
Abstract
A series of hydroxylic compounds (1–10, NK-154 and NK-168) have been assayed for the inhibition of three physiologically relevant carbonic anhydrase isozymes, the cytosolic isozymes I, II and tumor-associated isozyme IX. The investigated compounds showed inhibition constants in the range of 0.068–4003, 0.012–9.9 and 0.025–115 μm at the hCA I, hCA II and hCA IX enzymes, respectively. In order to investigate the binding mechanisms of these inhibitors, in silico studies were also applied. Molecular docking scores of the studied compounds are calculated using scoring algorithms, namely Glide/induced fit docking. The inhibitory potencies of the novel compounds were analyzed at the human isoforms hCA I, hCA II and hCA IX as targets and the KI values were calculated.
Introduction
Phenol (1) is the specific name for hydroxybenzene. It is an important industrial commodity as a precursor to many materials and useful compounds. Phenol and its chemical derivatives are key for building polycarbonates, detergents, herbicides and numerous pharmaceutical drugsCitation1. Catechol (2) is a natural organic compound with the formula 1,2-dihydroxybenzene. Resorcinol (3) and pyrogallol (4) are organic compounds. They also have antiseptic properties. Salicylic acid (5) is a beta-hydroxybenzoic acid. Salicylic acid and its derivatives are widely used for the treatment of various diseases. o-coumaric acid (6) is a 2-hydroxycinnamic acid, and it can be found in vinegarCitation2,Citation3. Dobutamine (7) is used to treat acute but potentially reversible heart failure, such as cardiac surgery or cases of septic or cardiogenic shock, on the basis of its positive inotropic actionCitation4. Curcumin (8), the yellow pigment isolated from the root of the plant Curcuma longa L., has several biological effects, the most important being its anticarcinogenic effect. Curcumin is a nutraceutical compound reported to possess therapeutic properties against a variety of diseases ranging from cancer to cystic fibrosisCitation5,Citation6.
The carbonic anhydrases (CAs, EC 4.2.1.1) are zinc-containing metalloenzymes that catalyze the reversible hydration of carbon dioxide in a two-step reaction, to yield bicarbonate and protonsCitation7. Among the 16 isoenzymes described up to now in mammalian organisms, CA I and CA II are present at high concentrations in the cytosol of erythrocytes and the gastrointestinal tractCitation7–11. CA IX is found in tumor cells and absent (or are present in very limited amount) in normal tissuesCitation12–15. CA isoenzymes constitute interesting targets for the design of pharmacological agents that are useful in the treatment or prevention of a variety of disorders such as glaucoma, epilepsy, as diuretics or antitumor agents/diagnostic toolsCitation7,Citation14.
Many hydroxy compound derivatives have been widely used as prodrugs or drugs. For instance, paracetamol is a widely used over-the-counter analgesic and antipyretic. Another hydroxy group containing molecule, amoxicillin is antibiotic. Our group recently investigated the interaction of CA isozymes I, II with salicylic acid derivatives, antioxidant phenolic compounds, hydroxy group containing compounds, etc.Citation2,Citation3,Citation6–11,Citation16 We wanted to extend these earlier investigations to some hydroxy and phenolic compounds in order to discover novel powerful CA inhibitors (CAIs) which might have implications in medicineCitation6–11,Citation16.
In this work, a series of hydroxylic compounds (1–10, NK-154 and NK-168) have been assayed for the inhibition of CA I, II and IX isozymes. Inhibition is reported as KI (μm) and the results are the average of at least three independent experiments. Since X-ray structures of these small molecules with CA targets are not yet available, in silico studies are performed for the investigation of their binding mechanisms.
Experimental
All chemicals were obtained commercially from Sigma-Aldrich (Bornem, Belgium). IR spectra were recorded on a PerkinElmer 100 FTIR spectrometer and all 1H-NMR and 13C-NMR spectra were recorded using an Oxford NMR 400 MHz spectrometer using TMS as the internal standard d values in ppm at ambient temperature. Melting points were measured on the variable heater.
Synthesis of NK-154 and NK-168
Detailed synthetic procedures for the preparation of N-(1-(2-hydroxyphenylamino)-3-methyl-1-oxobutan-2-ylcarbamothioyl) benzamide (NK-154) and N-(1-(2-hydroxyphenylamino)-4-methyl-1-oxopentan-2-ylcarbamothioyl) benzamide (NK-168) can be found in 17.
Purification of human carbonic anhydrase isozymes by affinity chromatography
Erythrocytes were purified from fresh human blood obtained from the Blood Center of the Research Hospital at Atatürk University. The blood samples were centrifuged at 1500 rpm for 15 min and the plasma and buffy coat were removed. The red cells were isolated and washed twice with 0.9% NaCl and hemolyzed with 1.5 volumes of ice-cold water. The ghost and intact cells were removed by centrifugation at 20 000 rpm for 30 min at 4 °C. The pH of the hemolysate was adjusted to 8.7 with solid TrisCitation2. First, benzoyl chloride was stirred for 4 h at room temperature in CH2Cl2 cellulose. After the spacer arm cellulose added as a benzyl group and finally diazotized sulfanilamide clamped to the para position of benzyl group as a ligand. The hemolysate was applied to the prepared cellulose-benzyl-sulfanilamide affinity column equilibrated with 25 mm Tris-HCl/0.1 M Na2SO4 (pH 8.7). The affinity gel was washed with 25 mm Tris-HCl/22 mm Na2SO4 (pH 8.7). The human carbonic anhydrase (hCA I and hCA II) isozymes were eluted with 1 M NaCl/25 mm Na2HPO4 (pH 6.3) and 0.1 M CH3COONa/0.5 M NaClO4 (pH 5.6), respectively. All procedures were performed at 4 °C.
CA IX purification protocol has been used for obtaining high amounts of hCA IX needed in these experiments. The cDNA of the catalytic domain of hCA IX (isolated as described by Pastorek et al.)Citation17 was amplified by using PCR and specific primers for the glutathione S-transferase (GST)-Gene Fusion Vector pGEX-3X. The obtained fusion construct was inserted in the pGEX-3X vector and then expressed in Escherichia coli BL21 Codon Plus bacterial strain (from Stratagene, San Diego, CA). The bacterial cells were sonicated, then suspended in the lysis buffer (10 mm Tris pH 7.5, 1 mm EDTA pH 8, 150 mm NaCl and 0.2% Triton X-100). After incubation with lysozyme (approx. 0.01 g/l), the protease inhibitors Complete™ were added to a final concentration of 0.2 mm. The obtained supernatant was then applied to a prepacked glutathione sepharose 4B column, extensively washed with buffer and the fusion (GST-CA IX) protein was eluted with a buffer consisting of 5 mm reduced glutathione in 50 mm Tris-HCl, pH 8.0. Finally, the GST part of the fusion protein was cleaved with thrombin. The obtained CA IX was further purified by sulfonamide affinity chromatography.
CA inhibition
An applied photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activityCitation18. Phenol red (at a concentration of 0.2 mm) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mm Hepes (pH 7.5) as buffer and 20 mm Na2SO4 (for maintaining a constant ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mm for the determination of the kinetic parameters and inhibition constants (five different substrate concentrations have been used). For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mm) were prepared in distilled-deionized water and dilutions up to 0.01 nm were done thereafter with distilled-deionized water. Experiments were done using six different inhibitor concentrations, varying from 100 pM to 0.1 nm. Inhibitor and enzyme solutions were preincubated together for 15 min to 24 h at room temperature (15 min) or 4 °C (all other incubation times) prior to assay, in order to allow for the formation of the E-I complex or for the eventual active site-mediated hydrolysis of the inhibitor. Data reported in shows the inhibition after 15 min incubation, as there were no differences of inhibitory power when the enzyme and inhibitors were kept for longer periods in incubationCitation19,Citation20. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3, as reported earlierCitation18,Citation20, and represent the mean from at least three different determinations. The three CA isozymes used in the experiments were recombinant ones, obtained as reported earlierCitation19–21.
Table 1. KI values for the in vitro inhibition of hCA I, hCA II and IX with compounds 1–10, NK-154, NK-168 and AZA.
In silico docking studies
Glide/SP and Glide/XP docking protocols are applied for the prediction of topologies as well as binding energies (i.e. docking scores) of used compounds at the active sites of CA I, CA II and CA IX enzymes. The active site of the enzyme is defined from the cocrystallized ligands from Protein Data Bank (PDB) files. Crystal structures of hCA I, II and IX are retrieved from PDB server (PDB codes: 2FW4, 5AML and 3IAI), respectivelyCitation22–24. Throughout the docking simulations, both partial flexibility and full flexibility around the active site residues are performed by Glide/SP/XP and induced fit docking (IFD) approachesCitation25. IFD procedure was established in three consequence stages which involve: (i) docking of all ligands into the generated model, (ii) refining of amino acid residues within 4 Å of docked poses and (iii) re-docking of docked ligands against the refined protein.
Results and discussion
Phenolic and polyphenolic compounds have in fact been evaluated as CAIs by our group earlier, being shown that many such derivatives are potent inhibitors of several physiologically relevant isoforms, such as CA I and IICitation8–11,Citation16. However, no X-ray crystallographic characterization of such adducts has been performed yet, the polyphenolic compounds being the first simple phenol (1) for which the X-ray crystal structure in complex with hCA II has been reported by Christianson’s groupCitation26. The rationale of investigating phenols as CAIs is due to the fact that the simple phenol (1) has been shown to be the only competitive inhibitor with CO2 as a substrate for the main isoform of CA, i.e. human CA II (hCA II)Citation26. Our group investigated the interactions of a few simple phenols (1–5), antioxidant phenolic compounds, some natural product and polyphenolic compounds with some mammalian isozymes ()Citation21,Citation27–32. Results for these molecules were in low micromolar/submicromolar inhibitions and derived molecules may assist to design isozyme selective CAIs. Indeed, the inhibition profile of various isozymes with this class of agents was diverse, with inhibition constants ranging from the millimolar to the submicromolar for many simple phenolsCitation19.
Figure 1. Chemical structures of some carbonic anhydrase inhibitor phenolic compounds.
We report here the first study on the inhibitory effects of compounds NK-154 and NK-168 on the CO2 hydration activity of hCA I, II and IX. These compounds were docked at the binding site of the targets (hCA I, hCA II and hCA IX). Glide SP/XP docking scores of docked inhibitors at hCA I, II and IX targets and corresponding binding interactions were tabulated in . The previous reports by Oztürk Sarikaya et al.Citation3 investigated pyrogallol (4) by using an esterase assay, for monitoring CA inhibition. Data of show the following regarding inhibition of hCA I, II and IX with compounds 1–10, NK-154 and NK-168, by using a stopped-flow, CO2 hydration assay for monitoring CA inhibition ( and ).
Figure 2. Chemical structures of tested compounds.
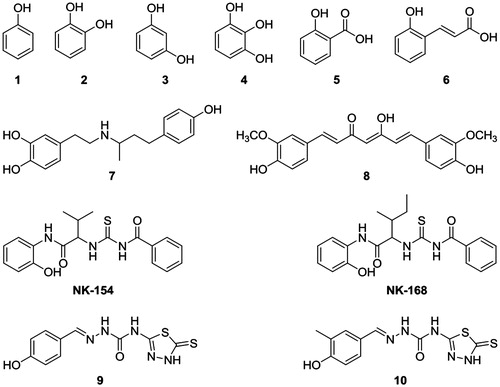
Against the slow cytosolic isozyme hCA I, catechol and resorcinol behave as moderate inhibitors, with KI values in the range of 795–4003 μm, similarly to the structurally related compounds 1, 4 and 5 (KIs of 7.41–10.2 μm). It is interesting to note that the compounds NK-154 and NK-168 were much better hCA I inhibitors as compared to the corresponding compounds 6–10 from which they were prepared. Kinetic investigations (Lineweaver–Burk plots, data not shown) indicate that similarly to sulfonamides and inorganic anionsCitation33–39, all the investigated natural compounds act as competitive inhibitors with CO2 as substrate (i.e. they bind in different regions of the active site cavity as compared to the substrate).
A better inhibitory activity has been observed with compounds dobutamine and curcumin investigated here for the inhibition of the rapid cytosolic isozyme hCA II (). Structure-activity relationship is thus quite sharp for this polyphenolic series of hydroxylic compounds: the N-(1-(2-hydroxyphenylamino) containing compounds NK-154 and NK-168 are ineffective leads, with monophenol ring-containing compounds 1–3, 5 and 6 is already a submicromolar hCA II inhibitor. The best hCA II inhibitor in this compounds was referans sulfonamide compound AZA, which with a KI of 0.012 μm.
In addition to partial flexible docking simulation, flexible receptor docking at the active site was also carried out to enhance the induce fitting treatment of amino acid residues within 4 Å of the ligand. The IFD results were tabulated in for CAI, II and IX receptors (top docking scores were considered). Revealed binding energies from IFD simulations are lower (i.e. higher docking scores) than the results of Glide/SP and XP results. In the case of compounds NK-154 and NK-168, lowest binding energies against CAI, II and IX receptors were connected to these ligands. 2D and 3D ligand binding interaction diagrams of docking poses of both novel inhibitors into the ligand binding site of CAI, II and IX receptors were represented in and . These ligand diagrams clearly identify key amino acid residues which participate in ligand-binding interactions for both receptors. Hydrophobicity and polarity of binding pockets were identified for each complex on diagrams. H-bonding and π–π stacking interactions were displayed by red and blue lines, respectively. Zinc metal ion in all complexes was located in the active site and the neighborhood of inhibitor. The His94 amino acid residue was constrained in all complexes, which plays a critical role in CAI and II inhibitory processes. In the case of CA-I, Gln92 amino acid residue forms an H-bond with the hydroxyl group of compounds 7 and 8. In the case of CA-II, Thr200 has participated in ligand-receptor interaction and hydroxyl groups of compounds 7 and 8 form H bonds. and display 3D docking poses of ligands 7 and 8 at the binding pockets of CAI and II, respectively. In order to determine active sites and orientations of ligands, positions of the docked ligand were zoomed graphically for each complex and the corresponded residues around 4 Å of ligands were labeled. According to the superimposition data of the complexes, in individual CAI, II and IX receptors, both inhibitors convulsively have shared the same positions in ligand-binding pockets.
Figure 3. 2D docking poses of NK-154 and NK-168 into the hCAI, hCA II and hCAIX receptors. Polar, hydrophobic and charged domains were highlighted by different colors as shown in diagram legend.
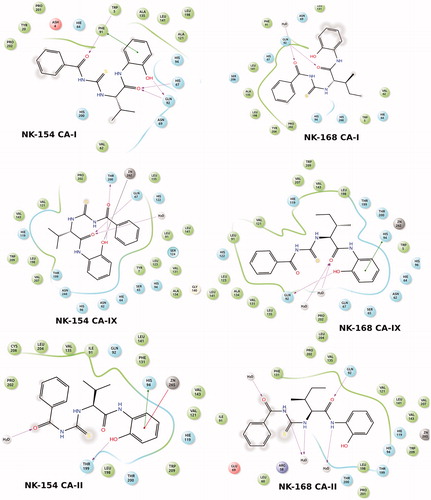
Figure 4. 3D docking poses of NK-154 and NK-168 into the hCAI, hCA II and hCA IX receptors. Key amino acid residues were determined in the 3D positions in active sites.
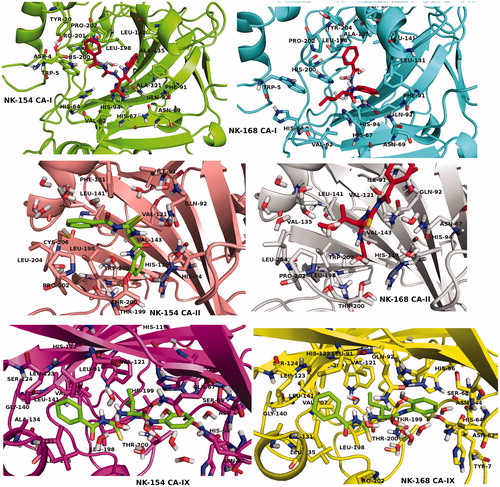
Table 2. Docking score values of selected ligands against hCA I, hCA II and hCA IX.
In a recent study, it was reported that catechol (13) and resorcinol (14)Citation18 as simple compounds which they not involve sulfonamide, sulfamate or related functional groups and these compounds are found as CAIs, and could represent the starting point for a new class of inhibitors that may have advantages for patients with sulfonamide allergiesCitation18,Citation28. The sulfonamide zinc-binding group is thus superior to the thiol one (from the thioxolone hydrolysis product) for generating CAIs with a varied and sometimes isozyme-selective inhibition profile against the mammalian enzymes. However, it is critically important to explore further classes of potent CAIs in order to detect compounds with a different inhibition profile as compared to the sulfonamides and their bioisosteres and to find novel applications for the inhibitors of these widespread enzymes.
Conclusion
Compounds NK-154 and NK-168 used in this study affect the activity of CA isozymes due to the presence of the different functional groups (OH, phenyl, benzyl and methyl) present in their aromatic and non-aromatic scaffold. Our findings here indicate thus another class of possible CAIs of interest, in addition to the well-known sulfonamides/sulfamates/sulfamides. Indeed, some hydroxylic compounds investigated here showed effective hCA I and hCA IX inhibitory activity, in the low micromolar range, by the CO2 hydrase method which usually gives KI-s. These findings point out that substituted hydroxylic compounds may be used as leads for generating potent CAIs eventually targeting other isoforms which have not been assayed yet for their interactions with such agents.
Declaration of interest
This study was financed by TUBITAK (The Scientific and Technological Research Council of Turkey) (Project no: 114Z731) for (MS), Ege University (Scientific Research Project 2011 Fen 050) for (NK and DA) and Ondokuz Mayis University Scientific Research Projects Council (Project no: 2013/PYO.ZRT.1901.13.004) for (DE). The authors report no conflicts of interest.
References
- Weber M, Weber M, Kleine-Boymann M. Ullmann’s encyclopedia of industrial chemistry. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA; 2004
- Bayram E, Senturk M, Kufrevioglu OI, Supuran CT. In vitro inhibition of salicylic acid derivatives on human cytosolic carbonic anhydrase isozymes I and II. Bioorg Med Chem 2008;16:9101–5
- Oztürk Sarikaya SB, Topal F, Sentürk M, et al. In vitro inhibition of α-carbonic anhydrase isozymes by some phenolic compounds. Bioorg Med Chem Lett 2011;21:4259–62
- Rang HP, Dale MM, Ritter JM, Flower RJ. Rang and Dale’s pharmacology. In: Shen H, ed. Illustrated pharmacology memory cards: pharmnemonics. 6th edn. London: Churchill Livingstone Elsevier; 2008:6
- Huang MT, Newmark HL, Frenkel K. Inhibitory effects of curcumin on tumorigenesis in mice. J Cell Biochem Suppl 1997;27:26–34
- Sentürk M, Gülçin I, Beydemir S, et al. In Vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Işık S, Vullo D, Durdagi S, et al. Interaction of carbonic anhydrase isozymes I, II, and IX with some pyridine and phenol hydrazinecarbothioamide derivatives. Bioorg Med Chem Lett 2015;23:5636–41
- Ekinci D, Cavdar H, Talaz O, et al. NO-releasing esters show carbonic anhydrase inhibitory action against human isoforms I and II. Bioorg Med Chem 2010;18:3559–63
- Sentürk M, Ekinci D, Göksu S, Supuran CT. Effects of dopaminergic compounds on carbonic anhydrase isozymes I, II, and VI. J Enzyme Inhib Med Chem 2012;27:365–9
- Yerlikaya E, Erdogan O, Demirdag R, et al. Expression of hCA IX isoenzyme by using sumo fusion partner and examining the effects of antitumor drugs. Turk J Biochem 2015;40:334–42
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77
- Robertson N, Potter C, Harris AL. Role of carbonic anhydrase IX in human tumor cell growth, survival, and invasion. Cancer Res 2004;64:6160–5
- Supuran CT, Scozzafava A, Conway J. Carbonic anhydrase – its inhibitors and activators. Boca Raton, New York, London: CRC Press; 2004
- Salmas RE, Senturk M, Yurtsever M, Durdagi S. Discovering novel carbonic anhydrase type IX (CA IX) inhibitors from seven million compounds using virtual screening and in vitro analysis. J Enzyme Inhib Med Chem 2015. [Epub ahead of print]. doi: 10.3109/14756366.2015.1036049
- Ekhteiari Salmas R, Mestanoglu M, Durdagi S, et al. Kinetic and in silico studies of hydroxy-based inhibitors of carbonic anhydrase isoforms I and II. J Enzyme Inhib Med Chem 2015. [Epub ahead of print]. doi: 10.3109/14756366.2014.1003216
- Pastorek J, Pastoreková S, Callebaut I, et al. Cloning and characterization of MN, a human tumor-associated protein with a domain homologous to carbonic anhydrase and a putative helix-loop-helix DNA binding segment. Oncogene 1994;9:2877–88
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73
- Innocenti A, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: inhibition of mammalian isoforms I-XIV with a series of substituted phenols including paracetamol and salicylic acid. Bioorg Med Chem 2008;16:7424–8
- Abdel-Hamid MK, Abdel-Hafez AA, El-Koussi NA, et al. Design, synthesis, and docking studies of new 1,3,4-thiadiazole-2-thione derivatives with carbonic anhydrase inhibitory activity. Bioorg Med Chem 2007;15:6975–84
- Innocenti A, Gülçin I, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Antioxidant polyphenols effectively inhibit mammalian isoforms I-XV. Bioorg Med Chem Lett 2010;20:5050–3
- Temperini C, Scozzafava A, Supuran CT. Carbonic anhydrase activators: the first X-ray crystallographic study of an adduct of isoform I. Bioorg Med Chem Lett 2006;16:5152–6
- Ivanova J, Leitans J, Tanc M, et al. X-ray crystallography-promoted drug design of carbonic anhydrase inhibitors. Chem Commun (Camb) 2015;51:7108–11
- Alterio V, Hilvo M, Di Fiore A, et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc Natl Acad Sci USA 2009;106:16233–8
- Farid R, Day T, Friesner RA, Pearlstein RA. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg Med Chem 2006;14:3160–73
- Nair SK, Ludwig PA, Christianson DW. Two-site binding of phenol in the active site of human carbonic anhydrase II: structural implications for substrate association. J Am Chem Soc 1994;116:3659–60
- Alp C, Özsoy Ş, Alp NA, et al. Sulfapyridine-like benzenesulfonamide derivatives as inhibitors of carbonic anhydrase isoenzymes I, II and VI. J Enzyme Inhib Med Chem 2012;27:818–24
- Özdemir ZÖ, Şentürk M, Ekinci D. Inhibition of mammalian carbonic anhydrase isoforms I, II and VI with thiamine and thiamine-like molecules. J Enzyme Inhib Med Chem 2013;28:316–19
- Güney M, Çavdar H, Şentürk M, Ekinci D. Synthesis and carbonic anhydrase inhibitory properties of novel uracil derivatives. Bioorg Med Chem Lett 2015;25:3261–3
- Balaydin HT, Durdagi S, Ekinci D, et al. Inhibition of human carbonic anhydrase isozymes I, II and VI with a series of bisphenol, methoxy and bromophenol compounds. J Enzyme Inhib Med Chem 2012;27:467–75
- Sentürk M, Talaz O, Ekinci D, et al. In vitro inhibition of human erythrocyte glutathione reductase by some new organic nitrates. Bioorg Med Chem Lett 2009;19:3661–3
- Talaz O, Cavdar H, Durdagi S, et al. Synthesis of 1,4-bis(indolin-1-ylmethyl)benzene derivatives and their structure-activity relationships for the interaction of human carbonic anhydrase isoforms I and II. Bioorg Med Chem 2013;21:1477–82
- Ekinci D, al-Rashida M, Abbas G, et al. Chromone containing sulfonamides as potent carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:744–7
- Ekinci D, Cavdar H, Durdagi S, et al. Structure-activity relationships for the interaction of 5,10-dihydroindeno[1,2-b]indole derivatives with human and bovine carbonic anhydrase isoforms I, II, III, IV and VI. Eur J Med Chem 2012;49:68–73
- Fidan I, Salmas RE, Arslan M, et al. Carbonic anhydrase inhibitors: design, synthesis, kinetic, docking and molecular dynamics analysis of novel glycine and phenylalanine sulphonamide derivatives. Bioorg Med Chem 2015. [Epub ahead of print]. doi: 10.1016/j.bmc.2015.10.009
- Gulcin I, Beydemir S, Coban A, Ekinci D. The inhibitory effect of dantrolene sodium and propofol on 6-phosphogluconate dehydrogenase from rat erythrocyte. Fresen Environ Bull 2008;17:1283–7
- Cakmak R, Durdagi S, Ekinci D, et al. Design, synthesis and biological evaluation of novel nitroaromatic compounds as potent glutathione reductase inhibitors. Bioorg Med Chem Lett 2011;21:5398–402
- Innocenti A, Maresca A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: thioxolone versus sulfonamides for obtaining isozyme-selective inhibitors? Bioorg Med Chem Lett 2008;18:3938–41
- Korkmaz N, Obaidi OA, Senturk M, et al. Synthesis and biological activity of novel thiourea derivatives as carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:75–80