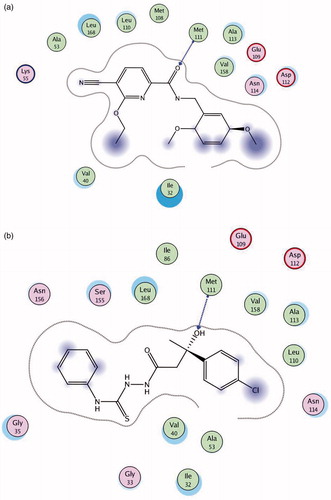
shows the binding mode of compound 4c docked and minimized in the JNK1 binding site. In this case, a hydrogen bond is observed between the oxygen of compound 4c and NH group of Met 111. In addition, compound 4c displayed hydrophobic interactions with the same amino acids Ile 32, Ile 86, Leu 168 and Val 158 bound to pyridine carboxamide molecule. Therefore, the docking study showed that compound 4c has a binding pattern which is close to the pattern observed in the structure of the lead fragment bound to JNK1. Compound 4c forms hydrogen bonds with the same residue (Met 111) as that observed in the structure of the inhibitor with JNK1. This may account for increased activity recorded for compound 4c.
Conclusion
The aim of the present study was to synthesize and investigate the anticancer activity of a group of new thiosemicarbazides and 1,3,4-oxadiazole derivatives in MCF-7 breast cancer cells. The results demonstrated that the thiosemicarbazide derivative 4c reduced viability and induced apoptosis in breast cancer cells and was less cytotoxic to normal breast epithelial cells than breast cancer cells. In addition, 4c modified apoptotic response and increased the expression levels of proapoptotic proteins Bad, Bax and ERK1/2 protein kinases, while it reduced expression levels of antiapoptotic proteins Bcl-2 and protein kinases Akt and STAT3. Additionally, 4c increased the phosphorylated SAPK/JNK which was completely inhibited by MEK inhibitor I. Moreover, 4c-induced apoptosis was abolished by JNK inhibitor. Thus, it seems plausible that JNK could play a role in induction of apoptosis by 4c. MOE-based molecular docking results suggested that compound 4c showed a binding pattern close to the pattern observed in the structure of the lead fragment bound to JNK1. Finally, 4c could be considered as a starting point for further optimization and derivatization in order to synthesize compounds with more potency and less toxicity.
Supplementary material available online
Supplemental Material.pdf
Download PDF (58.6 KB)Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing the paper. The authors would like to thank the Department of Health Sciences, Qatar University for financial support of the biological part of this work.
References
- Youlden DR, Cramb SM, Dunn NA, et al. The descriptive epidemiology of female breast cancer: an international comparison of screening, incidence, survival and mortality. Cancer Epidemiol 2012;36:237–48
- Siegel R, Naishadham D, Jemal A. Cancer statistics. CA Cancer J Clin 2013;63:11–30
- DeSantis C, Siegel R, Bandi P, Jemal A. Breast cancer statistics. CA Cancer J Clin 2011;61:408–18
- Wiebe JP, Zhang G, Welch I, Cadieux-Pitre HAT. Progesterone metabolites regulate induction, growth, and suppression of estrogen- and progesterone receptor-negative human breast cell tumors. Breast Cancer Res 2013;15:R38
- Anderson BO, Yip CH, Ramsey SD, et al. Breast cancer in limited-resource countries: health care systems and public policy. Breast J 2006;12:54–69
- Tinoco G, Warsch S, Glück S, et al. Treating breast cancer in the 21st century: emerging biological therapies. J Cancer 2013;4:117–32
- Wind NS, Holen I. Multidrug resistance in breast cancer: from in vitro models to clinical studies. Int J Breast Cancer 2011;2011:1–12
- Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 2010;9:447–64
- Lee J, Lim KT. Phytoglycoprotein (38 kDa) induces cell cycle (G/G) arrest and apoptosis in HepG2 cells. J Cell Biochem 2011;112:3129–39
- Kim JE, Kim JY, Lee KW, Lee HJ. Cancer chemopreventive effects of lactic acid bacteria. J Microbiol Biotechnol 2007;17:1227–35
- Kim MK, Min J, Choi BY, et al. Discovery of cyclin-dependent kinase inhibitor, CR229, using structure-based drug screening. J Microbiol Biotechnol 2007;17:1712–16
- Lim H, Oh HL, Lee CH. Effects of Aralia continentalis root extract on cell antiproliferation and apoptosis in human promyelocytic leukemia HL-60 cells. J Microbiol Biotechnol 2006;16:139–1404
- Lim H, Lim Y, Cho YH, Lee CH. Induction of apoptosis in the HepG2 cells by HY53, a novel natural compound isolated from bauhinia forficata. J Microbiol Biotechnol 2006;16:1262–8
- Jim JH, Sung KJ, Cho MC, et al. Antitumor effect of soluble beta-1,3-glucan from Agrobacterium sp. R259 KCTC 1019. J Microbiol Biotechnol 2007;17:1513–20
- Sah JF, Balasubramanian S, Eckert RL, Rorke EA. Epigallocatechin-3-gallate inhibits epidermal growth factor receptor signaling pathway. Evidence for direct inhibition of ERK1/2 and AKT kinases. J Biol Chem 2004;279:12755–62
- Olson JM, Hallahan AR. p38 MAP kinase: a convergence point in cancer therapy. Trends Mol Med 2004;10:125–9
- Cellurale C, Girnius N, Jiang F, et al. Role of JNK in mammary gland development and breast cancer. Cancer Res 2012;72:472–81
- Garcia CC, Brousse BN, Carlucci MJ, et al. Inhibitory effect of thiosemicarbazone derivatives on Junin virus replication in vitro. Antivir Chem Chemother 2003;14:99–105
- Sau DK, Butcher RJ, Chaudhuri S, Saha N. Spectroscopic, structural and antibacterial properties of Cu(II) complexes with bio-relevant 5-methy-3-formylpyrazole N(4)-benzyl-N(4)-methylthiosemicarbazone. Mol Cell Biochem 2003;253:21–9
- Đilović I, Rubčić M, Vrdoljak V, et al. Novel thiosemicarbazone derivatives as potential antitumor agents: synthesis, physicochemical and structural properties, DNA interactions and antiproliferative activity. Bioorg Med Chem 2008;16:5189–98
- Vandresen F, Falzirolli H, Batista SAA. Novel R-(+)-limonene-based thiosemicarbazones and their antitumor activity against human tumor cell lines. Eur J Med Chem 2014;79:110–16
- Hu K, Yang Z, Pan S-S, et al. Synthesis and antitumor activity of liquiritigenin thiosemicarbazone derivatives. Eur J Med Chem 2010;45:3453–8
- Murren J, Modiano M, Clairmont C, et al. Phase I and pharmacokinetic study of triapine, a potent ribonucleotide reductase inhibitor, administered daily for five days in patients with advanced solid tumors. Clin Cancer Res 2003;9:4092–100
- Abu-Zaied MAZ, Nawwar GAM, Swellem RH, El-Sayed SH. Synthesis and screening of new 5-substituted-1,3,4-oxadiazole-2-thioglycosides as potent anticancer agents. Pharmacol Pharm 2012;3:254–61
- Kumar D, Sundaree S, Johnson EO, Shah K. An efficient synthesis and biological study of novel in-dolyl-1,3,4-oxadiazoles as potent anticancer agents. Bioorg Med Chem Lett 2009;19:4492–4
- Zhang X-M, Qiu M, Sun J, et al. Synthesis, biological evaluation, and molecular docking studies of 1,3,4-oxadiazole derivatives possessing 1,4-benzodioxan moiety as potential anticancer agents. Bioorg Med Chem 2011;19:6518–24
- Ramazani A, Khoobi M, Torkaman Ar, et al. One-pot, four-component synthesis of novel cytotoxic agents1-(5-aryl-1,3,4-oxadiazol-2-yl)-1-(1H-pyrrol-2-yl)methanamines. Eur J Med Chem 2014;78:151–6
- Aboraia AS, Abdel-Rahman HM, Mahfouz NM, El-Gendy MA. Novel 5-(2-hydroxyphenyl)-3-substituted-2,3-dihydro-1,3,4-oxadiazole-2-thione derivatives: promising anticancer agents. Bioorg Med Chem 2006;14:1236–46
- Abdel Rahman DE. Synthesis, quantitative structure-activity relationship and biological evaluation of 1,3,4-oxadiazole derivatives possessing diphenylamine moiety as potential anticancer agents. Chem Pharm Bull 2013;61:151–9
- Guimarães CRW, Boger DL, Jorgensen WL. Elucidation of fatty acid amide hydrolase inhibition by potent α ketoheterocycle derivatives from Monte Carlo simulations. J Am Chem Soc 2005;127:17377–84
- O'Boyle NM, Greene LM, Bergin O, et al. Synthesis, evaluation and structural studies of antiproliferative tubulin-targeting azetidin-2-ones. Bioorg Med Chem 2011;19:2306–25
- Picotin G, Miginiac P. Activation of zinc by trimethylchlorosilane. An improved procedure for the preparation of beta-hydroxy esters from ethyl bromoacetate and aldehydes or ketones (Reformatsky reaction). J Org Chem 1987;52:4796–8
- Frankenfeld JW, Werner JJ. Improved procedure for the Reformatsky reaction of aliphatic aldehydes and ethyl bromoacetate. J Org Chem 1969;34:3689–91
- Newman MS, Kutner A. New reactions involving alkaline treatment of 3-nitroso-2-oxazolidones. J Am Chem Soc 1951;73:4199–204
- Krystof V, Cankar P, Frýsová I, et al. 4-Arylazo-3,5-diamino-1H-pyrazole CDK inhibitors:SAR study, crystal structure in complex with CDK2, selectivity, and cellular effects. J Med Chem 2006;49:6500–9
- Abdel-Rahman HM, Hussein MA. Synthesis of beta-hydroxypropanoic acid derivatives as potential anti-inflammatory, analgesic and antimicrobial agents. Arch Pharm (Weinheim) 2006;339:378–87
- Shrivastava HY, Ravikumar T, Shanmugasundaram N, et al. Cytotoxicity studies of chromium (III) complexes on human dermal fibroblasts. Free Radical Bio Med 2005;38:58–69
- Zhuang S, Schnellmann RG. A death-promoting role for extracellular signal-regulated kinase. J Pharmacol Exp Ther 2006;319:991–7
- Kim H, Khursigara G, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol 2001;21:893–901
- Haftchenary S, Avadisian M, Gunning PT. Inhibiting aberrant Stat3 function with molecular therapeutics: a progress report. Anticancer Drugs 2011;22:115–27
- Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 2009;9:537–49
- Zhao H, Serby MD, Xin Z, et al. Discovery of potent, highly selective, and orally bioavailable pyridine carboxamide c-Jun NH2-terminal kinase inhibitors. J Med Chem 2006;49:4455–8
- Molecular Operating Environment (MOE) 2009.10, Montréal, Canada: Chemical Computing Group Inc; 2009. Available from: http://www.chemcomp.com