
Abstract
Context: Thioredoxin reductase (TrxR) is up-regulated in a number of human malignant cells and becomes a promising target for anticancer drug development.
Objective: To evaluate N-acetyl-S-(p-chlorophenylcarbamoyl)cysteine (NACC), a potent anticancer agent against melanoma, as an inhibitor of mammalian TrxR1.
Material and methods: The mechanism of inhibition against TrxR1 was investigated using substrate protection, dialysis and liquid chromatography–tandem mass spectrometry.
Results: NACC inhibits TrxR1 in a time and concentration dependent manner. The Ki and kinact of NACC against TrxR1 were determined to be 80 μM and 0.178 min−1, respectively. The inhibition occurred only in the presence of NADPH and persisted after extensive dialysis. The tandem mass spectrometric analysis demonstrated that the selenocysteine rather than cysteine residue at the active site was p-chlorophenyl carbamoylated by NACC. Inhibition of intracellular TrxR by NACC in cultured melanoma cells was observed.
Discussion and conclusion: NACC which irreversibly inhibits TrxR1 by forming a covalent bond with selenocysteine can be an effective tool in the study of TrxR1.
Introduction
Mammalian thioredoxin reductases (TrxRs), a family of pyridine nucleotide-disulfide oxidoreductases with a special selenocysteine (Sec) residue, are responsible for the NADPH-dependent reduction of thioredoxin (Trx) and many other oxidized cellular constituentsCitation1–3. These enzymes possess an N-terminal redox motif (Cys59–Cys64) and a C-terminal cysteine–selenocysteine motif (Cys497–Sec498). During catalysis, electrons transferred from NADPH via enzyme-bound flavin adenine dinucleotide (FAD) are delivered from N-terminal active site to the oxidized C-terminal arm reducing the selenylsulfide (S-Se, formed by Cys497/Sec498)Citation4,Citation5. As a result, TrxR containing reduced Sec498 residue is able to catalyze the reduction of Trx and other substrates. Three isoforms of TrxR have been identified in mammals: (i) cytosolic TrxR1, (ii) mitochondrial TrxR2 and (iii) testis-specific TrxR3Citation5,Citation6. Accumulated evidence indicates that TrxR1 plays important roles in the development and progression of cancer. TrxR1 is overexpressed in many human malignant cells including melanoma, breast cancer, non-small cell lung carcinoma, thyroid, prostate cancer, etcCitation7–9. In some of the malignant cells, the overexpression of TrxR1 is 10-fold or even greater than those in normal cellsCitation10–13 resulting in remarkably increased TrxR1 activity, which is possibly associated with tumor aggressiveness, apoptosis inhibition, metastasis and increased resistance to chemotherapy. A decrease in TrxR1 activity in cancer cells reverses many characteristics of cancer malignancy and inhibits cancer cell growth and DNA replicationCitation10,Citation14,Citation15. Numerous pieces of evidence have suggested TrxR1 to be a promising target for anticancer drug developmentCitation2,Citation7,Citation10.
In recent years, various TrxR inhibitors have been developed or identified. Nitroureas, organochalcogenides (S, Se and Te), nitroaromatic compounds, polyphenols and metal complexes were identified as effective TrxR inhibitorsCitation1. However, by 2014, only two anticancer compounds targeting TrxR were under clinical trials: Motexafin gadolinium (MGd, Phase III) and ethaselen (Phase I). MGd targets both TrxR and ribonucleotide reductase and improves the therapeutic effect of whole-brain radiation therapy in non-small-cell lung cancer patients. Ethaselen shows significant activities against various cancer cell lines. It has also been used in combination with cisplatin in the treatment of colon adenocarcinomaCitation2,Citation10.
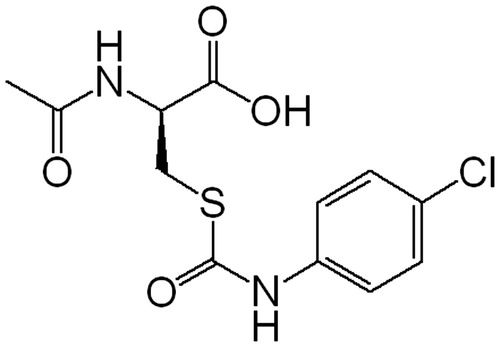
We have previously reported on the design, synthesis and biological evaluation of N-acetyl-S-(p-chlorophenylcarbamoyl)cysteine (NACC, ) and its analogs as a novel class of anticancer agentsCitation16. Of this class of compounds, NACC was determined to be the most potent anticancer agent against two melanoma cell lines and was shown to induce apoptosis in melanoma cellsCitation16. In this work, we found that NACC was also an irreversible and competitive inhibitor of mammalian TrxR1. The inhibitory activity of NACC against rat liver TrxR1 was comparable to some of metal-containing moleculesCitation17–19, including MGd and natural products such as curcuminCitation11, whose inhibition potency lies in the micromolar range. The characterization of NACC as an inhibitor of mammalian TrxR1 and its mechanism of action are presented.
Figure 1. Chemical structure of NACC.
Methods
Materials
All reagents for enzyme assays, including rat liver TrxR1, Escherichia coli Trx, 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), reduced form of nicotinamide adenine dinucleotide phosphate (NADPH), (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) ethylenediaminetetraacetic acid (EDTA), ammonium bicarbonate, guanidine·HCl, bovine serum albumin (BSA) and insulin were purchased from Sigma-Aldrich Chemical Co (St. Louis, MO). HPLC grade acetonitrile and trifluoroacetic acid (TFA) were purchased from Tedia Company, Inc (Fairfield, OH). Formic acid was obtained from ROE Scientific Inc (Newark, DE). Fetal bovine serum (FBS), RPMI 1640 growth medium, penicillin/streptomycin, phosphate buffered saline (PBS) and trypsin were purchased from Gibco (Grand Island, NY). The mass spectrometry grade trypsin was purchase from Promega Corporation (Madison, WI). NACC was synthesized in this laboratory according to the previously published methodCitation16 and prepared as a 50 mM stock solution in ethanol for all enzyme assays and a 100 mM stock solution in DMSO for cell-based assay.
Cell line and culture conditions
Human melanoma cell line UACC-62 was a kind gift from Dr. Xiangming Guan of South Dakota State University, USA. The cells were cultured at 37 °C in a humidified atmosphere of 5% CO2 in RPMI-1640 medium supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin.
TrxR assay
All enzyme activity assays and kinetic studies were carried out in at least three independent experiments at room temperature using a Multiskan Spectrum microplate reader (Thermo Scientific, Waltham, MA). DTNB and Trx reduction assays were performed to measure TrxR activity using the method described by Arner and HolmgrenCitation20 with minor modifications in pure enzyme system and cell extracts samples, respectively. In brief, in a 96-well plate, 10 μL of 0.34 U/mL mammalian TrxR1 was incubated with different concentrations of NACC in 40 μL of PE buffer (100 mM potassium phosphate, pH 7.0 with 2 mM EDTA) containing 100 μg/mL BSA in the presence of 250 μM NADPH for different incubation times at room temperature. For the DTNB reduction assay, 150 μL of a mixture solution (DTNB 5 mM final, NADPH 300 μM final) prepared in PE buffer was added at the end of incubation. The increase in absorbance at 412 nm was recorded for the initial 90 s. The TrxR activity in cell homogenates was determined by a Trx reduction assay. In brief, the cell homogenate was incubated with 7 μL (0.25 U) of E. coli Trx and a mixture solution (HEPES buffer 85 mM final, insulin 0.3 mM final, NADPH 660 μM final and EDTA 3 mM final) with a total volume of 50 μL at 37 °C for 30 min. Five hundred microliters of 1 mM DTNB in 6 M guanidine solution (pH 8.0) were added to the reaction mixture and allowed to stand at 37 °C for 5 min for a complete reaction of reduced insulin with DTNB. The absorbance at 412 nm was recorded. For the control experiment of each sample, the corresponding background reaction was measured in the same assay by omitting E. coli Trx from the reaction buffer. The TrxR activity in cell homogenate was calculated by subtracting the background reaction from the total reaction.
Kinetics of TrxR1 inhibition
The time and concentration dependence of TrxR1 inhibition by NACC was evaluated to determine the parameters of enzyme inhibition kinetics. 0.34 U/mL rat liver TrxR1 in PE buffer containing 100 μg/mL BSA was incubated with different concentrations of NACC (12.5, 25, 50 and 100 μM) in the presence of 250 μM NADPH at room temperature. Aliquots were withdrawn for determination of TrxR1 activity at 5, 10 and 15 min. Samples in the absence of NACC were conducted in parallel as a control. Master mixture (150 μL) was added to the TrxR1 assay solution (50 μL) in a 96-well plate, and enzyme activity was determined as described above. Inhibitory parameters, Ki and kinact, were determined by the method of Kitz and WilsonCitation21.
Dialysis of NACC-inhibited TrxR1 to determine the irreversibility of TrxR1 inhibition
Rat liver TrxR1 (0.34 U/mL) was incubated at room temperature with NACC (1 mM) in the presence of 250 μM NADPH at room temperature for 30 min to achieve a complete inhibition, followed by extensive dialysis in PE buffer (100 mM potassium phosphate, pH 7.0, 2 mM EDTA) in a Slide-A-Lyzer dialysis cassette (Thermo Scientific) with a molecular mass cutoff of 10 000 Da. Control samples were prepared in the absence of NACC. Aliquots were withdrawn at different time intervals, and the remaining TrxR1 activity was determined as described in the TrxR1 assay.
Substrate protection assay
NADPH-pretreated 0.34 U/mL rat liver TrxR1 was incubated with NACC (0.1 mM) in the presence or absence of DTNB (0.1, 0.2 and 0.4 mM) in PE buffer (100 mM potassium phosphate, pH 7.0, 2 mM EDTA) at room temperature for 15 min. The remaining TrxR1 activity was determined as described in the TrxR1 assay.
NADPH-dependent TrxR1 inhibition
Rat liver TrxR1 (0.34 U/mL) was incubated with NACC (0.1 mM) in the presence or absence of NADPH (0.1 mM) in PE buffer (100 mM potassium phosphate, pH 7.0, 2 mM EDTA) at room temperature for 15 min. After incubation, the remaining TrxR1 activity was determined as described for the TrxR1 assay.
Mass spectrometry analysis of NACC-treated TrxR1
TrxR1 protein samples for mass spectrometric analysis were prepared as described by Fang et al.Citation11 with minor modifications. In brief, 8.0 μg reduced rat liver TrxR1 was treated with 0.5 mM NACC in the presence of NADPH (0.1 mM) in PE buffer for 15 min at room temperature. The control sample in which no NACC was added was processed in the same way. After incubation, the remaining NACC was removed by a PD MiniTrap G-25 column (GE Healthcare Life Sciences). The TrxR1 samples were denatured in 6 M guanidine hydrochloride at 60 °C for 30 min, and then, the samples were digested with trypsin (1:50, w/w) in 25 mM ammonium bicarbonate solution at 37 °C for 2 h. The digested peptides were extracted and desalted by using Pierce® C18 Tips (ThermoFisher Scientific, Rockford, IL) following the manufactory protocol. The samples were dried and reconstituted in 0.1% TFA aqueous solution and 200 ng digested peptides were analyzed by a Q-Exactive Orbitrap high-resolution mass spectrometer equipped with an Easy-nLC™ 1000 ultra-high pressure nano-HPLC system. The samples were separated on a C18 Acclaim® PepMap 100 pre-column (100 mm× 2 cm, 5 μm, 100 Å) (Thermofisher Scientific) and a C18 Acclaim® PepMap RSLC column (50 μm × 15 cm, 2 μm, 100 Å) (Thermofisher Scientific), and then the eluted peptide was introduced into the mass spectrometer operating in tandem mass mode across a 60 min gradient (5% acetonitrile/0.1% formic acid to 40% acetonitrile/0.1% formic acid in 40 min, 40% acetonitrile/0.1% formic acid to 90% acetonitrile/0.1% formic acid in 10 min and held at 90% acetonitrile/0.1% formic acid for other 10 min).
Determination of intracellular TrxR inhibition in UACC-62 cells
UACC-62 cells were treated with different concentrations of NACC (50, 100, 200 and 500 μM) for 5 h. Control samples were processed in parallel without NACC treatment. After the treatment, the medium was collected and combined with trypsinized cells. The cell suspension was centrifuged at 1000g for 5 min and the cell pellets were washed twice with cold PBS. The cell pellets were suspended in hypotonic phosphate buffer (1 mM, pH 7.4) containing 1 mM EDTA, and homogenized over ice with an Omni homogenizer. The homogenate was centrifuged at 150 000 g for 30 min at 4 °C. The supernatant was collected and used to determine TrxR activity as described for the TrxR assay. Protein concentration was quantified by the bicinchoninic acid (BCA) method.
Results
Kinetics of TrxR1 inhibition
The rat liver TrxR1 was shown to be inhibited by NACC in a concentration and time-dependent manner as shown in . The figure presents a plot derived from the natural logarithm of the enzyme activity versus time at various concentrations of NACC. As shown in the figure, the rat liver TrxR1 lost activity over time, a characteristic of irreversible enzyme inhibition. By plotting the reciprocal of apparent rate constant of inhibition (kapp) (slopes obtained from ) against the reciprocal of inhibitor concentration, the inhibitory parameters Ki and kinact of NACC were determined to be 80 μM and 0.178 min−1, respectively ()Citation21.
Figure 2. Time- and concentration-dependent inhibition of TrxR1 by NACC. The natural logarithm of remaining TrxR1 activity is plotted against time. TrxR1 was incubated with various concentrations of NACC (♦, 12.5 μM; ▪, 25 μM; ▴, 50 μM; ○, 100 μM), and aliquots were withdrawn at different time intervals. The data were derived from one of triplicate experiments.
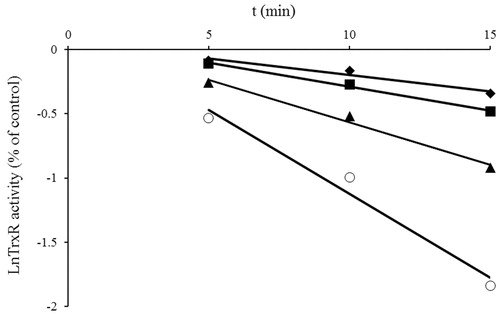
Figure 3. Double-reciprocal plot of the apparent rate constants of inhibition (kapp, slope from ) versus the reciprocal of inhibitor concentration ([I]). The Ki and kinact values were determined to be 80 μM and 0.178 min−1, respectively, based on the method of Kitz and WilsonCitation20.
![Figure 3. Double-reciprocal plot of the apparent rate constants of inhibition (kapp, slope from Figure 2) versus the reciprocal of inhibitor concentration ([I]). The Ki and kinact values were determined to be 80 μM and 0.178 min−1, respectively, based on the method of Kitz and WilsonCitation20.](/cms/asset/f2d3dcc0-d3cf-4f2a-9607-6b4a03f944ab/ienz_a_1016512_f0003_b.jpg)
Confirmation of the irreversible inhibition by dialysis
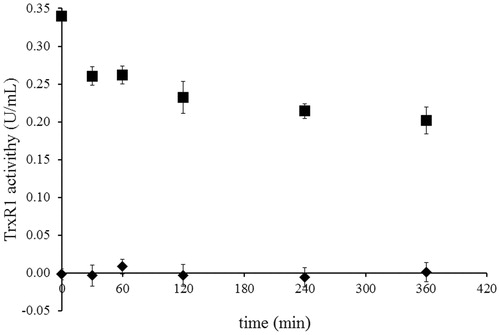
Dialysis of NACC-inhibited TrxR1 was conducted to confirm the irreversibility of the inhibition of NACC against TrxR1. The NACC-inhibited TrxR1 was extensively dialyzed in PE buffer. No recovery of enzyme activity was observed in NACC-treated TrxR1 after 6 h of dialysis, thus further confirming that the inhibition of TrxR1 by NACC was irreversible ().
Figure 4. Determination of the irreversibility of TrxR1 inhibition by NACC via dialysis. The activity of 0.34 unit/mL rat liver TrxR1 was completely inhibited by incubation at room temperature with NACC (1 mM) in the presence of 0.25 mM NADPH for 30 min. Then extensive dialysis was performed in a Slide-A-Lyzer dialysis cassette (Thermo) for 6 h. Aliquots were withdrawn at indicated time intervals, and the remaining TrxR1 activity was determined as described in the TrxR1 assay. No recovery of enzyme activity was observed after 6 h of dialysis for NACC-treated enzyme. The results are presented as the means ± S.D. of three independent experiments. ▪, control; ♦, NACC treated.
Substrate protection assay
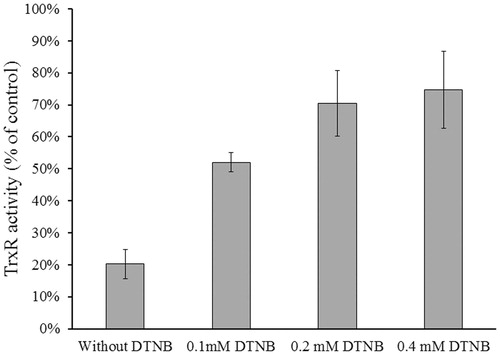
The inhibitory experiments were conducted in the absence and presence of substrate DTNB to determine whether NACC is a competitive inhibitor of TrxR1 and whether the inhibition occurs at the active site of the enzyme. The result showed that DTNB, a substrate of the enzyme, protected TrxR1 from inhibition by NACC. The inhibitory effect of NACC decreased with an increase in DTNB concentration revealing that the substrate protected the enzyme from inhibition by NACC (). This indicated that NACC and DTNB were competing for the same binding site of the enzyme.
Figure 5. Substrate protection of TrxR1 from NACC inhibition. Reduced rat liver TrxR1 (0.34 unit/mL) was incubated with NACC (0.1 mM) in the presence or absence of DTNB (0.1, 0.2 and 0.4 mM) in PE buffer (100 mM potassium phosphate, pH 7.0, 2 mM EDTA) at room temperature for 15 min. An aliquot was withdrawn and tested for the remaining TrxR1 activity as described for the TrxR assay. The results are presented as the means ± S.D. of three independent experiments.
NADPH-dependent TrxR1 inhibition
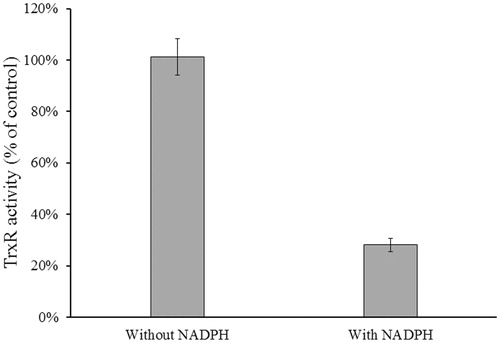
The reduction reaction catalyzed by TrxR1 requires reduction of the selenylsulfide bond (S-Se) (the oxidized form) at the enzyme active site to a selenothiol (–SeH) bond (the reduced form) by NADPHCitation5. To determine whether the selenothiol of selenocysteine (Sec) residue or the thiol group of Cys residue at the active site is involved in NACC inhibition, inhibitory experiments in the presence and absence of NADPH was conducted. In , no inhibition was observed in the absence of NADPH, indicating that only the selenolthiol or thiol in the active site was involved in the inhibition suggesting that it was the reduced form of the enzyme that was inhibited.
Figure 6. Determination of NADPH-dependent TrxR1 inhibition. Rat liver TrxR1 (0.34 unit/mL) was incubated with NACC (0.1 mM) in the presence or absence of NADPH (0.1 mM) in PE buffer at room temperature for 15 min. An aliquot was withdrawn and tested for the remaining TrxR1 activity as described for the TrxR assay. The results are presented as the means ± S.D. of three independent experiments.
Mass spectrometry analysis of NACC-treated TrxR1
Rat liver TrxR1 was incubated with 0.5 mM NACC in PE buffer in the presence of NADPH for 15 min at room temperature as described earlier for the enzyme assay. The NACC-treated and the untreated TrxR1 samples were denatured and followed by trypsin digestion. And then the desalted samples were subjected to a Q-Exactive Orbitrap high resolution mass spectrometer equipped with an Easy-nLC™ 1000 ultra-high pressure nano-HPLC system. The trypsin-digested peptides and their corresponding tandem mass fragment ions of TrxR1 protein sequence (www.uniprot.org; O89049) were predicted by Skyline software (MacCoss Lab, Department of Genome Sciences, University of Washington, Seattle, WA, USA). The predicted trypsin-digested peptide –SGGDILQSGCUG containing the active Cys497 and Sec498 is located in the C-terminal of rat liver TrxR1 sequence. The precursors of unmodified –SGGDILQSGCUG (m/z 1144.3842, M + H), and its corresponding mono- and bis-p-chlorophenyl carbamoylated adducts are m/z 572.69570 [(M + 2H)2+], 649.19475 [(M + 2H + 152.9981)2+] and 725.69380 [(M + 2H + 152.9981 * 2)2+], respectively. In the NACC-treated TrxR1 samples, only precursor ion at m/z 649.19475 ± 10 ppm rather than m/z 572.69570 and m/z 725.69380 was extracted from the mass spectrum () at retention time of 33.5 min indicating that the active site of TrxR1 was completely modified by NACC via monomer modification. By investigating the tandem mass of the monomer precursor ion at m/z 649.19475, a series of fragment ions were observed (, ). A y2 ion at m/z 379.99, which equals the addition of mono-p-chlorophenyl carbamoylation and the UG residue, was observed revealing that only the Sec498 was p-chlorophenyl carbamoylated by NACC (). The modification was further confirmed by the observation of ions at m/z 650.19421 () and m/z 381.99 () which showed the isotope peaks of 37Cl in the precursor and tandem mass spectrum, respectively. No adduct on Cys497 was observed through the tandem mass spectrum. As in the untreated TrxR1 sample, there was no p-chlorophenyl carbamoylation detected on any of Cys497 or Sec498.
Figure 7. Mass spectrum of trypsinized NACC-treated TrxR1. (A) Mass spectrum of mono-p-chlorophenyl carbamoylated peptide –SGGDILQSGCUG containing active site of the enzyme. (B) Fragmentation of precursor ion m/z 649.19475 eluting at 33.5 min from trypsin-digested NACC-treated TrxR1. A series of fragment ions were observed including the ion m/z 379.99 which is the p-chlorophenyl carbamoylated y2 ion. (C) The extended tandem mass spectrum of (B) shows the chlorine element on the ion m/z 379.99 through isotope peak at m/z 381.99 confirming the p-chlorophenyl carbamoyl adduct.
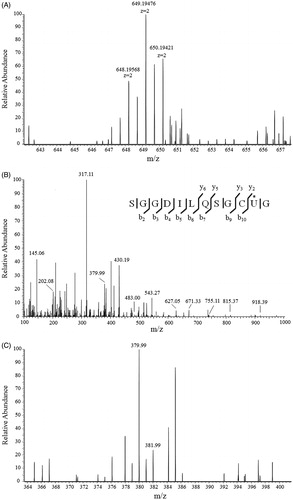
Figure 8. Proposed mechanism of TrxR1 inhibition by NACC. The tandem mass analysis of the NACC-inhibited TrxR1 revealed the selenothiols of Sec498 at the active site reacted with NACC to form mono-p-chlorophenyl carbamoylated enzymes, resulting in irreversible inhibition.
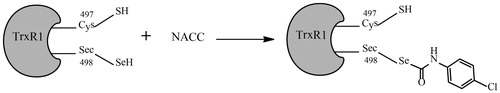
Table 1. Observed fragment ions of peptide –SGGDILQSGCU*G in the tandem mass spectrum of trypsin-digested NACC-treated TrxR1.
Determination of intracellular TrxR inhibition in UACC-62 cells
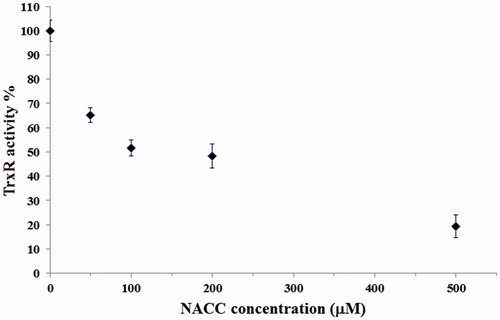
To evaluate whether NACC can inhibit intracellular TrxR, NACC at 50, 100, 200 and 500 μM concentrations was incubated with UACC-62. After a 5-h treatment, the intracellular TrxR activity was significantly attenuated by NACC (), indicating that NACC can also inhibit intracellular TrxR activity. The IC50 value of NACC is around 200 μM.
Figure 9. Inhibition of intracellular TrxR by NACC in UACC-62 cells. UACC-62 cells were incubated in the presence of 50, 100, 200 and 500 μM NACC for 5 h, and the enzyme activity was determined and presented as the percentage of the control. The results are shown as the means ± S.D. of three independent experiments.
Discussions
During last decades, various natural products and synthetic compounds have emerged as effective mammalian TrxR1 inhibitors for cancer treatment. In this work, we found that the anticancer agent NACC was a TrxR1 inhibitor. In vitro, the inhibition of mammalian TrxR1 by NACC was comparable to some of the existing TrxR inhibitors, such as Butyltin (IV) BenzoatesCitation3, quinone compoundCitation4 and anticancer drug laromustineCitation22. The Ki value of MGd, the TrxR inhibitor undergoing clinical trial, is even higher. The Kis of MGd against the thioredoxin system (combination of E. coli Trx and rat TrxR) were determined to be 180 and 70 μM in the presence of 0.3 and 0.1 mM insulin, respectivelyCitation17.
NACC exhibited a time and concentration dependent inhibition suggesting that the inhibition was irreversible. The irreversible inhibition was further confirmed by the dialysis experiment. When the completely inhibited enzyme was dialyzed extensively, no enzyme activity was recovered. The inhibition by NACC was prevented by the presence of substrate DTNB, indicating the inhibitor was acting at or near the active site of TrxR1. The inhibition also required the presence of the selenothiol or thiol group suggesting they probably were involved in the inhibition. All the evidence indicated the formation of a covalent bond between NACC and the reduced selenothiol or thiol group at the active site of the enzyme. Based on the mechanistic studies of other TrxR1 inhibitors, the irreversibly inhibitory effects were mainly based on their interactions with the Sec or Cys residues at the C-terminal active site of TrxRCitation5,Citation11,Citation23. Moreover, two cysteines in the N-terminal FAD domain (–Cys59–x–x–x–x–Cys64–) are conserved between mammalian TrxR and glutathione reductase and function as primary acceptors for electrons from NADPH via FAD, thus are also essential for the TrxR1 activityCitation24. To identify which residues between Cys59, Cys64, Cys497 and Sec498 are involved in the covalent bond with NACC, an liquid chromatography–tandem mass spectrometry analysis was performed. Interestingly, the tandem mass spectrometric analysis showed that only the Sec498 residue rather than Cys59, Cys64 and Cys497 was modified by NACC through a p-chlorophenyl carbamoyl covalent bond. This finding indicated that NACC inhibits TrxR1 by selectively p-chlorophenyl carbamoylating the selenothiol group at Sec498 residue while the thiol at Cys497 was unaffected ().
Conclusions
In summary, we have characterized an irreversible inhibition mechanism of TrxR1 by NACC. In addition, NACC significantly inhibited TrxR activity in a melanoma cell line. TrxR1 plays a critical role in regulation of thiol redox state as well as cancer progression. Selectivity of an inhibitor is one of the major hurdles in the development of a successful TrxR inhibitor. Most existing TrxR inhibitors inhibit the enzyme through the Sec/Cys residuesCitation25. Some of the inhibitors including ethaselen and curcuminCitation5,Citation11 form covalent bonds with both selenothiol and thiol groups at the active site of TrxR1. The fact that NACC inhibits TrxR1 through selenothiol may enable NACC to selectively inhibit TrxR1. The Cys thiols are essential in many enzymes, but very few mammalian enzymes were found containing selenothiols at their active sitesCitation26. Thus, NACC which only targets selenolthiol of TrxR1 could be used as a lead compound to develop selective inhibitors of TrxR1. Moreover, the potent activity, easy preparation and excellent water solubility of NACC make it an effective tool in studying the biochemical process of TrxR1.
Declaration of interest
The authors confirm that this article’s contents have no conflict of interest.
This work was supported by National Natural Science Foundation of China (81202553, 81302840), Medical and Health Science Foundation of Zhejiang Province (2012KYA034) and Zhejiang Key Laboratory of Diagnosis and Treatment Technology on Thoracic Oncology (Lung and Esophagus).
References
- Xiao GQ, Liang BX, Chen SH, et al. 3-Nitro-2H-chromenes as a new class of inhibitors against thioredoxin reductase and proliferation of cancer cells. Arch Pharm (Weinheim) 2012;345:767–70
- Liu Y, Li Y, Yu S, Zhao G. Recent advances in the development of thioredoxin reductase inhibitors as anticancer agents. Curr Drug Targets 2012;13:1432–44
- Navakoski de Oliveira K, Andermark V, von Grafenstein S, et al. Butyltin(IV) benzoates: inhibition of thioredoxin reductase, tumor cell growth inhibition, and interactions with proteins. ChemMedChem 2013;8:256–64
- Xu J, Arner ES. Pyrroloquinoline quinone modulates the kinetic parameters of the mammalian selenoprotein thioredoxin reductase 1 and is an inhibitor of glutathione reductase. Biochem Pharmacol 2012;83:815–20
- Wang L, Yang Z, Fu J, et al. Ethaselen: a potent mammalian thioredoxin reductase 1 inhibitor and novel organoselenium anticancer agent. Free Radic Biol Med 2012;52:898–908
- Arner ES. Focus on mammalian thioredoxin reductases – important selenoproteins with versatile functions. Biochim Biophys Acta 2009;1790:495–526
- Cai W, Zhang B, Duan D, et al. Curcumin targeting the thioredoxin system elevates oxidative stress in HeLa cells. Toxicol Appl Pharmacol 2012;262:341–8
- Yoon BI, Kim DY, Jang JJ, Han JH. Altered expression of thioredoxin reductase-1 in dysplastic bile ducts and cholangiocarcinoma in a hamster model. J Vet Sci 2006;7:211–16
- Berggren M, Gallegos A, Gasdaska JR, et al. Thioredoxin and thioredoxin reductase gene expression in human tumors and cell lines, and the effects of serum stimulation and hypoxia. Anticancer Res 1996;16:3459–66
- He J, Li D, Xiong K, et al. Inhibition of thioredoxin reductase by a novel series of bis-1,2-benzisoselenazol-3(2H)-ones: organoselenium compounds for cancer therapy. Bioorg Med Chem 2012;20:3816–27
- Fang J, Lu J, Holmgren A. Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anticancer activity. J Biol Chem 2005;280:25284–90
- Grogan TM, Fenoglio-Prieser C, Zeheb R, et al. Thioredoxin, a putative oncogene product, is overexpressed in gastric carcinoma and associated with increased proliferation and increased cell survival. Human Pathol 2000;31:475–81
- Smart DK, Ortiz KL, Mattson D, et al. Thioredoxin reductase as a potential molecular target for anticancer agents that induce oxidative stress. Cancer Res 2004;64:6716–24
- Yoo MH, Xu XM, Carlson BA, et al. Thioredoxin reductase 1 deficiency reverses tumor phenotype and tumorigenicity of lung carcinoma cells. J Biol Chem 2006;281:13005–8
- Yoo MH, Xu XM, Carlson BA, et al. Targeting thioredoxin reductase 1 reduction in cancer cells inhibits self-sufficient growth and DNA replication. PLoS One 2007;2:e1112
- Chen W, Seefeldt T, Young A, et al. Design, synthesis, and biological evaluation of N-acetyl-S-(p-chlorophenylcarbamoyl)cysteine and its analogs as a novel class of anticancer agents. Bioorg Med Chem 2011;19:287–94
- Hashemy SI, Ungerstedt JS, Zahedi Avval F, Holmgren A. Motexafin gadolinium, a tumor-selective drug targeting thioredoxin reductase and ribonucleotide reductase. J Biol Chem 2006;281:10691–7
- Mura P, Camalli M, Bindoli A, et al. Activity of rat cytosolic thioredoxin reductase is strongly decreased by trans-[bis(2-amino-5- methylthiazole)tetrachlororuthenate(III)]: first report of relevant thioredoxin reductase inhibition for a ruthenium compound. J Med Chem 2007;50:5871–4
- Casini A, Gabbiani C, Sorrentino F, et al. Emerging protein targets for anticancer metallodrugs: inhibition of thioredoxin reductase and cathepsin B by antitumor ruthenium(II)-arene compounds. J Med Chem 2008;51:6773–81
- Arner ES, Holmgren A. Measurement of thioredoxin and thioredoxin reductase. Current protocols in toxicology/editorial board, Mahin D Maines. 2001; chapter 7:Unit 7 4
- Kitz R, Wilson IB. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J Biol Chem 1962;237:3245–9
- Rice KP, Klinkerch EJ, Gerber SA, et al. Thioredoxin reductase is inhibited by the carbamoylating activity of the anticancer sulfonylhydrazine drug laromustine. Mol Cell Biochem 2012;370:199–207
- Chew EH, Lu J, Bradshaw TD, Holmgren A. Thioredoxin reductase inhibition by antitumor quinols: a quinol pharmacophore effect correlating to antiproliferative activity. FASEB J 2008;22:2072–83
- Fujiwara N, Fujii T, Fujii J, Taniguchi N. Roles of N-terminal active cysteines and C-terminal cysteine-selenocysteine in the catalytic mechanism of mammalian thioredoxin reductase. J Biochem 2001;129:803–12
- Cai W, Zhang L, Song Y, et al. Small molecule inhibitors of mammalian thioredoxin reductase. Free Radic Biol Med 2012;52:257–65
- Wessjohann LA, Schneider A, Abbas M, Brandt W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol Chem 2007;388:997–1006