
Abstract
Malaria is a major public health problem all over the world, particularly in tropical and subtropical countries due to the development of resistance and most deadly infection is caused by Plasmodium falciparum. There is a direct need for the discovery of new drugs with unique structures and mechanism of action to treat sensitive and drug-resistant strains of various plasmodia for radical cure of this disease. Traditional compounds such as quinine and related derivatives represent a major source for the development of new drugs. This review presents recent modifications of 4-aminoquinoline and 8-aminoquinolone rings as leads to novel active molecules which are under clinical trials. The review also encompasses the other heterocyclic compounds emerged as potential antimalarial agents with promising results such as acridinediones and acridinone analogues, pyridines and quinolones as antimalarials. Miscellaneous heterocyclics such as tetroxane derivatives, indole derivatives, imidazolopiperazine derivatives, biscationic choline-based compounds and polymer-linked combined antimalarial drugs are also discussed. At last brief introduction to heterocyclics in natural products is also reviewed. Most of them have been under clinical trials and found to be promising in the treatment of drug-resistant strains of Plasmodium and others can be explored for the same purpose.
Introduction
Malaria is a disease caused by vermin of the genus Plasmodium and it is transmitted via the bites of infected female Anopheles Mosquitoes. Plasmodium falciparum and Plasmodium vivax are the rampant species but the former is the most lethal. Malaria is the most fatal human parasitic infection. At present, there are 250 million estimated cases of malaria worldwide. The vast majority of these cases (86%) are in the African region, followed by the South-East Asia (9%) and Eastern Mediterranean regions (3%). There were an estimated 0.881 million deaths worldwide in 2006, of which 90% were in the African region and 4% in each of the South-East Asia and the Eastern Mediterranean regionsCitation1. The individual most at risk of significant morbidity and mortality owing to malaria is the children under the age of 5 years and the pregnant womenCitation2,Citation3. However, as a result of varying transmission intensity, population flux and trans-migration, adults lacking acquired immunity may also develop severe malaria. Human malaria is caused by four species of protozoan parasites of the genus Plasmodium. These are P. falciparum, P. vivax, Plasmodium ovale and Plasmodium malariae, each of which differs in geographical distribution, microscopic appearance, clinical features and potential for the development of resistance to the antimalarial drugs. Malaria prevention and treatment currently relies on the vector control and chemotherapy. The vector control is achieved by interrupting their life cycle, and creating a barrier between the human host and the mosquitoes.
New empirical estimates put the number of episodes of clinical P. falciparum malaria in the region of half a billion per yearCitation4. The mortality rate from malaria has been estimated at approximately 2.7 million per year, with over 75% of these deaths occurring in African childrenCitation5. Unfortunately, these staggering figures are on the increase largely as a result of parasite multi-drug resistanceCitation6. The introduction of chloroquine (CQ) in the 1940s had a marvelous impact on global health; however, today resistance to the drug has been observed in every region where P. falciparum occursCitation7. Over the next 20 years resistance to chloroquine gradually spread throughout South America and Southeast Asia. Thereafter, the resistance still continued and finally arrived even in East Africa in the late 1970s. CQ resistance has since spread across all of Sub-Saharan Africa. As a result, many countries switched their first-line antimalarial drug to sulfadoxine–pyrimethamine (SP); however, resistance to SP grew and spread quickly, especially in Southeast Asia, South AmericaCitation8 and, more recently, in many areas of AfricaCitation9. Clinical failure to newer antimalarial drugs has also occurred at an alarming rate; for example, resistance to mefloquine was reported as early as five years after its introduction as a prophylactic treatment in parts of Thailand, whereas resistance to atovaquone was even more rapid, emerging in the same year as its launchCitation10. The increasing spread of malaria together with the emergence of resistance against conventional drugs has put enormous pressure on public health systems to introduce new malaria treatments.
Structural modification of 4-aminoquinoline
Quinoline containing compounds are still attractive models for treatment of malaria. The success of the antimalarial aminoquinoline drug, chloroquine 1 (CQ), has been based on its excellent clinical efficacy, limited host toxicity, ease to use and simple cost-effective synthesis. However, the use of this drug has been seriously eroded in recent years, mainly as a result of the development of parasite resistance to CQ. Amodiaquine (AQ) 2, another drug based on 7-chloro-4-aminoquinoline nucleus, is effective against many CQ-resistant strains of P. falciparum. However, its clinical use has been restricted because of associations with hepatotoxicity and agranulocytosis due to the toxic quinone–imine metabolite ethyl 2-(1,4-dihydro-4-oxoquinolin-2-yl)acetate 3Citation11.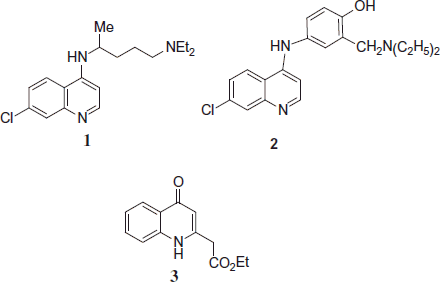
Novel bisquinolines as antimalarials
The potential role of bisquinolines as novel antimalarials against CQ-resistant malaria has been reviewedCitation12. Ayad et al. synthesized the novel bisquinoline compounds comprising 4-(4-diethylamino-1-methylbutyl)aminoquinoline units joined through the 2-position by a (CH2)n linker. They are found to inhibit the growth of both CQ-sensitive (D10) and CQ-resistant (K1) strains of P. falciparum. These compounds appear to act by the hydrogen peroxide-mediated pathway for decomposition of heme, and the conversion of haem to β-haematin. The activity was affected by the length of the linker and the most active (11, n = 12) showed effects similar to CQ in three of the assays. However, it was even more active against the resistant strain [IC50, 17 nM (K1); 43 nM (D10)], much superior to CQ (IC50, 540 nM) and slightly better than mefloquine (4) (IC50, 30 nM) in this regard. Compounds 5 (X = O, n = 2) and 6 (X = NH, n = 3) are recent examples of the class 4-aminoquinoline units which is linked by 4-position and therefore can also be regarded as related to CQCitation13,Citation14.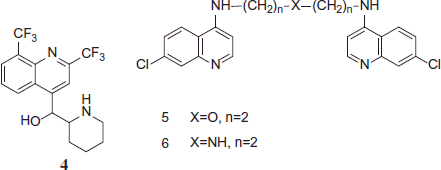

The most active of these 9 (n = 8) appeared to have overcome the CQ-resistance mechanism. All three compounds 9, 10 and 11 displayed markedly high activity against the resistant strain, while the activity against the sensitive (D10) strain showed a steady increase with increase in length of the linker.
The data suggest that the bisquinolines exert their antimalarial activity in a manner similar to that of CQ, while the interesting antimalarial activity of 11 (n = 12), especially against the CQ-resistant strain of parasite, warrants further exploration of this compound.
Isoquinuclidine analogs of CQ as novel antimalarial
A number of isoquinuclidine-based pharmacophores prepared over the years has shown interesting pharmacological activities. Several reviews of the antiaddictive properties of analogs of the iboga alkaloids as well as a review of iboga alkaloids and their role as precursors of antineoplastic bisindole Catharanthus alkaloids have appeared. Recently, the synthesis and biological evaluation of 18-methoxycoronaridine congeners as potential antiaddictive agents have been reportedCitation15.
Isoquinuclidine (2-azabicyclo[2.2.2]octane) ring system which may be viewed as a semi-rigid boat form of the piperidine ring and, when properly substituted, a scaffold for rigid analogs of biologically active ethanolamines and propanolamines. It is present in natural products (such as ibogaine and dioscorine) that flaunt remarkable pharmacological properties. Khan et al. synthesized various semirigid analogs of CQ by incorporating isoquinuclidine (2-azabicyclo[2.2.2]octane) ring system (12–17) in numerous pharmacophores to get better pharmacological activity. The analogs were tested in vitro against P. falciparum strains and Leishmania donovani promastigote cultures. Compounds 12 and 15 [R = CH2N(CH3)2] displayed potent antimalarial activity against both CQ-susceptible D6 and CQ-resistant W2 strains of P. falciparum. The synthesized and tested molecules act by the mechanism similar to CQ against both the sensitive as well as resistant strains, i.e. inhibition of conversion of free heme to hemozoineCitation16.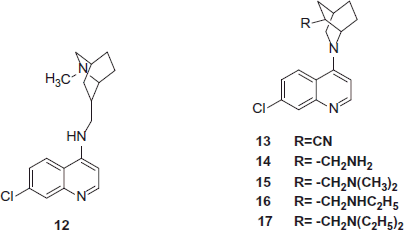
Novel CQ and thiazolidenone scaffolds as antimalarials
The success of the antimalarial aminoquinoline drug, CQ, has been based on its excellent clinical efficacy, limited host toxicity, ease to use and simple cost-effective synthesis. However, the use of this drug has been seriously eroded in recent years, mainly as a result of the development of parasite resistance to CQ. Currently, double drug development and/or multi-therapeutic strategies, which utilize new chemical entities with two (or more than two) different N-heterocyclic skeletons, are also valid perspective to create new antimalarial drugs. These strategies have the potential to overcome the drug resistant parasites problem. So, the design and synthesis of aminoquinoline-containing dual inhibitors or “double drugs” that would potentially inhibit haemozoin formation and another target within P. falciparum, and will not be recognized by the proteins involved in drug efflux, are very productive in the generation of new chemical entities that are effective against drug-resistant parasites in the long term. The success of this hybridization approach has magnificent examples as trioxaquines or artemisinin–quinine hybrid.
Among these nitrogen containing heterocyclic skeletons, thiazolidin-4-ones, rigid molecules, are considered as a biologically privileged scaffold well-tolerated in human subjects and could be perspective models for the designing of new antimalarial CQ-hybridsCitation17.
Ruiz et al. synthesized a series of compounds, four of them with benzyl amino fragment, showed an excellent activity, up to 3-fold more active than CQ. All active compounds inhibited the ferriprotoporphyrin IX biocrystalization (FBIT) process in concentrations around similar to CQ. In vivo preliminary results have shown that at least two compounds are as active as CQ against Plasmodium berghei ANKA.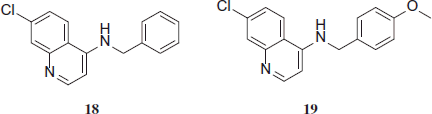
Initially, all 21 compounds were tested for their activity against CQ-sensitive 3D7 strain of P. falciparum, under in vitro conditions. Subsequently, compounds that showed an IC50 value less than 2 µM were evaluated against multidrug-resistance Dd2 strain. No compound more active than CQ against 3D7 strain was found. In fact, the activity shown by the most active compounds was 9–15-fold minor than CQ. Nonetheless, against Dd2 strain, 4-benzylamino-7-chloroquinolines 1, 2, 3, and 4-amino-7-chloroquinoline-thiazolidinone hybrid 33 were more active than CQ (IC50 = 0.50 µM), with IC50 values between 0.30 and 0.44 µMCitation18,Citation19.
The activity showed by 4-benzylamino-7-chloroquinolines 18–21 was greater than 4-amino-7-chloroquinoline-thiazolidinone hybrids 22–38.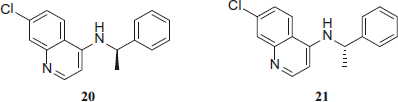
Compounds 18, 19, 20 and 21 were the most active on FBIT as well, showing IC50 values between 39.32 and 46.52 µM, which are only 1.76–2.1-fold lower than CQ (IC50 = 22.29 µM). Moreover, a direct relationship between the activity against the parasite and the capability to inhibit β-hematine formation was observed.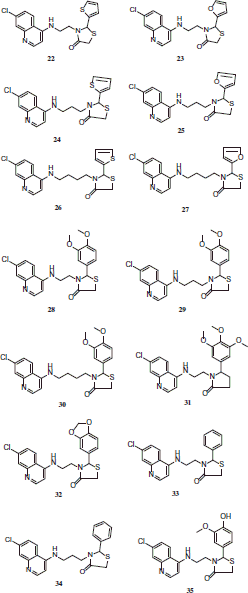
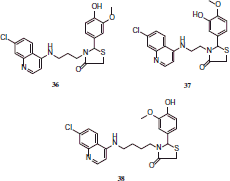
Compounds 18, 33 and 37 showed excellent in vitro and in vivo antimalarial activity and low cytotoxicity levels to be considered as potential antimalarial agents.
Novel ferroquine-based antimalarials
Ferroquine (FQ; SSR97193), a ferrocene-containing 4-aminoquinoline derivative, has potent in vitro efficacy against CQ-resistant P. falciparum and CQ-sensitive P. vivax. FQ showed tremendous activity against CQ-sensitive and -resistant P. falciparum and P. vivax (median 50% inhibitory concentrations [IC50], 9.6nM and 18.8 nM, respectively), there was significant cross-susceptibility with the quinoline-based drugs CQ, amodiaquine and piperaquine (for P. falciparum, r = 0.546–0.700, p < 0.001; for P. vivax, r = 0.677–0.821, p < 0.001). The observed ex vivo cross-susceptibility is likely to reflect similar mechanisms of drug uptake/efflux and modes of drug action of this drug class. However, the potent activity of FQ against resistant isolates of both P. falciparum and P. vivax highlights a promising role for FQ as a lead antimalarial against CQ-resistant Plasmodium and a valuable partner drug for artemisinin-based combination therapyCitation20.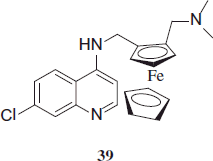 Marfurt et al. synthesized the 4-aminoquinoline FQ, 39 an unusual ferrocene moiety which is inserted into the side chain. FQ shows high activity against CQ-resistant malaria parasites. In a dose-ascending study, FQ was generally well tolerated up to 1600 mg as single dose and up to 800 mg as repeated dose in an asymptomatic young male with P. falciparum infectionCitation21,Citation22.
Marfurt et al. synthesized the 4-aminoquinoline FQ, 39 an unusual ferrocene moiety which is inserted into the side chain. FQ shows high activity against CQ-resistant malaria parasites. In a dose-ascending study, FQ was generally well tolerated up to 1600 mg as single dose and up to 800 mg as repeated dose in an asymptomatic young male with P. falciparum infectionCitation21,Citation22.
Novel 4-aminoquinoline analogs as antimalarials
O'Neill et al. synthesized the 4-aminoquinoline AQ-13, 40 which is a short side-chain analogue of CQ, since it shows high activity against CQ-resistant strains of P. falciparum (IC50 values of 40–60 nM against the K1 strain).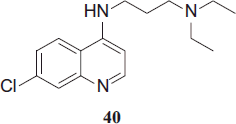
It has already undergone Phase I clinical trials, which indicate a similar profile to CQ. Dose adjustment is required because of increased clearance compared with CQ. Efficacy studies are being conducted in Mali (studies of a candidate aminoquinoline antimalarial (AQ-13); Clinical Trials. Gov; Identifier: NCT01614964)Citation23.
Novel trioxaquine derivatives
SAR116242 (PA1103), 41 is a so-called trioxaquine, a hybrid structure composed of an aminoquinoline and a 1,2,4-trioxane. The dual mode of action of SAR116242 has been demonstrated by evaluating its iron(II)-heme-alkylating capacity in vitro and observing several heme drug adducts, as well as by confirming its inhibition of heme aggregation.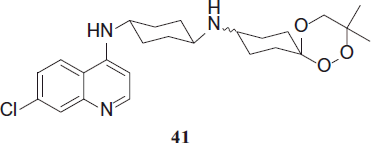
SAR116242, 41 showed high activity against P. falciparum in vitro (e.g. IC50 against W2 strain = 15 nM) and in vivo. Once-daily administration of 32 mg (70 µmol)/kg SAR115242 for 4 days led to 100% parasitemia reduction on day 4 in humanized mice infected with P. falciparumCitation24. The cis-SAR116242 and trans-SAR116242 showed similar activity against the FcM29 P. falciparum strain (IC50 = 12 and 8 nM, respectively)Citation25.
Novel isoquines as antimalarials
To prevent the formation of the toxic quinone imine from amodiaquine, numerous analogs have been designed as a part of a public–private partnership between Liverpool School of Tropical Medicine, Medicines for Malaria Venture (MMV) and GSK pharmaceuticals, one of them being N-tert-butyl-isoquine (GSK369796), 42.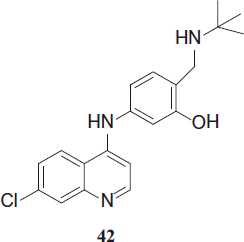
Due to its tert-butyl amino moiety, the compound shows enhanced metabolic stability. IC50 values against CQ-resistant strains range from 11 to 17 nM. In P. berghei-infected mice an ED50 of 2.8 mg (7.8 µmol)/kg is obtainedCitation26. Due to problems with inadequate exposure levels occurring in tests with humans, development is currently on hold.
Novel cinnamic acid/4-aminoquinoline conjugates
In the year 2012, Perez et al. synthesized a series of cinnamic acid/4-aminoquinoline conjugates obtained after linking through a proper retro-enantio dipeptide, a heterocyclic scaffold known to prevent hemozoin formation, to a trans-cinnamic acid motif capable of inhibiting enzyme catalytic Cys residues, as potential dual-action antimalarials. The effect of amino acid configuration and the absence of the dipeptide spacer were also assessed.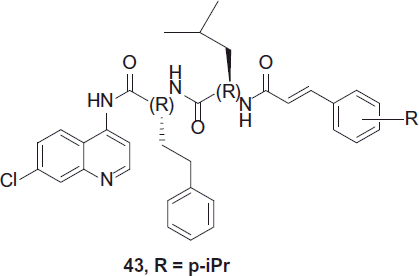
The replacement of the d-aminoacids by their natural l counterparts led to a decrease in both antiplasmodial and falcipain-2 inhibitory activity, indicating that the former be better approach. Molecules with such spacer were active against blood-stage P. falciparum, in vitro, and hemozoin formation, implying that the dipeptide played a key role in mediating these two activities. While, compounds without spacer were better falcipain-2 inhibitors, on account of being smaller and have their vinyl bonds in closer vicinity to the catalytic Cys, as evidenced by molecular modeling calculations. Among the synthesized analogues 43 was found to be the most potent antiplasmodial agent. These novel conjugates provide promising leads for the development of new antimalarials targeted at blood-stage malaria parasitesCitation27.
Structure modification of 8-aminoquinoline
The 8-aminoquinolines exemplified by primaquine (PQ) 44 exhibit activities against all four species of Plasmodium that infect human. PQ, a tissue schizontocidal antimalarial, is the only clinical drug available for the treatment of relapsing cases of malaria. Although PQ is effective against all life cycle stages of human malaria parasite, its use is often associated with serious adverse effects as a consequence of its toxic metabolites. Its clinical use as a tissue-schizontocide is limited by side effects such as severe hematotoxicities in patients with the glucose-6-phosphate dehydrogenase (G6PD) deficiency. PQ is also known to have poor pharmacokinetic properties with a short half life of 4–6 h. It has been shown that in rodents, the PQ is rapidly metabolized, and two major metabolites start appearing in the blood in about 30 min, one of which has been identified as 4-(6-methoxyquinolin-8-ylamino)pentanoic acid (carboxyprimaquine), a product also reported by the microbiological degradation of PQ. The modulation of PQ structure has resulted in less toxic and promising blood-schizontocides. Further, appropriate substitution on the quinoline ring and on the side-chain resulted in the PQ analogues exhibiting promising antimicrobial and antileishmanial activities, in addition to potent antimalarial activitiesCitation28.

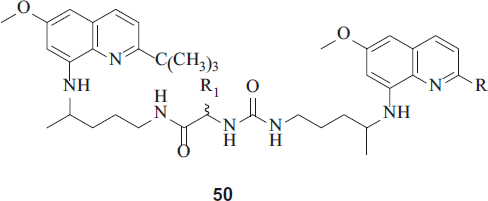
Kaur et al. synthesized potent antimalarials based on 8-aminoquinoline structural scaffold, three series of novel bis(8-aminoquinolines) using expedient one to four steps synthetic procedures were synthesized. Several compounds exhibited finer antimalarial activities compared to parent drug PQCitation29. Selected compounds (45, 49 and 50) when tested for in vivo blood-schizontocidal antimalarial activity (P. berghei) displayed potent blood-schizontocidal activities. Determination of in vitro antimalarial activity was based on the evaluation of plasmodial lactate dehydrogenase (LDH) activity. The antimalarial activities of all the synthesized analogues are reported as IC50 values against CQ-sensitive (D6) and CQ-resistant (W2) strains of P. falciparum.
Analogue 45, displayed IC50 of 2.6 µg/mL and 1.5 µg/mL for D6 and W2 strains, respectively.

Of these analogues, 46, 47 and 48 were most potent with IC50 values of 3.5, 2.9 and 2.99 µg/mL and IC90 of 7.0, 7.8 and 27 µg/mL, respectively, compared to the IC50 of 1 µg/mL and IC90 of 3.8 µg/mL for standard drug, pentamidine, 48.
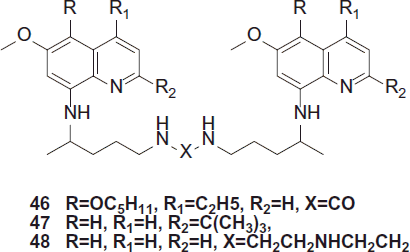
The analogue, 49 exhibited significantly high activity and cured 100% mice at a dose of 25 mg/kg, and were suppressive at the lowest tested dose of 10 mg/kg.
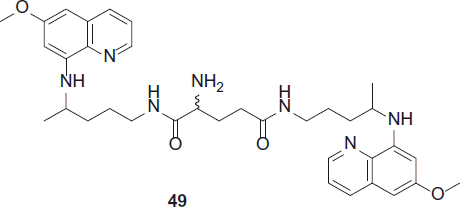
50 [R = C(CH3)3, R1 = Ornithine] was the most active and exhibited IC50 values of 0.34 and 0.30 µg/mL against D6 and W2 strains, respectivelyCitation29,Citation30.
NPC 1161 B, 51 is the (−)-enantiomer of the racemic 8-aminoquinoline. NPC 1161 C (DN3-27-1) (US patent No.: US6376511 B2)Citation31. It displayed significantly higher activity than the (+)-enantiomer NPC 1161 A against P. berghei in miceCitation32. After orally administering three doses of 4 mg (9 µmol)/kg/day, all mice had 0% parasitemia at day 6, and all mice were still alive at day 34. Recently NPC 1161 B was found to be practically insoluble in the physiological pH rangeCitation33.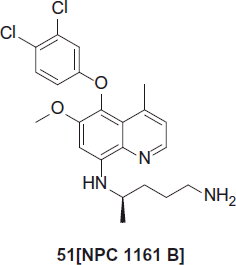
Novel naphthyridine as antimalarial
PQ is the drug of choice for the fundamental cure of P. vivax malaria, but possesses severe side effects. Novel PQ analogues were synthesized and lower toxicity was achieved by reducing or eliminating the tendency of forming chemically reactive and toxic intermediates and metabolites. In vitro and in vivo studies found that synthesized compounds were less toxic than the parent compound PQ, while preserving the desired antimalarial activity. Some of these compounds possess a therapeutic index over 10 times superior to that of the commonly used antimalarial drug CQ. These compounds, as well as the underlying design rationale, may find usefulness in the discovery and development of new antimalarial drugsCitation34. In vitro antimalarial activities of compounds 52–63 as IC50 values for the inhibition of CQ-resistant P. falciparum strain W2 were determined. Most compounds in this class exhibited potent antimalarial activity (IC50 ≤ 0.11 µM), superior to that of CQ (IC50 = 0.31 µM). All new compounds (52–63) exhibited potent in vitro antimalarial activity against CQ-resistant P. falciparum strain W2Citation35,Citation36.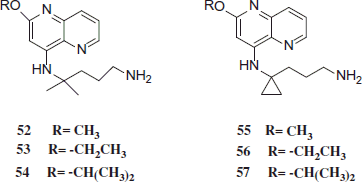
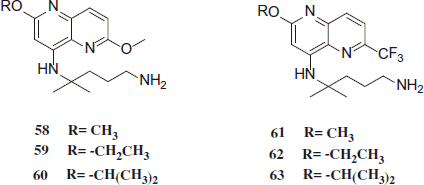
Jain et al. assumed that changing the structure of the aromatic mainstay of PQ will change the mode of action, hence portrait the compound active in vitro against P. falciparum. Some compounds 52–63 also revealed remarkable in vivo blood-schizontocidal antimalarial activity against both P. berghei (sensitive strain) and Plasmodium yoelii nigeriensis (multidrug-resistant strain). They were over three times less toxic than the parent compound PQ, as precise by the in vivo LD50. These compounds, as well as the causal design rationale, may find usefulness in the discovery and development of new antimalarial drugsCitation37.
Tafenoquine as antimalarials
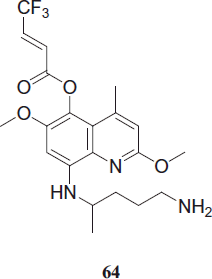
Nasveld et al. synthesized Tafenoquine, 64 which is an 8-aminoquinoline structurally related to PQ but displaying a considerably longer half-life. During the last two decades, tafenoquine has been evaluated as a radical cure against P. vivax infections. In one clinical trial, vortex keratopathy was observed, which was not associated with any effect on visual acuity and was fully resolved in all subjects within one yearCitation38.
In a further trial, Leary et al. observed no such adverse effectsCitation39.
Modification of acridinone and acridinediones ring
Structural modifications of acridinone derivatives
Acridinone derivatives have newly demonstrated to be active against malaria parasite.
New derivatives of 10-alyl- (65–68), 10-(3-methyl-2-butenyl)- (69–72) and 10-(1,2-propadienyl)-9(10H) (73–76) acridinone were evaluated for their antimalarial activity against P. falciparum. One compound, 69 (1-fluoro-10-(3-methyl-2-butenyl)-9(10H)-acridinone), can be classified as hit for antimalarial drug development exhibiting IC50 less than 0.2 µg/mL with SI greater than 100Citation40.
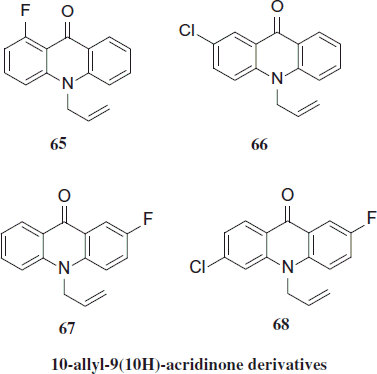
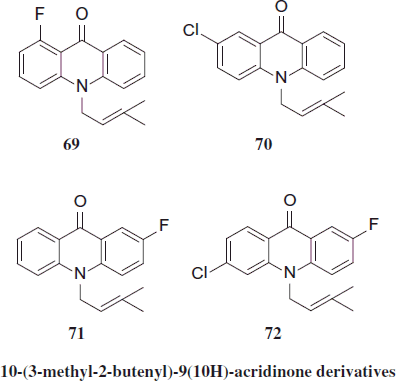
Derivatives of 10-(1,2-propadienyl)-9(10H)-acridinone were inactive or marginally active showing IC50 values equal or higher than 64 µM. Calienes et al. observed that three 10-allyl-9(10H)-acridnone derivatives exhibited antimalarial activity (IC50 < 64 µM). No cytotoxicity against MRC-5 fibroblast was detected for tested compounds. Three compounds (66, 67 and 69) exhibited the most potent and selective antimalarial activity showing IC50 less than 0.5 µg/mL and SI greater than 39. Inhibition of β-hematin formation test showed IC50 values over 20 Meq for all acridinones whereas CQ, the reference compound, exhibited IC50 of 1.2 ± 0.5 MeqCitation41.
Structural modifications of acridinediones
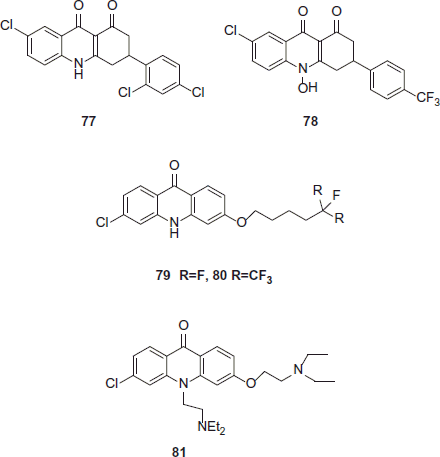
Acridinediones are the potent antimalarials and their mechanism of action involves heme binding and also prevention of crystallization to hemozoin. In comparison with mode of action of two synthesized analogues hydroacridinediones 78 (floxacrine) and 77 (WR249685) showed that 77 (WR249685) had higher in vitro antimalarial activity than 78 (floxacrine) (IC50 = 15 nM versus IC50 = 140 nM for 79 [floxacrine]) however, 78 (floxacrine) bound to heme 20-times betterCitation42,Citation43. Riscoe et al. have synthesized a number of highly potent acridone ether derivatives (compounds 79 and 80) to achieve a better perceptive of the antimalarial structure–activity relationship (SAR)Citation44. Compound 81 (T3.5) is heme targeting scaffold and a chemosensitization functionality into one moleculeCitation45.
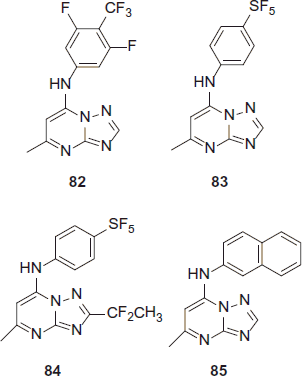
Novel triazolopyrimidine as antimalarials
Compounds 82 and 83 are the two new triazolopyrimidines with the substitution of phenyl moieties which results into good plasma exposure and also better efficacy against P. berghei mouse modelCitation46. Compound 84 was recognized as a preclinical applicant and is presently in the MMV preclinical pipelineCitation47. On the other hand, compound 85 was found to exhibit poor in vivo activity in the mice model (P. berghei)Citation48. The activity of lead compound 85 was improved by including various phenyl substituted groups in the place of naphthyl. This modification amplified the metabolic stability of the compound and also concealed the growth of P. berghei in mice after oral administration.
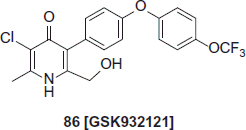
Novel pyridones as antimalarials
Pyridines are the new class of antimalarial which targeting the bc1 complexCitation49. With the collaboration of Yeates et al. GlaxoSmithKline (GSK) synthesized a series of disubstituted derivatives of clopidolCitation50. Though, development of the compound was terminated by the MMV due to unexpected side effects as cardiotoxicityCitation51. Further modifications of the template results into compound 86 (GSK932121) which was found to be highly potent both in vitro and against multidrug resistant strains of P. falciparum in a novel murine P. falciparum modelCitation52,Citation53.
Novel quinolones as antimalarials
Next class of antimalarial is quinolones targeting the Plasmodium bc1 complex. Several alkyl and alkoxy 4(1H)-quinolones were synthesized in an effort to determine the SAR around these compounds that would results into a potent and selective inhibitorCitation54. Compound 87 (ELQ-300) is a selective potent inhibitor of the Plasmodium bc1 complex and it has been selected as a preclinical candidate by the MMVCitation55. Cowley et al. synthesized quinolone esters and by broad study of SAR, it have been proven that by targeting the P. falciparum bc1 complex, highly potent antimalarial compounds synthesized with the lead compound 88 (RCQ) having an IC50 value of 0.46 nMCitation56. Biagini et al. synthesized analogue 90 (hydroxy-2-dodecyl-4-(1H)-quinolone [HDQ]) targets the Pf NDH2 inhibitorCitation57. The synthesized compound 90 (HDQ) was used in HTS technique. Analogue 89 (CK-2-68) was selected as a lead compound for further developmentCitation58.
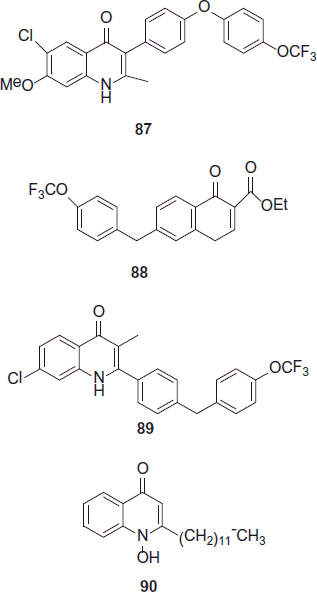
Miscellaneous heterocyclics as antimalarial under clinical trials
Novel tetroxane derivatives as antimalarials
The tetroxane derivative [RKA 182], 91 is a synthetic peroxide. Heme alkylation has been suggested as its mode of action. IC50 values against 3D7 and K1 strains were below 1 nM. In P. berghei-infected mice, it showed ED50/ED90 values of 1.33/4.18 mg (1.6/4.9 µmol)/kg.

Even against 11 Southeast Asian isolates from patients having failed ACT combination chemotherapy, RKA 182 showed superior in vitro activity (IC50 values: 2–5 nM) compared to mefloquine, artesunate and artemisinin. Toxicity was found to be acceptable. With the formation of the ditosylate salt, an increased oral bioavailability in rat (38%) and mouse (42%) models was achievedCitation59.
Novel indole derivatives as antimalarials
Barker et al. synthesized novel indole derivative Genz-668764, 92 which is one out of several hundred derivatives developed by both Genzyme and the Broad Institute. The compound originates from a screen at the Broad Institute and displays IC50 values of 28–65 nM against various strains of P. falciparum as well as an acceptable pharmacokinetic profile. In a P. berghei model, 3 out of 5 animals were cured when treated with 200 mg (620 µmol)/kg/day, twice a dayCitation60.
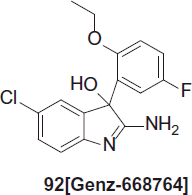
Novel imidazolopiperazine as antimalarial
Wu et al. synthesized GNF156, 93 an imidazolopiperazine, which has been optimized from a hit obtained by a cell-based proliferation assay of P. falciparum from a GNF (Global Nature Fund) compound library collection conducted by Novartis. This imidazolopiperazine seems to inhibit neither protein biosynthesis nor cytochrome bc1, but arrests the parasite in its early stages of developmentCitation61,Citation62.
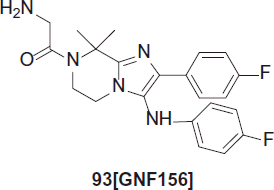
GNF156 reveals in vitro activity for blood and liver stage infections. Moreover, it blocks transmission at 500 nM and is potent against different strains of P. falciparum: EC50 = 6 nM for W2, 10 nM for 3D7. In a P. berghei mouse model, ED50 is 0.48 mg (1.17 µmol)/kg and ED99 is 2.2 mg (5.35 µmol)/kg. Oral bioavailability is 74% for mice, 20% for rats, 89% for dogs and is estimated to be 67% for humansCitation63. GNF156 recently entered Phase I studies.
Novel biscationic choline based compounds as antimalarials
The design, synthesis and biological screening of a series of 15 disulfide prodrugs as precursors of albitiazolium bromide [(SAR97276, compound 94, a choline analogue under clinical trials (phase II)], has been accomplished for the treatment of severe malaria.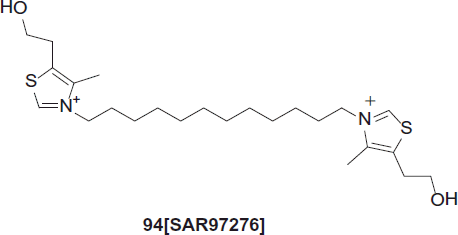
The corresponding prodrugs are expected to revert back to the active bis-thiazolium salt through an enzymatic reduction of the disulfide bond. To enhance aqueous solubility of these prodrugs, an amino acid residue (valine or lysine) or a phosphate group was introduced on the thiazolium side chain. Most of the novel derivatives exhibited potent in vitro antimalarial activity against P. falciparum. After oral administration the cyclic disulfide prodrug showed the best improvement of oral efficacy in comparison to the parent drugCitation64.
Le Roche et al. synthesized SAR97276, compound 94 also known as T3, is a member of the class of biscationic compounds designed based on the choline structure. T3 inhibits choline uptake and synthesis, presumably with several targets. Due to its permanently charged nature, T3 is not orally bioavailable and is, therefore, evaluated as a parenteral formulation for the treatment of severe malariaCitation65.
Novel heterocyclics as antimalarial from natural sources
Novel artemisinin analogs as antimalarials
The presence of widespread drug resistance is hampering the effectiveness of most of the available drug arsenalCitation66–71 with the prominent exception of artemisinin-based derivatives. Artemisinin-based combination therapies (ACTs) are the recommended first-line treatments for falciparum malaria in all countries with rampant diseaseCitation72. Though, ACTs remains effective, there are recent concerns that the efficacy of artemisinin-based derivatives has declined on the Thai-Cambodian borderCitation73–75. Potential for clinical resistance is closely monitored in areas of Southeast Asia where artemisinin had been used as monotherapy for decades and where antimalarial drug resistance previously developed. The potential development of artemisin in resistance by the parasite and loss of ACTs would be a catastrophe, as ACTs have been recommended worldwide and all new agents currently in phase 3 clinical trials are based on ACTs. In the scuffle to fight malaria, introduction of new drugs with novel mechanisms of action is decisive.

Nagelschmitz et al. synthesized Artemisone, 95 which is a second generation artemisinin derivative with high efficacy, improved bioavailability and negligible neuro- and cytotoxicity in vitro and in vivo, proofed to be a safe, well tolerated, and potent drug in Phase I studies. Its IC50 values in vitro are 1.49 nM (K1 strain) and 1.59 nM (TM90-C2A strain)Citation76. Waknine-Grinberg et al. found that artemisone also shows efficacy against cerebral malaria in P. berghei-infected miceCitation77.
Novel indoloquinoline derivatives as antimalarial drugs
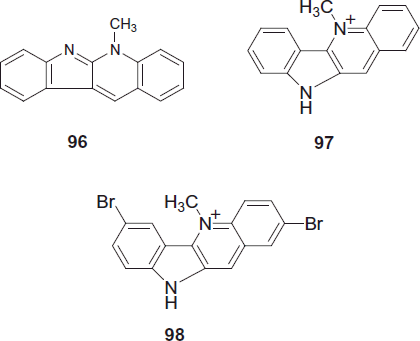
Wright et al. synthesized the most promising analogue up to date is 2,7-dibromocryptolepine 98, which is approximately nine-fold more potent than cryptolepine against CQ-resistant P. falciparum (strain K1) and when tested in mice infected with P. berghei 25 mg/kg/day given i.p., parasitemia was suppressed by 90% with no apparent toxicity to the mice. Recent studies also suggested that the antimalarial mode of action of this and other cryptolepine analogues involves other mechanisms in addition to the inhibition of hemozoin formationCitation78,Citation79. The cryptolepine 97 isomer, neocryptolepine 96, a minor constituent of Cryptolepis sanguinolenta, is also currently being pursued as a lead to new antiprotozoal agents.
Novel quassinoid analogues as antimalarials
One group of compounds that has been explored further is the quassinoids which are degraded triterpenes found in various species of Simaroubaceae, some of which are used traditionally for the treatment of malaria and other protozoal diseasesCitation80. Although quassinoids are generally cytotoxic, a few compounds such as glaucarubinone 99, from Simarouba amara are relatively selective against P. falciparum in vitro but the latter was found to be toxic in vivo when tested in mice infected with P. berghei. Structure–activity relationship studies of a series of quassinoids as well as structural modifications of quassin 100 from Quassia amara, were carried out in an attempt to improve selectivity against P. falciparum. The antiplasmodial and cytotoxic properties of the quassinoids are both due to protein synthesis inhibition and it is likely that parasite and host cell ribosomes are too similar to allow for the development of selective inhibitors.

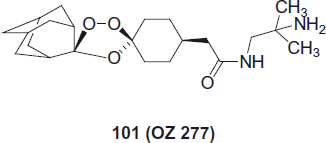
OZ 277 compound 101, consists of a trioxane ring attached to an adamantane ring which provides stability to the endoperoxide group without declining its antimalarial action and the molecule also includes a tosylate side chain that confers superior water solubility and formulation propertiesCitation81. This compound, which was developed initially with the support of the WHO Tropical Diseases Research Programme and later by the Medicines for Malaria Venture is now undergoing Phase I clinical trials in the UK.
Brusatol, 102 a constituent of Brucea javanica, was carried out in an attempt to improve selectivity against P. falciparum but these strategies were not successful. The antiplasmodial and cytotoxic properties of the quassinoids are both due to protein synthesis inhibition and it is likely that parasite and host cell ribosomes are too similar to allow for the development of selective inhibitors.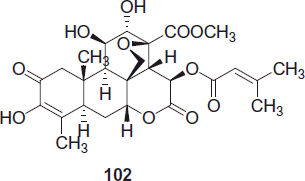
Polonsky et al. refer quassinoids to the group of compounds, which are the bitter principles of the Simaroubaceae family. According to their basic skeleton; quassinoids are categorized into five different groups, 103 (C-18), 104 (C-19), 105 (C-20), 106 (C-25), 107 (C-22) and 108 (C-22) typesCitation82,Citation83.
It has been considered as an enormous discovery that several quassinoids possess potent antimalarial activity, especially against CQ-resistant P. falciparumCitation84,Citation85. In the malaria parasite, quassinoids are rapid and potent inhibitors of protein synthesis, most likely due to effects upon the ribosome rather than upon nucleic acid metabolismCitation86. Studies have revealed that the chance of cross-resistance between quassinoids and CQ is less, since CQ did not affect protein synthesis. Thus, quassinoids may be presumed to act upon the malaria parasite through a fundamentally different mechanism to that of CQ. Some quassinoids have shown greater selectivity against P. falciparum than against KB cellsCitation87.
Novel benzo-1,3-dioxole based antimalarials
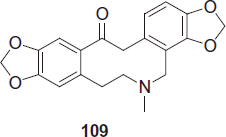
Different alkaloids berberine, allocryptopine and protopine 109 are obtained from Argemone mexicana. Although these compounds showed in vitro antimalarial activity (IC50 of protopine against the W2-strain: 0.32 µg/mL = 0.91 µM)Citation88, berberine is merely absorbed, and the aqueous decoction of A. mexicana was not active against P. berghei in the mouse modelCitation89.
Novel pyranoindolopiperidine-based antimalarials
The letters PR 259 CT1, compound 110 represent an herbal medicinal product comprising an ethanol extract from the bark of Nauclea pobeguinii, which is being developed by the University of Antwerp and the Democratic Republic of Congo. Nauclea pobeguinii is an evergreen tree native to the paleotropics, of which the Democratic Republic of Congo is a part.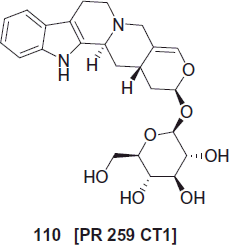
PR 259 CT1, compound 110 contains 5.6% strictosamide as the putative active compound and has been shown to be well tolerated in healthy volunteers. The drug enduringly cured 10 out of 11 patients from the diseaseCitation90.
Novel 1,2,4-trioxolane and morpholine-based antimalarials
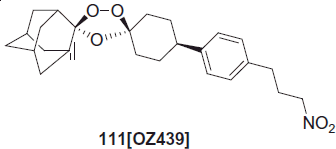
Charman et al. has been recently designed OZ439 compound 111 to overcome the instability of OZ277. The enhanced stability of OZ439 in infected blood has been attributed to the steric effect of the phenyl substituent in the 8-position. A single dose of OZ439 cures P. berghei-infected miceCitation91.
In preclinical studies the arterolane (also known as OZ277 or RBx11160 compound 113) proved to be a highly active rapidly acting antimalarial. In malaria patients, however, it showed significantly lower bioavailability when compared to healthy subjects. This is most probably due to higher Fe2+ loads resulting in rapid iron mediated degradation of OZ277. In a clinical study using OZ277 as a single therapeutic, recrudescence rates up to 30% were observed. OZ277 is now evaluated in combination with piperaquineCitation92.
Novel polymer-linked combined heterocyclic antimalarial drugs
Recently, our research group has synthesized polymer-linked combined antimalarial drugs conjugates by linking PQ as 8-aminoquinoline derivative and artemisinin derivatives like dihydroartemisinin obtained from derivatives of Artemisia annua employing substituted polyphosphazene as polymeric backbone and converting them to nanoparticles formulations and tested in P. berghei infected mice model. Among the synthesized derivatives, the polymer-linked combined antimalarial drugs conjugate 113 was found to be the most potent.
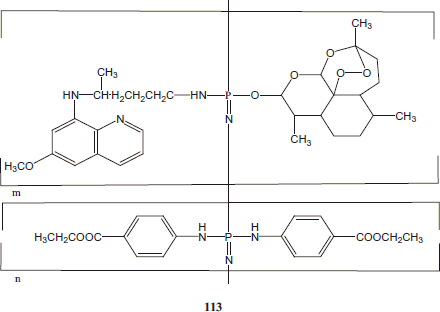
The combination therapy exhibited promising antimalarial efficacy at lower doses in comparison to the standard drug combination. Further, this combination therapy provided protection over 35 days without any recrudescence, thus proving to be effective against resistant malaria. The study provides an alternative combination regimen found to be effective in the treatment of resistant malariaCitation93.
Conclusion
As discussed in this review, several novel heterocyclic compounds containing unique structural composition have been discussed which are being evaluated for antimalarial potential for drug sensitive and resistant strains. These novel heterocyclic compounds have exhibited promising antimalarial activities in vitro and in vivo. The nature has also provided novel heterocyclic leads, which can be developed into safe drugs by synthetic strategies as exemplified by novel artemisinin analogs, quassinoids, indoloquinoline derivatives, benzo-1,3-dioxole-based antimalarials, pyranoindolopiperidine, 1,2,4-trioxolane, morpholine-based antimalarials, polymer-linked combined antimalarials. Therefore, compounds described herein provide useful pharmacophores, which could be modulated to obtain potential antimalarials active against not only drug-sensitive, but also drug-resistant and multi-drug resistant strains of Plasmodium. To conclude, the structural modifications in novel heterocyclic have been generous in providing advancements in remedies for the treatment of malaria. However, various new heterocyclic systems are still there to be explored, which when systematically developed will provide additional new leads, pharmacophores and drugs for the treatment and radical cure of malaria.
Declaration of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors acknowledge the Life Science Research Board (LSRB) of Defence Research and Development Organization (DRDO), New Delhi, project no. DL/81/48222/LSRB-232/SH & DD/2011. We are also thankful to Sh. Parveen Garg, Chairman, I.S.F. College of Pharmacy, Moga for his continuous support. We also acknowledge Department of Research, Innovation and Consultancy, Punjab Technical University, Jalandhar, India.
References
- World Malaria Report. Geneva: World Health Organization; 2008
- Ashley E, Gready RM, Proux S, et al. Malaria. Travel Med Infect Dis 2006;4:159–73
- Lalloo DG. Malaria in adolescence: burden of disease, consequences and opportunities for intervention. Lancet Infect Dis 2006;6:780–93
- Snow RW, Trape JF, Marsh K. The past, present and future of childhood malaria mortality in Africa. Trends Parasitol 2001;17:593–7
- Breman JG. The ears of the hippopotamus: manifestations, determinants, and estimates of the malaria burden. Am J Trop Med Hyg 2001;64:1–11
- Wongsrichanalai C, Pickard AL, Wernsdorfer WH, et al. Epidemiology of drug-resistant malaria. Lancet Infect Des 2002;2:209–18
- White NJ. Antimalarial drug resistance: the pace quickens. J Antimicrob Chemother 1992;30:571–85
- Sibley CH, Hyde JE, Sims PF, et al. Pyrimethamine–sulfadoxine resistance in Plasmodium falciparum. Trends Parasitol 2001;17:582–8
- Ridley RG. Malaria: to kill a parasite. Nature 2002;415:686–93
- Ruiz FA, Sanchez RNG, Estupinan SV, et al. Synthesis and antimalarial activity of new heterocyclic hybrids based on chloroquine and thiazolidinone scaffolds. Bioorg Med Chem 2011;19:4562–73
- Kouznetsov VV, Gómez BA. Recent developments in the design and synthesis of hybrid molecules based on aminoquinoline ring and their antiplasmodial evaluation. Eur J Med Chem 2009;44:3091–113
- Raynes KR. Bisquinoline antimalarials: their role in malaria chemotheraphy. Int J Parasitol 1999;29:367–79
- Ayad F, Tilley L, Deady LW. Synthesis, antimalarial activity and inhibition of haem detoxification of novel bisquinolines. Bioorg Med Chem Lett 2001;11:2075–7
- Vennerstrom JL, Ager AL, Dorn A, et al. Peroxides as antimalarials. Med Chem 1998;41:4360–4
- Nagata K, Aistrup GL, Honda H, et al. Modulation of the nicotinic acetylcholine receptor by dioscorine in clonal rat Phaeochlomocytoma (Pc12) cells. Biochem Physiol 1999;64:157–65
- Khan MOF, Levi MS, Tekwani BL. Synthesis of isoquinuclidine analogues of chloroquine, antimalarial and antileishmanial activity. Bioorg Med Chem 2007;15:3919–25
- Madrid PB, Liou AP, DeRisi JL, et al. Incorporation of an intramolecular hydrogen-bonding motif in the side chain of 4-aminoquinolines enhances activity against drug resistant P. falciparum. Med Chem 2006;49:4535–43
- Ruiz FAR, Sanchez RNG, Estupinan SV, et al. Synthesis and antimalarial activity of new heterocyclic hybrids based on chloroquine and thiazolidinone scaffolds. Bioorg Med Chem 2011;19:4562–73
- Solomon VR, Haq W, Srivastava K, et al. Synthesis and antimalarial activity of side chain modified 4-aminoquinoline derivatives. Med Chem 2007;50:394–8
- Jutta M, Ferryanto C, Pak P, et al. Ex vivo drug susceptibility of ferroquine against chloroquine-resistant isolates of P. falciparum and P. vivax. Antimicrob Agents Chemother 2011;55:4461–4
- Marfurt J, Chalfein F, Prayoga P, et al. Ex vivo drug susceptibility of ferroquine against chloroquine-resistant isolates of P. falciparum and P. vivax. Antimicrob Agents Chemother 2011;55:4461–4
- Ngoma GM, Supan CC, Bianco MPD, et al. Phase I randomized dose-ascending placebo controlled trials of ferroquine a candidate anti-malarial drug in adults with asymptomatic Plasmodium falciparum infection. Malaria J 2011;10:53. doi: 10.1186/1475-2875-10-53
- O'Neill PM, Barton VE, Ward SA, et al. 4-Aminoquinolines: chloroquine amodiaquine and next-generation analogues. In: Staines HM, Krishna S, eds. Treatment and prevention of malaria. Springer Basel AG; 2012:19–44
- Coslédan F, Fraisse L, Pellet A, et al. Selection of a trioxaquine as an antimalarial drug candidate. PNAS 2008;105:79–84
- Garah FB, Stigliani JL, Cosledan F, et al. Docking studies of structurally diverse antimalarial drugs targeting PfATP6: no correlation between in silico binding affinity and in vitro antimalarial activity. ChemMedChem 2009;4:1469–79
- O'Neill PM, Park BK, Shone AE, et al. Candidate selection and preclinical evaluation of N-tert-butyl isoquine (GSK369796), an affordable and effective 4-aminoquinoline antimalarial for the 21st century. J Med Chem 2009;52:1408–15
- Perez BC, Teixeira C, Figueiras M, et al. Novel cinnamic acid/4-aminoquinoline conjugates bearing non-proteinogenic amino acids: towards the development of potential dual action antimalarials. EJMC 2012;54:887–99
- Jain R, Jain S, Gupta RC, et al. Synthesis of aminoacid derivatives of 8-[(4-amino-1-methylbutyl)amino]-6-methoxy-4-substituted/4,5-disubstituted-quinolines as potential antimalarial agents. Indian J Chem 1994;33B:251–4
- Kaur K, Jain M, Khan SI, et al. Synthesis, antiprotozoal, antimicrobial, β-hematin inhibition, cytotoxicity and methemoglobin formation activities of bis(8-aminoquinolines). Bioorg Med Chem 2011;19:197–210
- Makler MT, Hinrichs DJ. Measurements of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am J Trop Med Hyg 1993;48:205–10
- Tekwani BL, Walker LA. 8-Aminoquinolines: future role as antiprotozoal drugs. Curr Opin Infect Dis 2006;19:623–31
- Chesney JM, Nanayakkara NPD, Bartlett M, et al. 8-Aminoquinolines, US Patent No. US 6376511 B2. 2008
- Dutta AK, Stodghill SP, Wyandt CM. Physicochemical characterization of NPC 1161C, a novel antimalarial 8-aminoquinoline, in solution and solid state. AAPS Pharm Sci Tech 2011;12:177–91
- Zhu S, Zhang Q, Gudise C, et al. Synthesis and evaluation of Naphthyridine compounds as antimalarial agents. Bioorg Med Chem Lett 2007;17:6101–6
- Desjardins RE, Canfield CJ, Haynes DE, et al. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother 1979;16:710–18
- Chulay JD, Haynes JD, Diggs CL. Plasmodium falciparum: assessment of in vitro growth by hypoxanthine incorporation. Exp Parasitol 1983;55:138–46
- Jain M, Vangapandu S, Sachdeva S, et al. 8-Quinolinamines conjugated with amino acids are exhibiting potent blood-schizontocidal antimalarial activities. Med Chem 2004;47:239–47
- Nasveld PE, Edstein MD, Reid M, et al. Tafenoquine Study Team. Randomized, double-blind study of the safety, tolerability, and efficacy of tafenoquine versus mefloquine for malaria prophylaxis in nonimmune subjects. Antimicrob Agents Chemother 2010;54:792–8
- Leary KJ, Riel MA, Roy MJ, et al. A randomized, doubleblind, safety and tolerability study to assess the ophthalmic and renal effects of tafenoquine 200 mg weekly versus placebo for 6 months in healthy volunteers. Am J Trop Med Hyg 2009;81:356–62
- Kelly JX, Smilkstein MJ, Cooper RA, et al. Design, synthesis, and evaluation of 10-N-substituted acridones as novel chemosensitizers in Plasmodium falciparum. Antimicrob Agents Chemother 2007;51:4133–40
- Calienes AF, Pellon R, Docampo M, et al. Antimalarial activity of new acridinone derivatives. Biomed Pharma 2011;65:210–14
- Waffo AF, Coombes PH, Crouch NR, et al. Acridone and furoquinoline alkaloids from Teclea gerrardii (Rutaceae: Toddalioideae) of southern Africa. Phytochem 2007;68:663–7
- Biagini GA, Fisher N, Berry N. Acridinediones: selective and potent inhibitors of the malaria parasite mitochondrial bc1 complex. Mol Pharmacol 2008;73:1347–55
- Riscoe M, Winter R, Kelly J, et al. Evaluation and lead optimization of antimalarial aromatic ketones. Am J Trop Med Hyg 2006;75:97–8
- Kelly JX, Smilkstein MJ, Brun R. Discovery of dual function acridones as a new antimalarial chemotype. Nature 2009;459:270–3
- Gujjar R, Mazouni FE, White KL. Lead optimization of aryl and aralkyl amine-based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with antimalarial activity in mice. J Med Chem 2011;54:3935–49
- Coteron JM, Marco M, Esquivias J. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J Med Chem 2011;54:5540–61
- Gujjar R, Marwaha A, Mazouni FE. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J Med Chem 2009;52:1864–72
- Xiang H, Freed JM, Moorthy GS. Preclinical drug metabolism and pharmacokinetic evaluation of GW844520, a novel anti-malarial mitochondrial electron transport inhibitor. J Pharm Sci 2006;95:2657–72
- Yeates CL, Batchelor JF, Capon EC. Synthesis and structure–activity relationships of 4-pyridones as potential antimalarials. J Med Chem 2008;51:2845–52
- Bueno JM, Manzano P, Garcia MC. Potent antimalarial 4-pyridones with improved physico-chemical properties. Bioorg Med Chem Lett 2011;21:5214–18
- Diaz MBJ, Mulet T, Viera S. Improved murine model of malaria using Plasmodium falciparum competent strains and non-myelodepleted NOD-scid IL2Rgnull mice engrafted with human erythrocytes. Antimicrob Agents Chemother 2009;53:4533–6
- Bueno JM, Herreros E, Barturen IA. Exploration of 4(1H)-pyridones as a novel family of potent antimalarial inhibitors of the plasmodial cytochrome bc1. Future Med Chem 2012;4:2311–23
- Winter RW, Kelly JX, Smilkstein MJ, et al. Antimalarial quinolones: synthesis, potency and mechanistic studies. Exp Parasitol 2008;118:487–97
- Nilsen A, Lacrue AN, White KL, et al. Quinolone-3-diarylethers: a new class of antimalarial drug. Sci Transl Med 2013;5:1–25
- Cowley R, Leung S, Fisher N. The development of quinolone esters as novel antimalarial agents targeting the Plasmodium falciparum bc1 protein complex. Med Chem Comm 2012;3:39–44
- Biagini GA, Fisher N, Shone AE. Generation of quinolone antimalarials targeting the Plasmodium falciparum mitochondrial respiratory chain for the treatment and prophylaxis of malaria. Proc Natl Acad Sci USA 2012;109:8298–303
- Pidathala C, Amewu R, Pacorel B. Identification, design and biological evaluation of bisaryl quinolones targeting Plasmodium falciparum type II NADH:quinone oxidoreductase (PfNDH2). J Med Chem 2012;55:1831–43
- O'Neill PM, Amewu RK, Nixon GL, et al. Identification of a 1,2,4,5-tetraoxane antimalarial drug-development candidate (RKA182) with superior properties to the semisynthetic artemisinins. Angew Chem Int Ed 2010;49:5693–7
- Barker RH, Urgaonkar S, Mazitschek R, et al. Aminoindoles, a novel scaffold with potent activity against Plasmodium falciparum. Antimicrob Agents Chemother 2011;55:2612–22
- Wu T, Nagle A, Kuhen K, et al. Imidazolopiperazines: hit to lead optimization of new antimalarial agents. J Med Chem 2011;54:5116–30
- Meister S, Plouffe DM, Kuhen KL, et al. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 2011;334:1372–7
- Weaver M, Han X, Francese G, et al. Discovery of novel antimalarials through cell based medicinal chemistry optimization of HTS hits. Med Chem Symp 2011;1–28
- Caldarelli SA, Fangour S, Sharon W, et al. New bis-thiazolium analogues as potential antimalarial agents: design, synthesis and biological evaluation. Med Chem 2013;56:496–509
- Roche KL, Johnson J, Ahiboh H, et al. A systematic approach to understand the mechanism of action of the bisthiazolium compound T4 on the human malaria parasite, Plasmodium falciparum. BMC Genomics 2008;9:513
- Valderramos SG, Fidock DA. Transporters involved in resistance to antimalarial drugs. Trends Pharmacol Sci 2006;27:594–601
- Ekland EH, Fidock DA. In vitro evaluations of antimalarial drugs and their relevance to clinical outcomes. Int J Parasitol 2008;38:743–7
- Nkrumah LJ, Riegelhaupt PM, Moura P. Probing the multifactorial Basisium falciparum quinine resistance: evidence for a strain-specific contribution of the sodium proton exchanger PfNHE. Mol Biochem Parasitol 2009;165:122–31
- Sidhu AB, Valderramos SG, Fidock DA. pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol Microbiol 2005;57:913–26
- Sidhu AB, Uhlemann AC, Valderramos SG, et al. Decreasing pfmdr1 copy number in Plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin. J Infect Dis 2006;194:528–35
- Sidhu AB, Pinard DV, Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science 2002;298:210–13
- Eastman RT, Fidock DA. Artemisinin-based combination therapies: a vital tool in efforts to eliminate malaria. Nat Rev Microbiol 2009;7:864–74
- Dondorp AM, Nosten F, Yi P. Artemisinin resistance in Plasmodium falciparum Malaria. N Engl J Med 2009;361:455–67
- Lim P, Alker AP, Khim N. Pfmdr1 copy number and arteminisin derivatives combination therapy failure in falciparum malaria in Cambodia. Malaria J 2009;8:11. doi: 10.1186/1475-2875-8-11
- White NJ. Qinghaosu (Artemisinin): the price of success. Science 2008;320:330–4
- Nagelschmitz J, Voith B, Wensing G, et al. First assessment in humans of the safety, tolerability, pharmacokinetics, and ex vivo pharmacodynamic antimalarial activity of the new artemisinin derivative artemisone. Antimicrob Agents Chemother 2008;52:3085–91
- Waknine-Grinberg JH, Hunt N, Marciano AB, et al. Artemisone effective against murine cerebral malaria. Malaria J 2010;9:227. doi: 10.1186/1475-2875-9-227
- Wright CW, Kyereme JA, Breen AG, et al. Synthesis and evaluation of cryptolepine analogues for their potential as new antimalarial agents. J Med Chem 2001;44:3187–94
- Onyeibor O, Croft SL, Dodson HI, et al. Synthesis of some cryptolepine analogues, assessment of their antimalarial and cytotoxic activities and consideration of their antimalarial mode of action. J Med Chem 2005;48:2701–9
- Phillipson JD, Wright CW, Kirby GC, et al. Phytochemistry of some plants used in traditional medicine for the treatment of protozoal diseases. Oxford: Oxford Science Publications; 1995:95–135
- Vennerstrom JL, Barnes SA, Brun R, et al. Identification of an antimalarial synthetic trioxolane drug development. Nature 2004;430:900–4
- Polonsky J, Fortschr. Quassinoid bitter principles. Chem Org Naturst 1973;30:101–50
- Polonsky J, Fortschr. Antimalarial activity of quassinoids against chloroquine resistant Plasmodium falciparum in vitro. Chem Org Naturst 1985;47:221
- Ang HH, Chan KL, Mak JW. In vitro antimalarial activity of quassinoids from Eurycoma longifolia against Malaysian chloroquine-resistant Plasmodium falciparum isolates. Planta Med 1995;61:177–8
- O”Neill MJ, Bray DH, Boaedman P, et al. Plants as sources of antimalarial activities of some quassinoids. Antimicrob Agents Chemother 1986;30:101–4
- Kirby GC, O'Neill MJ, Phillipson JD, et al. In vitro studies on the mode of action of quassinoids with activity against chloroquine-resistant Plasmodium falciparum. Biochem Pharmacol 1989;38:4367–74
- Anderson MM, O'Neill MJ, Phillipson JD, et al. In vitro cytotoxicity of a series of quassinoids from Brucea javanica, fruits against KB cells. Planta Med 1991;57:62–4
- Pires CAS. Investigation of antiplasmodial compounds from various plant extracts, These de doctorat. Univ Geneve no Sc 2009;4129:1–226
- Willcox ML, Graz B, Falquet J, et al. A reverse pharmacology approach for developing an anti-malarial phytomedicine. Malaria J 2011;10:8. doi: 10.1186/1475-2875-10-S1-S8
- Mesia K, Tona L, Mampunza MM, et al. Antimalarial efficacy of a quantified extract of Nauclea pobeguinii stem bark in human adult volunteers with diagnosed uncomplicated falciparum malaria: Part 1: a clinical phase II A trial. Planta Med 2012;78:211–18
- Charman SA, Barnes SA, Bathurst IC, et al. Vennerstrom, synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. PNAS 2011;108:4400–5
- Valecha N, Martensson S, Looareesuwan. Arterolane, a new synthetic trioxolane for treatment of uncomplicated Plasmodium falciparum malaria: a phase II, multicenter, randomized, dose-finding clinical trial. Clin Infect Dis 2010;51:684–91
- Kumar S, Singh RK, Sharma R, et al. Design, synthesis and evaluation of antimalarial potential of polyphosphazene linked combination therapy of primaquine and dihydroartemisinin. Eur J Pharm Sci 2015;66:123–37