
Abstract
Novel 1,5-diphenyl-6-substituted-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-ones were synthesized and characterized. All compounds were screened for their anti-proliferative activities in five different cancer cell lines. The results showed that compounds 7a and 7b comprising aminoguanidino or guanidino moiety at position 6 inhibited proliferation of RKO colon cancer cells with IC50 of 8 and 4 μM, respectively. Compounds 7a and 7b induced apoptosis in RKO cells, which was confirmed by TUNEL and annexin V-FITC assays. Flow cytometric analysis indicated that compounds 7a and 7b arrested RKO cells in the G1 phase and the most active compound 7b increased levels of p53, p21, Bax, ERK1/2 and reduced levels of Bcl2 and Akt. Compound 7b also activates release of cytochrome c, which is consistent with activation of caspase-9. Additionally, compound 7b increased caspase-3 activity and cleaved PARP-1 in RKO cells. Collectively, these findings could establish a molecular basis for the development of new anti-cancer agents.
Introduction
Colorectal cancer (CRC) is the fourth most common form of cancer and the second leading cause for cancer-related deathsCitation1. The prevalence of CRC is still increasing despite enhanced understanding of the pathogenesis of this disease and establishment of improved screening strategies for this malignancy. Current data suggest that epidemiological risk factors for colon cancer, other than genetic risk factors, include smoking, physical inactivity, obesity and food with high fat and carbohydrates that could promote tumor growthCitation2. Although several drugs have been developed for the management of cancer, the high systemic toxicity and drug resistance remain significant obstacles to their success in many cancersCitation3. Thus, the discovery of new therapeutic targets for cancer and development of novel, more effective therapeutic agents remain major challenges in the field of cancer chemotherapy.
Programmed cell death (PCD), or apoptosis, is an essential physiological process that leads to cellular self-destruction and plays essential roles in development and maintenance of tissue homeostasis. Deregulation of apoptosis is implicated in a variety of pathological conditions such as cancer, neurodegenerative disorders and autoimmune diseasesCitation4. Therefore, inhibition of cell cycle progression and induction of apoptotic cancer cell death represents a key strategy for the prevention and treatment of cancer. Apoptosis is triggered by two major apoptosis-initiating pathways: the extrinsic and intrinsic pathways. The extrinsic pathway is regulated by a sub-group of tumor necrosis factor receptors (TNFR) superfamily that includes TNFR, FAS and TRAIL. The intrinsic pathway is initiated from within the cell and depends on the balance of the pro- and anti-apoptotic members of Bcl-2 family proteinsCitation5–7. The most widely studied form of intrinsic apoptosis is initiated by the stress-mediated release of cytochrome c from the mitochondria that results in the release of apoptogenic proteins into the cytosol. This is followed by activation of caspase-9 and caspase-3 and apoptosis can be executed through protease activity targeting substrate proteinsCitation8.
The pyrazolo[3,4-d]pyrimidine nucleus is considered as an isostere to the purine nucleus and hence exhibits promising anti-proliferative activities. Several members of this family were found to induce apoptosis and/or reduce cell proliferation in many cancer cell lines such as breast, colon, lung and liver cell linesCitation9–17. Different mechanisms account for the cytotoxic effect of this class of compounds, where they have been reported to act as inhibitors of different enzymes such as CDK 2Citation14, epidermal growth factorCitation15, Src or dual Src/AbI inhibitorsCitation16–19, mammalian target of rapamycin inhibitorsCitation20 or modulators of oxygen stress in cancer cellsCitation9. Over the past two decades, considerable attention has been devoted to pyrazolo[3,4-d]pyrimidin-4-one as a prime target for the synthesis of anti-tumor agents. In this context, several pyrazolo[3,4-d]pyrimidin-4-ones were reported to possess anti-proliferative activity against human colon cancer cell line HCT116 and other cell lines (e.g. compounds A–D; )Citation12,Citation21,Citation22.
Figure 1. Chemical structure of previously reported pyrazolo[3,4-d]pyrimidines endowed with anti-cancer and apoptosis inducing activities (A–D) and the synthesized compounds (E).
![Figure 1. Chemical structure of previously reported pyrazolo[3,4-d]pyrimidines endowed with anti-cancer and apoptosis inducing activities (A–D) and the synthesized compounds (E).](/cms/asset/a2ab83b3-d4ab-4630-9c0e-be98963c0602/ienz_a_1118686_f0001_c.jpg)
Encouraged by the above-mentioned findings and in continuation of our efforts linked with discovering and exploring novel lead structures as potent chemotherapeutic agentsCitation23–26, it was attempted to synthesize new pyrazolo[3,4-d]pyrimidin-4-ones comprising a variety of substituents at position 6 (E; ) to be evaluated for their anti-cancer activity. The substituents at position 6 include various pharmacophores and functionalities that are believed to be responsible for the biological significance of some relevant anti-cancer agents such as alkylaminoCitation22,Citation27 together with other pharmacophoric groups that would confer different electronic and lipophilic environment, which would influence the targeted anti-cancer activity. Since many guanidines were reported to possess cytotoxic and apoptosis inducing activitiesCitation28–30, we deemed it interesting to explore the effect of introducing guanidino and aminoguanidino moieties at position 6. Moreover, due to the reported anti-tumor and apoptosis inducing activities of some hydrazonesCitation12,31–35 it was considered worthwhile to extend substitution at position 6 to include the structural elements of hydrazones. These combinations were suggested in an attempt to investigate the possible synergistic influence of such structure hybridizations on the anticipated biological activity, hoping to discover and develop apoptosis inducers as potential new anti-cancer agents.
Methods and materials
Chemistry
All reagents and solvents were purchased from commercial suppliers and were dried and purified when necessary by standard techniques. Melting points were determined in open glass capillaries using Stuart capillary melting point apparatus (Stuart Scientific Stone, Staffordshire, UK) and were uncorrected. Infrared (IR) spectra were recorded on Perkin Elmer 1430 infrared spectrophotometer (Perkin Elmer, Beaconsfield, UK) and measured by υ cm−1 scale using KBr cell. 1H NMR spectra were scanned on Varian Mercury VX-300 MHz (San Diego, CA) or Brüker avance 400 MHz (Bruker, UK), using tetramethylsilane as an internal standard and DMSO-d6 as the solvent (chemical shifts are given in δ ppm). Splitting patterns were designated as follows: s: singlet; br s: broad singlet; d: doublet; t: triplet; q: quartet; m: multiplet. 13C NMR spectra were recorded on a Varian Mercury VX-300 MHz or Brüker 400 MHz spectrometer in DMSO-d6 and measured in δ scale. Mass spectra were run on a gas chromatograph/mass spectrometer Shimadzu GCMS-QP 2010 Plus (70 eV) (Kyoto, Japan). Microanalyses were performed at the regional Center for Mycology and Biotechnology, Alazhar University and the found values were within ±0.4% of the theoretical values. Follow-up of the reactions and checking the purity of the compounds was performed by thin-layer chromatography on silica gel-precoated aluminum sheets (Type 60 GF254; Merck, Germany) and the spots were detected by exposure to UV lamp at λ 254 nm for few seconds.
Ethyl 5-amino-1-phenyl-1H-pyrazole-4-carboxylate (1)36 and 1,5-diphenyl-6-thioxo-6,7-dihydro-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (2)37 were prepared according to previously published reaction conditions.
6-(Methylsulfanyl)-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (3)
To a stirred solution of the thione 2 (6.4 g, 20 mmol) in dry dimethyl formamide (20 ml) containing anhydrous potassium carbonate (2.8 g, 20 mmol), methyl iodide (3.4 g, 1.5 ml, 24 mmol) was slowly added. The reaction mixture was left stirred at room temperature for 6 h then poured onto crushed ice. The separated solid product was filtered, washed with water, dried and crystallized from ethanol. Yield: 5.21 g, 78%; m.p.: 170–172 °C. IR (KBr, cm−1): 3055, 2930 (CH), 1706 (C = O), 1594 (C = N), 1529, 1503, 1477 (C = C), 1276, 1064 (C–S–C). 1H NMR (300 MHz, DMSO-d6) δ 2.45 (s, 3H, SCH3), 7.32–7.40 (m, 3H, phenyl C3,4,5–H), 7.50–7.60 (m, 5H, phenyl-H), 8.1 (d, J = 7.8 Hz, 2H, phenyl C2,6–H), 8.28 (s, 1H, pyrazolopyrimidine C3–H). 13C NMR (DMSO-d6) δ 16.05 (CH3), 104.58 (pyrazolopyrimidine C3a), 121.76, 127.31, 129.72, 129.92, 130.04, 130.41, 136.26, 136.98 (phenyl-C), 138.76 (pyrazolopyrimidine C3), 150.76 (pyrazolopyrimidine C7a), 157.29 (C = O), 164.34 (pyrazolopyrimidine C6). Anal. Calcd. for C18H14N4OS (334.39): C, 64.65; H, 4.22; N, 16.75. Found: C, 64.48; H, 4.36; N, 16.59.
6-(Methylsulfonyl)-1,5-diphenyl-1H-pyrazolo[3,4-d] pyrimidin-4(5H)-one (4)
To an ice-cold well-stirred solution of the methylsulfanyl derivative 3 (6.7 g, 20 mmol) in dimethyl formamide (50 ml), potassium hydrogenpersulfate (36.9 g, 60 mmol) dissolved in water (140 ml) was slowly added over a period of 2 h. The reaction mixture was further stirred at room temperature for 16 h then filtered to remove inorganic material. The filterate was poured into ice-cold water and the obtained solid product was filtered, dried and crystallized from dioxane. Yield: 5.43 g, 74%; m.p.: 215–217 °C. IR (KBr, cm−1): 3062, 2962 (CH), 1715 (C = O), 1594 (C = N), 1563, 1527, 1504, 1488, 1458 (C = C), 1346, 1162 (SO2), 1291, 1093 (C–S–C). 1H NMR (400 MHz, DMSO-d6) δ 2.76 (s, 3H, SO2CH3), 7.40–7.73 (m, 8H, phenyl-H), 8.19 (d, J = 7.8 Hz, 2H, phenyl C2,6–H), 8.53 (s, 1H, pyrazolo pyrimidine C3–H). 13C NMR (DMSO-d6) δ 31.12 (CH3), 103.74 (pyrazolopyrimidine C3a), 121.44, 127.21, 129.09, 129.48, 129.75, 130.42, 135.51, 137.11 (phenyl-C), 138.86 (pyrazolopyrimidine C3), 151.11 (pyrazolopyrimidine C7a), 156.51 (C = O), 157.66 (pyrazolopyrimidine C6). MS (m/z, %): 366 (M+, 5.1), 287 (100). Anal. Calcd. for C18H14N4O3S (366.39): C, 59.01; H, 3.85; N, 15.29. Found: C, 58.88; H, 4.16; N, 15.39.
General procedure for the synthesis of 1,5-diphenyl-6-(substituted amino)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-ones (5a–i)
A mixture of 4 (0.73 g, 2 mmol) and the selected substituted amine (2 mmol) in dry dioxane (10 ml) was heated under reflux for 6–8 h. The reaction mixture was concentrated under reduced pressure and the remaining residue was triturated with diethyl ether whereupon a white solid was separated. It was filtered, washed with diethyl ether and crystallized from the proper solvent.
6-(Cyclohexylamino)-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (5a). Yield: 0.57 g, 74%; m.p.: 180–182 °C (methylene chloride/petroleum ether 60/80) (2:1). IR (KBr, cm−1): 3332 (NH), 3064, 2930, 2849 (CH), 1700 (C = O), 1597 (C = N), 1556, 1492, 1454 (C = C). 1H NMR (400 MHz, DMSO-d6) δ 1.06–1.30 (m, 6H, cyclohexyl C3,4,5–H2), 1.53–1.82 (m, 4H, cyclohexyl C2,6–H2), 3.83–3.85 (m, 1H, cyclohexyl C1–H), 5.43 (d, J = 7.2 Hz, 1H, NH, D2O exchangeable), 7.17–7.63 (m, 8H, phenyl-H), 8.10 (s, 1H, pyrazolopyrimidine C3–H), 8.20 (d, J = 7.6 Hz, 2H, phenyl C2,6-H). 13C NMR (DMSO-d6) δ 25.10 (cyclohexyl C3,5), 25.54 (cyclohexyl C4), 31.66 (cyclohexyl C2,6), 51.83 (cyclohexyl C1), 101.13 (pyrazolopyrimidine C3a), 120.62, 124.88, 127.36, 128.73, 129.43, 130.67, 134.85, 136.94 (phenyl-C), 139.43 (pyrazolopyrimidine C3), 153.07 (pyrazolopyrimidine C7a), 153.19 (C = O), 157.96 (pyrazolopyrimidine C6). MS (m/z, %): 385 (M+, 55), 288 (100). Anal. Calcd. for C23H23N5O (385.46): C, 71.67; H, 6.01; N, 18.17. Found: C, 71.83; H, 6.09; N, 18.35.
6-(Benzylamino)-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (5b). Yield: 0.58 g, 74%; m.p.: 201–203 °C (dioxane/water 4:1). IR (KBr, cm−1): 3280 (NH), 3107, 2925 (CH), 1700 (C = O), 1600 (C = N), 1551, 1492, 1458 (C = C). 1H NMR (300 MHz, DMSO-d6) δ 4.45 (d, J = 6.9 Hz, 2H, CH2), 7.01–7.64 (m, 13H, aromatic-H and NH), 7.93 (d, J = 7.2 Hz, 2H, phenyl C2,6–H), 8.08 (s, 1H, pyrazolopyrimidine C3–H). 13C NMR (DMSO-d6) δ 45.71 (CH2), 101.29 (pyrazolopyrimidine C3a), 120.75, 126.32, 127.18, 127.44, 128.36, 128.92, 129.37, 129.65, 130.71, 136.95, 139.31, 139.77 (phenyl and benzyl-C), 140.07 (pyrazolopyrimidine C3), 153.10 (pyrazolopyrimidine C7a), 153.91 (C = O), 157.93 (pyrazolopyrimidine C6). MS (m/z, %): 393 (M+, 56.9), 314 (100). Anal. Calcd. for C24H19N5O (393.44): C, 73.27; H, 4.87; N, 17.80. Found: C, 73.51; H, 4.92; N, 18.02.
6-(2-(3,4-dimethoxyphenyl)ethylamino)-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (5c). Yield: 0.5 g, 54%; m.p.: 84–86 °C (methylene chloride/petroleum ether 60/80) (2:1). IR (KBr, cm−1): 3384 (NH), 3061, 2997, 2932, 2834 (CH), 1703 (C = O), 1597 (C = N), 1554, 1498, 1458 (C = C), 1260, 1236, 1063, 1026 (C–O–C). 1H NMR (400 MHz, DMSO-d6) δ 2.78 (t, J = 6.8 Hz, 2H, C-CH2), 3.48 (t, J = 6.8 Hz, 2H, N-CH2), 3.70, 3.73 (2s, each 3H, 2OCH3), 6.22 (t, J = 6.8 Hz, 1H, NH, D2O exchangeable), 6.68 (d, J = 8.1 Hz, 1H, dimethoxyphenyl C6-H), 6.72 (s, 1H, dimethoxy phenyl C2-H), 6.87 (d, J = 8.1 Hz, 1H, dimethoxyphenyl C5-H), 7.27–7.61 (m, 8H, phenyl-H), 8.14 (s, 1H, pyrazolopyrimidine C3-H), 8.24 (d, J = 8.0 Hz, 2H, phenyl C2,6-H). 13C NMR (DMSO-d6) δ 34.66 (C–CH2), 44.05 (NCH2), 55.91, 56.06 (OCH3), 101.23 (pyrazolopyrimidine C3a), 112.57, 113.05, 120.25, 120.86, 126.38, 128.76, 129.43, 129.84, 130.56, 132.10, 135.03, 136.98, 139.59, 147.89, 149.22 (phenyl-C, dimethoxyphenyl-C and pyrazolopyrimidine C3), 153.39 (pyrazolopyrimidine C7a), 153.87 (C = O), 157.93 (pyrazolopyrimidine C6). MS (m/z, %): 467 (M+, 51), 151 (100). Anal. Calcd. for C27H25N5O3 (467.52): C, 69.36; H, 5.39; N, 14.98. Found: C, 69.72; H, 5.41; N, 15.16.
1,5-Diphenyl-6-(propylamino)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (5d). Yield: 0.44 g, 64%; m.p.: 144–146 °C (methylene chloride/petroleum ether 60/80) (2:1). IR (KBr, cm − Citation1): 3280 (NH), 3062, 2925 (CH), 1700 (C = O), 1594 (C = N), 1551, 1504, 1492, 1458 (C = C). 1H NMR (400 MHz, DMSO-d6) δ 0.84 (t, J = 6.9 Hz, 3H, CH3), 1.53–1.62 (m, 2H, C-CH2), 3.26 (q, J = 6.9 Hz, 2H, N–CH2), 6.29 (t, J = 6.9 Hz, 1H, NH, D2O exchangeable), 7.30–7.62 (m, 8H, phenyl-H), 8.15 (s, 1H, pyrazolopyrimidine C3-H), 8.22 (d, J = 8.0 Hz, 2H, phenyl C2,6-H). 13C NMR (DMSO-d6) δ 11.63 (CH3), 21.97 (CH2), 43.86 (N–CH2), 100.96 (pyrazolopyrimidine C3a), 120.66, 126.65, 129.46, 129.81, 130.09, 130.67, 134.95, 136.92 (phenyl-C), 139.41 (pyrazolopyrimidine C3), 154.01 (pyrazolopyrimidine C7a), 157.46 (C = O), 158.16 (pyrazolopyrimidine C6). MS (m/z, %): 345 (M+, 52.8), 317 (100). Anal. Calcd. for C20H19N5O (345.4): C, 69.55; H, 5.54; N, 20.28. Found: C, 69.48; H, 5.76; N, 20.39.
6-(2-Morpholinoethylamino)-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (5e). Yield: 0.43 g, 52%; m.p.: 187–189 °C (ethanol). IR (KBr, cm−1): 3288 (NH), 3063, 2946, 2902, 2845 (CH), 1702 (C = O), 1596 (C = N), 1553, 1474 (C = C). 1H NMR (400 MHz, DMSO-d6) δ 2.24–2.30 (m, 4H, morpholine C3,5-H2), 2.43 (t, J = 6.6 Hz, 2H, NCH2), 3.38–3.47 (m, 6H, morpholine C2,6–H2 and NCH2), 5.97 (s, 1H, NH, D2O exchangeable), 7.31–7.71 (m, 8H, phenyl-H), 8.12 (s, 1H, pyrazolopyrimidine C3-H), 8.20 (d, J = 8.0 Hz, 2H, phenyl C2,6-H). 13C NMR (DMSO-d6) δ 38.65 (NCH2), 53.22 (morpholine C3,5), 55.90 (NCH2), 66.61 (morpholine C2,6), 101.31 (pyrazolopyrimidine C3a), 120.88, 126.43, 129.47, 129.81, 129.92, 130.71, 135.20, 137.00 (phenyl-C), 139.51 (pyrazolopyrimidine C3), 153.33 (pyrazolopyrimidine C7a), 153.86 (C = O), 157.80 (pyrazolopyrimidine C6). MS (m/z, %): 416 (M+, 0.1), 100 (100). Anal. Calcd. for C23H24N6O2 (416.48): C, 66.33; H, 5.81; N, 20.18. Found: C, 66.59; H, 5.86; N, 20.43.
6-(2-(Pyrrolidin-1-yl)ethylamino)-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (5f). Yield: 0.41 g, 52%; m.p.: 106–107 °C (methylene chloride/petroleum ether 60/80) (2:1). IR (KBr, cm−1): 3318 (NH), 3062, 2950, 2924, 2850 (CH), 1721 (C = O), 1597 (C = N), 1557, 1493 (C = C). 1H NMR (400 MHz, DMSO-d6) δ 1.56–1.60 (m, 4H, pyrrolidine C3,4-H2), 2.36–2.40 (m, 4H, pyrrolidine C2,5–H2), 2.57 (t, J = 6.8 Hz, 2H, NCH2), 3.39 (m, 2H, NCH2), 6.13 (t, J = 6.8 Hz, 1H, NH, D2O exchangeable), 7.32–7.60 (m, 8H, phenyl-H), 8.15 (d, J = 7.4 Hz, 2H, phenyl C2,6-H), 8.32 (s, 1H, pyrazolopyrimidine C3-H). 13C NMR (DMSO-d6) δ 22.56 (pyrrolidine C3,4), 34.63, 51.03 (NCH2), 53.64 (pyrrolidine C2,5), 104.48 (pyrazolopyrimidine C3a), 121.78, 128.50, 129.15, 129.71, 129.89, 130.46, 136.14, 136.90 (phenyl-C), 138.66 (pyrazolopyrimidine C3), 150.71 (pyrazolopyrimidine C7a), 157.37 (C = O), 164.32 (pyrazolopyrimidine C6). MS (m/z, %): 400 (M+, 48), 85 (100). Anal. Calcd. for C23H24N6O (400.48): C, 68.98; H, 6.04; N, 20.99. Found: C, 69.07; H, 6.11; N, 21.24.
6-(2-Hydroxyethylamino)-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (5g). Yield: 0.44 g, 64%; m.p.: 172–173 °C (dioxane/diethyl ether) (1:1). IR (KBr, cm−1): 3436 (OH), 3291 (NH), 3098, 3044, 2935, 2868 (CH), 1687 (C = O), 1596 (C = N), 1554, 1495 (C = C). 1H NMR (400 MHz, DMSO-d6) δ 3.40 (t, J = 6.9 Hz, 2H, NCH2), 3.43 (t, J = 6.9 Hz, 2H, OCH2), 4.70 (s, 1H, OH, D2O exchangeable), 6.01 (s, 1H, NH, D2O exchangeable), 7.14–7.61 (m, 8H, phenyl-H), 8.01 (s, 1H, pyrazolopyrimidine C3-H), 8.21 (d, J = 7.4 Hz, 2H, phenyl C2,6–H). 13C NMR (DMSO-d6) δ 44.40 (NCH2), 59.10 (OCH2), 101.14 (pyrazolopyrimidine C3a), 120.53, 126.48, 129.71, 130.01, 130.48, 130.70, 134.84, 136.92 (phenyl-C), 139.34 (pyrazolopyrimidine C3), 153.22 (pyrazolopyrimidine C7a), 153.98 (C = O), 158.12 (pyrazolopyrimidine C6). MS (m/z, %): 347 (M+, 36.67), 302 (100). Anal. Calcd. for C19H17N5O2 (347.37): C, 65.69; H, 4.93; N, 20.16. Found: C, 65.87; H, 5.01; N, 20.43.
6-(1-Hydroxy-2-methylpropan-2-ylamino)-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (5h). Yield: 0.46 g, 62%; m.p.: 286–287 °C (ethanol). IR (KBr, cm−1): 3405 (OH), 3329 (NH), 3066, 3037, 2965, 2862 (CH), 1667 (C = O), 1604 (C = N), 1556, 1494, 1456 (C = C). 1H NMR (400 MHz, DMSO-d6) δ 1.60 (s, 6H, 2CH3), 3.60 (s, 2H, CH2), 4.50 (s, 1H, OH, D2O exchangeable), 6.13 (s, 1H, NH, D2O exchangeable), 7.13–7.50 (m, 8H, phenyl-H), 8.28 (d, J = 7.2 Hz, 2H, phenyl C2,6-H), 7.95 (s, 1H, pyrazolopyrimidine C3-H). 13C NMR (DMSO-d6) δ 29.12 (CH3), 50.37 (C(CH3)2), 66.80 (OCH2), 99.71 (pyrazolopyrimidine C3a), 120.34, 124.98, 126.84, 128.72, 129.11, 130.10, 136.73, 139.87 (phenyl-C), 140.60 (pyrazolopyrimidine C3), 157.71 (pyrazolopyrimidine C7a), 158.68 (C = O), 160.95 (pyrazolopyrimidine C6). MS (m/z, %): 375 (M+, 31.8), 69 (100). Anal. Calcd. for C21H21N5O2 (375.42): C, 67.18; H, 5.64; N, 18.65. Found: C, 67.35; H, 5.73; N, 18.91.
6-(1,3-Dihydroxy-2-(hydroxymethyl)propan-2-ylamino)-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (5i). Yield: 0.34 g, 42%; m.p.: >300 °C (ethanol). IR (KBr, cm−1): 3280 (NH), 3062, 2925 (CH), 1700 (C = O), 1594 (C = N), 1551, 1504, 1492, 1458 (C = C). 1H NMR (400 MHz, DMSO-d6) δ 3.50 (s, 6H, 3CH2), 4.40 (s, 1H, OH, D2O exchangeable), 6.3 (s, 1H, NH, D2O exchangeable), 7.30–7.78 (m, 8H, phenyl-H), 8.08–8.22 (m, 2H, phenyl C2,6–H), 8.4 (s, 1H, pyrazolopyrimidine C3–H). 13C NMR (DMSO-d6) δ 60.12 (C(CH2OH)3), 65.98 (OCH2), 103.73 (pyrazolopyrimidine C3a), 121.42, 127.13, 129.07, 129.46, 129.72, 129.93, 135.51, 137.08 (phenyl-C), 138.36 (pyrazolopyrimidine C3), 151.09 (pyrazolopyrimidine C7a), 156.46 (C = O), 157.64 (pyrazolopyrimidine C6). MS (m/z, %): 407 (M+, 96.8), 107 (100). Anal. Calcd. for C21H21N5O4 (407.42): C, 61.91; H, 5.20; N, 17.19. Found: C, 61.68; H, 5.46; N, 17.39.
6-(2-Aminoethylamino)-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (5j). Yield: 0.36 g, 52%; m.p.: 199–201 °C (methylene chloride/petroleum ether 60/80) (2:1). IR (KBr, cm−1): 3357, 3302 (NH), 3063, 2957, 2917, 2858 (CH), 1694 (C = O), 1596 (C = N), 1557, 1495 (C = C). 1H NMR (400 MHz, DMSO-d6) δ 2.72 (t, J = 6.2 Hz, 2H, NCH2), 3.32 (t, J = 6.2 Hz, 2H, NCH2), 6.30 (br s, 2H, NH2, D2O exchangeable), 6.40 (br s, 1H, NH, D2O exchangeable), 7.31–7.62 (m, 8H, aromatic-H), 8.15 (s, 1H, pyrazolopyrimidine C3–H), 8.20 (d, J = 8.4 Hz, 2H, phenyl C2,6–H). 13C NMR (DMSO-d6) δ 44.58, 45.96, (NCH2), 101.15 (pyrazolopyrimidine C3a), 120.79, 126.43, 129.45, 129.61, 129.93, 130.65, 135.0, 136.95 (phenyl-C), 139.40 (pyrazolopyrimidine C3), 154.08 (pyrazolopyrimidine C7a), 158.09 (C = O), 162.06 (pyrazolopyrimidine C6). MS (m/z, %): 346 (M+, 5.75), 304 (100). Anal. Calcd. for C19H18N6O (346.39): C, 65.88; H, 5.24; N, 24.26. Found: C, 66.02; H, 5.27; N, 24.53.
6-Ethoxy-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (6). Yield: 0.54 g, 82%; m.p.: 143–144 °C (ethanol). IR (KBr, cm−1): 3052, 2979, 2930 (CH), 1714 (C = O), 1571 (C = N), 1550, 1531, 1493, 1421 (C = C), 1265, 1056 (C–O–C). 1H NMR (400 MHz, DMSO-d6) δ 1.17 (t, J = 6.9 Hz, 3H, CH3), 4.39 (q, J = 6.9 Hz, 2H, CH2), 7.30–7.70 (m, 8H, phenyl-H), 8.10 (d, J = 7.8 Hz, 2H, phenyl C2,6–H), 8.35 (s, 1H, pyrazolopyrimidine C3–H). 13C NMR (DMSO-d6) δ 14.22 (CH3), 66.0 (CH2), 103.75 (pyrazolopyrimidine C3a), 121.58, 127.21, 129.05, 129.43, 129.93, 130.04, 135.51, 137.12 (phenyl-C), 138.36 (pyrazolopyrimidine C3), 151.11 (pyrazolopyrimidine C7a), 156.50 (C = O), 157.66 (pyrazolopyrimidine C6). MS (m/z, %): 332 (M+, 87), 287 (100). Anal. Calcd. for C19H16N4O2 (332.36): C, 68.66; H, 4.85; N, 16.86. Found: C, 68.38; H, 5.06; N, 17.09.
General procedure for the synthesis of compounds 7a, b
Method A. A mixture of 4 (0.36 g, 1.0 mmol), aminoguanidine or guanidine HCl (1.2 mmol) and anhydrous sodium acetate (0.1 g, 1.2 mmol) in dry dioxane/dimethyl sulfoxide (5:1) (25 ml) was heated under reflux for 8 h. The reaction mixture was then concentrated under vacuum then poured into cold water. The separated solid was filtered, dried and crystallized from ethanol.
Method B. A mixture of 4 (0.36 g, 1.0 mmol), aminoguanidine or guanidine bicarbonate (1.2 mmol) in dry dioxane/dimethyl sulfoxide (5:1) (15 ml) was heated under reflux for 8 h. The reaction mixture was then concentrated under vacuum then poured into cold water. The separated solid was filtered, dried and crystallized from ethanol.
(4-Oxo-1,5-diphenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-ylamino)guanidine (7a). Yield: 0.17 g, 48%, method A; 0.25 g, 70% method B, m.p.: 265–266 °C (ethanol). IR (KBr, cm−1): 3280 (NH), 3062, 2925 (CH), 1700 (C = O), 1594 (C = N), 1551, 1504, 1492, 1458 (C = C). 1H NMR (400 MHz, DMSO-d6) δ 4.46 (s, 2H, NH2, D2O exchangeable), 7.14–7.58 (m, 11H, 8 phenyl-H and 3NH), 7.93 (d, J = 7.6 Hz, 2H, phenyl C2,6–H), 8.07 (s, 1H, pyrazolopyrimidine C3–H). 13C NMR (DMSO-d6) δ 101.92 (pyrazolopyrimidine C3a), 121.80, 126.82, 127.04, 128.93, 129.33, 129.81, 129.90, 135.87 (phenyl-C), 138.80 (pyrazolopyrimidine C3), 152.49 (pyrazolopyrimidine C7a), 158.31 (C = O), 159.04 (pyrazolopyrimidine C6), 160.90 (C = NH). MS (m/z, %): 360 (M+, 56), 288 (100). Anal. Calcd. for C18H16N8O (360.37): C, 59.99; H, 4.48; N, 31.09. Found: C, 59.68; H, 4.46; N, 31.19.
(4-Oxo-1,5-diphenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)guanidine (7b). Yield: 0.17 g, 50%, method A; 0.25 g, 73% method B, m.p.: 275–277 °C. IR (KBr, cm−1): 3401, 3350, 3190 (NH), 3057, 2893, 2836 (CH), 1676 (C = O), 1611 (C = N), 1574, 1493 (C = C). 1H NMR (400 MHz, DMSO-d6) δ 7.08–7.76 (m, 12H, 8 phenyl-H and 4NH), 7.94 (d, J = 7.6 Hz, 2H, phenyl C2,6-H), 8.10 (s, 1H, pyrazolopyrimidine C3–H). 13C NMR (DMSO-d6) δ 102.15 (pyrazolopyrimidine C3a), 121.97, 127.52, 128.03, 128.78, 128.93, 129.88, 129.94, 136.93 (phenyl-C), 139.21 (pyrazolopyrimidine C3), 152.33 (pyrazolopyrimidine C7a), 157.76 (C = O), 158.0 (pyrazolopyrimidine C6), 159.92 (C = NH). MS (m/z, %): 345 (M+, 100). Anal. Calcd. for C18H15N7O (345.36): C, 62.60; H, 4.38; N, 28.39. Found: C, 62.87; H, 4.33; N, 28.62.
6-Hydrazino-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (8)
A mixture of 4 (7.3 g, 20 mmol) and hydrazine hydrate 98% (2.0 g, 1.94 ml, 40 mmol) in ethanol (40 ml) was stirred at room temperature for 6 h. The separated white solid was filtered, washed with ethanol, dried and crystallized from dioxane. Yield: 4.95 g, 78% (reported 38%); m.p.: 268–270 °C (as reported)Citation38.
General procedure for the synthesis of N′-(4-oxo-1,5-diphenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)substituted sulfohydrazides 9a, b and N′-(4-oxo-1,5-diphenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)substituted hydrazides (10a, b)
To a suspension of 8 (0.7 g, 2.2 mmol) in dry pyridine (7 ml), the appropriate aryl sulfonyl chloride or aroyl chloride (2.6 mmol) was slowly added at ice-cold temperature. The reaction mixture was stirred at room temperature over night then poured into ice-cold water. The obtained precipitate was filtered, washed with water, dried and crystallized from the proper solvent.
N′-(4-oxo-1,5-diphenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)benzenesulfohydrazide (9a). Yield: 0.54 g, 54%; m.p.: 216–218 °C (ethanol). IR (KBr, cm−1): 3380, 3244 (NH), 3099, 3052, 2917 (CH), 1704 (C = O), 1566 (C = N), 1525, 1495, 1459, 1426 (C = C), 1341, 1167 (SO2). 1H NMR (400 MHz, DMSO-d6) δ 7.30–7.95 (m, 13H, phenyl-H), 8.10 (d, J = 7.6 Hz, 2H, phenyl C2,6–H), 8.45 (s, 1H, pyrazolopyrimidine C3–H), 8.90, 9.98 (2s, each 1H, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ 101.79 (pyrazolopyrimidine C3a), 121.37, 121.61, 121.70, 122.53, 127.04, 129.36, 129.50, 129.93, 132.30, 134.05, 138.93, 139.10 (phenyl-C), 139.19 (pyrazolopyrimidine C3), 152.69 (pyrazolopyrimidine C7a), 153.33 (C = O), 157.65 (pyrazolopyrimidine C6). MS (m/z, %): 458 (M+, 94), 289 (100). Anal. Calcd. for C23H18N6O3S (458.49): C, 60.25; H, 3.96; N, 18.33. Found: C, 60.41; H, 4.02; N, 18.47.
N′-(4-oxo-1,5-diphenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)-4-methoxy benzenesulfohydrazide (9b). Yield: 0.56 g, 54%; m.p.: 238–240 °C (ethanol). IR (KBr, cm − Citation1): 3271, 3158 (NH), 3061, 2951 (CH), 1707 (C = O), 1594 (C = N), 1556, 1493, 1458, 1416 (C = C), 1342, 1162 (SO2), 1262, 1024 (C–O–C). 1H NMR (400 MHz, DMSO-d6) δ 3.75 (s, 3H, CH3), 7.05–7.93 (m, 12H, phenyl-H and methoxyphenyl C3,5–H), 8.0 (d, J = 7.6 Hz, 2H, methoxyphenyl C2,6–H), 8.07 (s, 1H, pyrazolopyrimidine C3-H), 8.80, 9.84 (2s, each 1H, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ 56.50 (OCH3), 101.80 (pyrazolopyrimidine C3a), 114.63 (methoxyphenyl C3,5), 121.66, 126.76, 128.02, 128.34, 129.75, 129.92, 130.43, 132.45, 134.05, 136.40, 143.34, 151.48, 154.12 (phenyl-C, methoxyphenyl C1,2,4,6 and pyrazolopyrimidine C3,7a), 155.24 (C = O), 157.44 (pyrazolopyrimidine C6). MS (m/z, %): 488 (M+, 34.19), 185 (100). Anal. Calcd. for C24H20N6O4S (488.52): C,59.01; H, 4.13; N, 17.20. Found: C, 59.19; H, 4.31; N, 17.02..
N′-(4-Oxo-1,5-diphenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)benzohydrazide (10a). Yield: 0.7 g, 75%; m.p.: 228–230 °C (dioxane). IR (KBr, cm − Citation1): 3374, 3246 (NH), 3098, 3047, 2916 (CH), 1680, 1702 (C = O), 1565 (C = N), 1528, 1490, 1465, 1429 (C = C). 1H NMR (400 MHz, DMSO-d6) δ 7.03–7.74 (m, 13H, phenyl-H), 7.94 (d, J = 7.6 Hz, 2H, phenyl C2,6–H), 8.42 (s, 1H, pyrazolopyrimidine C3–H), 8.91, 9.77 (2s, each 1H, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ 101.76 (pyrazolopyrimidine C3a), 121.42, 121.68, 121.69, 122.57, 127.65, 127.84, 129.90, 129.33, 132.44, 133.94, 134.11, 138.90 (phenyl-C), 139.22 (pyrazolopyrimidine C3), 152.65 (pyrazolopyrimidine C7a), 153.40 (C = O), 157.67 (pyrazolopyrimidine C6), 164.0 (C = O). MS (m/z, %): 422 (M+, 75), 289 (100). Anal. Calcd. for C24H18N6O2 (422.44): C, 68.24; H, 4.29; N, 19.89. Found: C, 68.45; H, 4.13; N, 19.55.
N′-(4-Oxo-1,5-diphenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)-4-methoxybenzohydrazide (10b). Yield: 0.7 g, 70%; m.p.: 245–247 °C (dioxane/water). IR (KBr, cm−1): 3270, 3164 (NH), 3058, 2949 (CH), 1677, 1705 (C = O), 1590 (C = N), 1554, 1490, 1450, 1415 (C = C), 1260, 1025 (C–O–C). 1H NMR (400 MHz, DMSO-d6) δ 3.70 (s, 3H, OCH3), 6.90 (d, J = 8.4 Hz, 2H, methoxyphenyl C3,5–H), 7.06–7.68 (m, 10H, phenyl-H), 7.88 (d, J = 8.4 Hz, 2H, methoxyphenyl C2,6–H), 8.10 (s, 1H, pyrazolopyrimidine C3–H), 8.82, 9.76 (2s, each 1H, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ 55.61 (OCH3), 101.84 (pyrazolopyrimidine C3a), 114.24 (methoxyphenyl C3,5), 121.12, 121.69, 126.22, 126.73, 128.83, 128.74, 129.74, 130.45, 134.12, 136.45, 143.36, 151.52, 156.22 (phenyl-C, methoxyphenyl C1,2,4,6 and pyrazolopyrimidine C3,7a), 156.44 (C = O), 158.41 (pyrazolopyrimidine C6), 163.5 (C = O). MS (m/z, %): 452 (M+, 33), 289 (100). Anal. Calcd. for C25H20N6O3 (452.46): C, 66.36; H, 4.46; N, 18.57. Found: C, 66.18; H, 4.30; N, 18.22.
General procedure for the synthesis of 6-(2-(substituted benzylidene)hydrazino)-1,5-diphenyl-1H-pyrazolo[3,4-d] pyrimidin-4(5H)-ones (11a–c)
A mixture of 6 (0.7 g, 2.2 mmol) and the appropriate aldehyde (2.2 mmol) in absolute ethanol (20 ml) containing two drops glacial acetic acid was heated under reflux for 4 h. The reaction mixture was allowed to attain room temperature and the separated solid product was filtered, washed with ethanol, dried and crystallized from the proper solvent.
6-(2-(4-Methoxybenzylidene)hydrazino)-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (11a). Yield: 0.79 g, 82%; m.p.: 238–239 °C (ethanol) (reported m.p.: 239–240 °C)Citation38.
6-(2-(3,4-Dimethoxybenzylidene)hydrazino)-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (11b). Yield: 0.86 g, 84%; m.p.: 225–226 °C (dioxane) (reported m.p.: 215–216 °C)Citation38.
6-(2-(3,4,5-Trimethoxybenzylidene)hydrazino)-1,5-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (11c). Yield: 0.92 g, 84%; m.p.: 236–237 °C (ethanol) (reported m.p.: 240–241 °C)Citation38.
Biological screening
In vitro anti-tumor activity
MTT bromide mitochondrial activity assay
Human cancer cell lines, lung cancer cells (H1299), liver cancer cells (HepG2), colon cancer cells (RKO), human vulvar epidermoid carcinoma cells (A431) and breast cancer cells (MCF-7) were obtained from ATCC (Manassas, VA). Cells were grown in RPMI 1640 medium containing 10% fetal bovine serum at 37 °C and 5% CO2. FHC cells were cultured in DMEM-F12 containing 25 mM HEPES, 10 ng/ml cholera toxin, 5 μg/ml insulin, 5 μg/ml transferrin, 100 ng/ml hydrocortisone and 10% fetal bovine serum. The effect of compounds on the proliferation of cancer cells was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT; Sigma, St. Louis, MO) assay. Cancer cells were seeded into 96-well plates at a concentration of 5000 cells per well with a culture medium exposed to each derivative at different concentrations. After 24 h incubation, MTT was added into each well. The absorbance of formazan product of MTT was measured at 570 nm using a microtiter plate reader. The percentage of cell viability was determined as the ratio of the absorbance of the sample versus the control. Each assay was performed in triplicate and standard deviation was determined. The IC50 of each derivative was calculated as the concentration showing 50% cell growth inhibition during 24 h of compound treatment.
Enzyme-linked immunosorbent apoptosis assay. Cells were seeded at a density of 2 × 104/well in a 96-well plate and incubated for 24 h. Media were changed to media containing compounds 7a and 7b (8 and 4 μM, respectively). Cells were then incubated for 24 h. An ELISA assay was performed using Cell Death Detection ELISA PLUS kit (Roche-Applied Science, Indianapolis, IN) for the quantitative in vitro determination of cytoplasmic histone-associated DNA fragments (mono- and oligonucleosomes) in apoptosing cells. Briefly, cells were lysed with 200-μl lysis buffer for 30 min at room temperature. The lysate was centrifuged at 200g for 10 min. One hundred and fifty microliters of supernatant was collected, of which 20 μl was incubated with anti-histone biotin and anti-DNA peroxidase at room temperature for 2 h. After washing with incubation buffer three times, 100 μl of substrate solution (2,2′-azino-di(3-ethylbenzthiazolin-sulphuric acid) was added to each well and incubated for 15–20 min at room temperature. The absorbance was measured using Spectra Max Plus reader (Sunnyvale, CA) at 405 nm. Cells without compounds treatment were used as control group. Each assay was done in triplicate and the standard deviation was determined.
Detection of apoptosis by TUNEL method. For in situ detection of apoptotic cells, TUNEL (terminal-deoxynucleotidyl transferase meditated nick end labeling) assay was performed using DeadEndTM fluorimetric tunnel system (Promega , Radnor, PA). Cells were cultured on four-chamber slides (VWR, Radnor, PA) at a density of 2 × 104 cells/chamber. After treatment with compounds 7a and 7b (8 and 4 μM, respectively), cells were washed with phosphate buffer saline (PBS) and fixed by incubation in 4% paraformaldehyde for 20 min at 4 °C, then permeabilized with 0.05% triton X-100 for 5 min at 4 °C. The fixed cells were then incubated with digoxigenin-conjugated dUTP in terminal deoxynucleotide transferase recombinant (rTdT)-catalyzed reaction and nucleotide mixture for 60 min at 37 °C in a humidified atmosphere and then immersed in stop/wash buffer for 15 min at room temperature. Cells were then washed with PBS to remove unincorporated fluorescein-12-dUTP. After washing with PBS, cells were incubated in 1 μg/ml 2-(4-amidinophenyl)-6-indole carbamidine dihydrochloride (DAPI) and fluorescin isothiocyanate (FITC) solution for 15 min in dark (data not shown). Cells were observed with fluorescence microscopy (RT slider Spot, Diagnostic Instruments, Inc.) and photographed at 100 × magnification.
Annexin V staining assay. The assay is used to measure and compare changes in apoptosis, 1 × 106 cells were seeded in 100-mm dishes and treated as described above, immediately trypsinized, counted for trypan blue exclusion, washed in PBS, and resuspended in 100 μl binding buffer containing Annexin V (Annexin V-Fluos kit, Roche, Switzerland). Cells were analyzed by flow cytometry using FACScan Flow Cytometer (Becton Dickson, Franklin Lakes, NJ) according to the manufacturer’s protocol after the addition of propidium iodide. Cells in the early stages of apoptosis with intact cell membranes and surface exposed phosphatidylserine will stain positive for Annexin V-FITC. Late apoptotic and necrotic cells, which have lost plasma membrane integrity, will become dually labeled with Annexin V-FITC and Propidium Iodide (green and red fluorescence). Live cells with intact plasma membranes will exclude propidium iodide (PI) and will remain unstained by the Annexin V-FITC probe.
Flow cytometric analysis. Cells were seeded at a density of 3–5 × 105/10 cm plate and incubated for 24 h before treatment. Media were changed to media containing compounds 7a and 7b. After 24 h, cells were harvested by trypsinization. The cells were washed with PBS and fixed with ice-cold 70% ethanol while vortexing. Finally, the cells were washed and resuspended in PBS containing 5 μg/ml RNase A (Sigma) and 50 μg/ml propidium iodide (Sigma) for analysis. Cell cycle analysis was performed using FACScan Flow Cytometer (Becton Dickson, Franklin Lakes, NJ) according to the manufacturer’s protocol. Windows multiple document interfaces (WinMDI, Scripps, CA) software was used to calculate the cell-cycle phase distribution from the resultant DNA histogram, and data were expressed as a percentage of cells in the G0/G1 and G2/M phases. The apoptotic cells were identified on the DNA histogram as a subdiploid peak.
Western blot analysis. Total protein was extracted from treated and untreated cells using lysis buffer (10 mM Tris–HCl (pH 7.5), 1 mM EDTA, 1% triton X-100, 150 mM NaCl, 1 mM dithiothretol, 10% glycerol, 0.2 mM phenylmethysulphonyl fluoride and protease inhibitors) for 30–50 min on ice. The extracts were centrifuged at 13 000g for 15 min at 4 °C to remove cell debris. Folin Lowry (Pierce, Grand Island, NY) protein assay was used to figure the protein concentration in the cell lysates. Proteins were resolved by electrophoresis on 8–10% sodium dodecyl sulfate polyacrylamide gel loading equal amount of proteins per lane. The resolved proteins were transferred onto polyvinylidene difluoride membrane and then probed with primary antibody against the protein of interest prepared in 5% milk/PBS-T. The membrane was washed using PBS with Tween 20 (PBS-T) and then appropriate secondary antibody conjugated to horseradish peroxidase was used for visualization of the bands using ECL chemiluminescence kit (GE, Pittsburgh, PA). Anti-(p21), anti-(p53), anti-Bax, anti-ERK1/2, anti-Akt, and anti-Bcl-2, anti-cytochrome-c, anti-caspase-9, anti-caspase-3, anti-PARP1, were purchased from SantaCruz, Dollas, TX. Pixel density of the proteins studied was calculated using Image J, version 1.41o, NIH (Scripps, North Torrey, CA). The values obtained were first normalized to loading control β-actin and the fold change was measured by normalizing to that of the control (0 h) value. At least two independent experiments were performed.
Caspase-3 activation assay. Caspase-3 activity was assayed according to the manufacturer’s protocol. Briefly, 5 × 106 cells were lysed in 100 μl lysis buffer containing 10 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 7.4, 2 mM EDTA, 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 350 μg/ml phenylmethylsulfonyl fluoride and 5 mM dithiothreitol. Cells were homogenized by three cycles of freezing and thawing and then centrifuged to remove the cellular debris. Each sample was then incubated in buffer containing 10 mM HEPES, pH 7.4, 2 mM EDTA, 0.1% CHAPS, 5 mM EDTA supplemented with Ac-DEVD-AFC (acetyl-Asp-Glu-Val-Asp-7-amino-4-trifluoromethylcoumarin) for 1 h at room temperature and then reaction was stopped with 1 N HCl. The optical density at 405 nm (OD 405) was measured using a spectrophotometer (Spectra Max Plus, Sunnyvale, CA).
Caspase-9 activation assay. The assay is based on the ability of the active enzyme to cleave the chromophore from the enzyme substrate LEHD-pNA (Ac-Leu-Glu-His-Asp-pNA). Cell lysates were incubated with peptide substrate in assay buffer (100 mM NaCl, 50 mM HEPES, 10 mM dithiothreitol, 1 mM EDTA, 10% glycerol and 0.1% CHAPS, pH 7.4) for 2 h at 37 °C. The release of p-nitroaniline was monitored at 405 nm. Results are represented as the percentage of change of activity compared with the untreated control. The assay was repeated with and without caspase-9 inhibitor (LEDH-CHO, Ac-Ala-Ala-Val-Ala-Leu-Leu-Pro-Ala-Val-Leu-Leu-Ala-Leu-Leu-Ala-Pro-Leu-Glu-His-Asp-CHO). For inhibition studies, cells were pre-incubated with 20 mM LEHD-CHO for1 h before the addition of 4 μM 7b for an additional 24 h. Each value was expressed as mean ± SD of three determinations.
Statistical analysis
The quantitative ratios of different groups were compared using Student’s t-test. Probability values of p<0.05 were regarded as statistically significant. All statistical tests were two sided.
Results and discussion
Chemistry
The synthetic strategies adopted for the synthesis of the intermediate and final compounds are depicted in Schemes 1 and 2. In Scheme 1, the amino ester 1Citation36 was readily cyclized to the corresponding 6-thioxo-6,7-dihydro-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one 2Citation37 through reaction with methyl N-phenyldithiocarbamate in DMF containing 1 N NaOH adopting previously reported procedure. Stirring 2 with methyl iodide in dry DMF containing anhydrous potassium carbonate gave rise to the methylsulfanyl derivative 3. At this stage, it was decided to oxidize the methylsulfanyl moiety to the corresponding methylsulfonyl group to be utilized in the subsequent nucleophilic substitution reactions for preparation of the target compounds. Oxidation of 3 to the corresponding methylsulfonyl derivative 4 was achieved by reaction with potassium hydrogenpersulfate (oxone) following reaction conditions previously reported for the synthesis of analogous compoundsCitation39. IR spectrum of the latter compound displayed absorption bands of SO2 at 1346 and 1162 cm−1, while its 1H NMR spectrum showed a relatively deshielded singlet at 2.76 ppm attributed to SO2CH3 protons. Moreover, the mass spectrum showed a molecular ion peak at m/z = 366, which was in accordance with its molecular formula. Refluxing the methylsulfonyl derivative 4 with cycloalkyl, aryl, aralkyl or alkyl amines in dry dioxane resulted in the target 6-substituted amino-1H pyrazolo[3,4-d]pyrimidin-4(5H)ones 5a–j in a good yield. In the first trial to prepare the aminoguanidino and guanidino derivatives (7a, b) the methylsulfonyl derivative 4 was reacted with the corresponding aminoguanidine or guanidine HCl salts in ethanol in presence of anhydrous sodium acetate but the 6-ethoxy derivative 6 was separated as a major product indicating nucleophilic replacement of the methylsulfonyl moiety by the solvent. However, when a mixture of dry dioxane/dimethyl sulfoxide (5:1) was used as solvent (method A) instead of ethanol it was possible to isolate compounds 7a, b in a moderate yield. In another trial to prepare these compounds, the bicarbonate salts of aminoguanidine and guanidine were refluxed with the methylsulfonyl derivative 4 in a mixture of dry dioxane/dimethyl sulfoxide (5:1) (method B), and it was possible to isolate compounds 7a, b in a better yield. IR spectra of compounds 7a, b displayed NH absorption bands in the range 3401–3190 cm−1, while 1H NMR spectra revealed D2O exchangeable singlets characteristic for aminoguanidine and guanidine NH at their expected chemical shifts. Moreover, 13C NMR showed resonance for the aminoguanidino and guanidino carbon at 160.90 and 159.92 ppm, respectively.
Scheme 1. Synthetic pathways for the target compounds 5–7.

Scheme 2. Synthetic pathways for the target compounds 9–11.
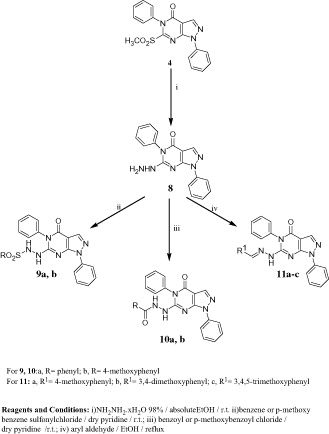
In Scheme 2, the methylsulfonyl derivative 4 was utilized for the preparation of the starting hydrazino compound 8. Thus, stirring an ethanolic suspension of 4 with hydrazine hydrate at room temperature afforded the hydrazine derivative 8 in an excellent yield. It should be noted here that compound 8 was differently prepared in a low yieldCitation38 from the corresponding thione 2. Reacting 8 with benzene or p-methoxybenzene sulfonylchloride in dry pyridine resulted in the arylsulfonyl derivatives 9a, b in a good yield. IR spectra of these compounds showed the characteristic SO2 absorption bands at 1341, 1342 and 1162, 1167 cm−1, while their 1H NMR spectra revealed two D2O-exchangeable signals at 8.80, 8.90 and 9.84, 9.98 ppm attributed to two NH groups. Similarly, reacting 8 with benzoyl or p-methoxybenzoyl chloride in dry pyridine resulted in the aroyl derivatives 10a, b. Finally, it was planned to synthesize some arylhydrazones incorporating methoxy groups in order to study the effect of magnification of compounds’ lipophilicityCitation40,Citation41 on the anticipated biological activity. Consequently, condensation of 8 with methoxylated aromatic aldehydes in refluxing ethanol furnished the target azomethine derivatives 11a–c in a good yield as previously reportedCitation38.
Biological screening
Cytotoxicity test
Cytotoxicity of the test compounds was assayed in five cancer cell lines, namely lung cancer cells (H1299), liver cancer cells (HepG2), colon cancer cells (RKO), human vulvar epidermoid carcinoma cells (A431) and breast cancer cells (MCF-7) by a tetrazolium based (MTT) colorimetric assay and utilizing cisplatin as a reference standard. The results of in vitro cytotoxic activity are expressed as IC50, which is the concentration of compound (in μM) that inhibits survival of the cells by 50% (). The effect of the synthesized compounds on the growth of the five cancer cell lines revealed that HepG2 cell line was the least sensitive cell line toward the test compounds, whereas the RKO cell line was the most sensitive. Compounds 7a and 7b emerged with IC50 values against RKO cells (IC50 = 8 and 4 μM, respectively) lower than cisplatin (IC50 = 17 μM), whereas they showed IC50 values against A431 (IC50 = 17 and 20 μM, respectively) comparable to that of cisplatin (IC50 = 20 μM). Moreover, compounds 10b and 11c exhibited the lowest IC50 values in this study against MCF-7 cells (IC50 = 10 and 12 μM, respectively) compared to the reference standard (IC50 = 13 μM). Since compounds 7a and 7b showed the lowest IC50 values in this study against RKO colon cancer cells, they were further investigated for their cytotoxicity against normal colon cells (FHC). The MTT cell viability assay indicated that compounds 7a and 7b were less cytotoxic to FHC cells () and they reduced cell viability to approximately 50% in RKO colon cancer cells compared to approximately 76% and 87% in FHC normal colon cancer cells after treatment of cells with 8 and 4 μM of 7a and 7b, respectively ().
Figure 2. Compounds 7a and 7b reduced cell viability and induced apoptosis in RKO cells. (A) A dose–response curve for compounds 7a and 7b on FHC normal colon cancer cells was constructed. Each data point was an average of results from three independent experiments performed in triplicate and presented as mean ± SD. (B) Comparison of 7a and 7b cytotoxicity in FHC and RKO cells. Both cells were treated with 8 and 4 μM of compounds 7a and 7b, respectively. Untreated cells were used as control. (C) Comparison of 7a and 7b induced apoptosis in FHC and RKO cells. FHC and RKO cells were treated with 8 and 4 μM of compounds 7a and 7b, respectively, for 24 h. Untreated cells were used as control. ELISA assay was applied for apoptotic cell detection. Each experiment was performed in triplicate. Data are presented as mean ± SD (n = 3). Significant differences between the control and test compounds are indicated by * (*p < 0.05). (D) TUNEL assay was used to confirm induction of apoptosis in RKO cells. Cells were treated with 8 and 4 μM of 7a and 7b, respectively, for 24 h. Untreated cells act as control. Lack of staining in control (untreated) cells, represents that the cells are actively proliferating, i.e. without apoptotic cell death and induction of apoptosis in treated cells was confirmed by appearance of TUNEL positive cells. Images were acquired for each fluorescence channel, using suitable filters at 200 × magnification.
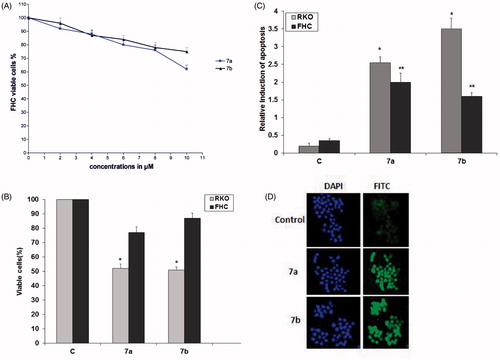
Table 1. IC50 (μM)Table Footnote* of the synthesized 6-substituted 1H-pyrazolo[3,4-d]pyrimidin-4(5H)-ones against five cancer cell lines.
Based on the results in , the following preliminary structure–activity relationships could be tentatively deduced. Compounds comprising an amino group at position 6 substituted with cyclohexyl, aralkyl, alkyl or substituted alkyl moieties (5a–g) exhibited mild activity against the five cell lines except for compound 5b (benzyl derivative) that displayed significant activity against A431 (IC50 = 15 μM) and 5f (pyrrolidinoethyl derivative), which demonstrated moderate activity against H1299 and A431 (IC50 = 19 and 15 μM, respectively). Substitution at C-1 of 5g (2-hydroxyethyl derivative) by two methyl groups or two hydroxymethyl groups resulted in compounds 5h and 5i with improved anti-proliferative activity toward H1299 (IC50 = 28 and 16 μM, respectively), HepG2 (IC50 = 36 and 28 μM, respectively), RKO (IC50 = 22 and 18 μM, respectively), and A431 (IC50 = 35 and 25 μM, respectively). However, substitution of the hydroxy group in 5g by an amino group (compound 5j) or replacement of the 2-hydroxyethylamino group in 5g by aminoguanidino or guanidino moieties (compounds 7a and 7b) potentiated the anti-proliferative activity toward H1299, RKO, A431 and MCF-7 cell lines. In this context, compounds 7a and 7b emerged with the highest activity in this study toward RKO cells as they showed IC50 values of 8 and 4 μM, respectively. Replacement of the substituted amino group by phenyl; or 4-methoxyphenyl sulfonylhydrazino (compounds 9a and 9b), benzo; or 4-methoxybenzohydrazide (10a and 10b) or azomethine moieties (11a–c) did not potentiate the activity against the five cancer cell lines except for the moderate activity of 9a, 10b and 11c against MCF7 cell line (IC50 = 22, 10 and 12 μM, respectively).
Finally, it could be concluded that presence of substituents such as aminoguanidino or guanidino at position 6 of the pyrazolo[3,4-d]pyrimidinone scaffold (compounds 7a and 7b) potentiated the cytotoxic activity especially against RKO colon cancer cell line. Accordingly, compounds 7a and 7b were chosen for further investigations.
Compounds 7a and 7b induced apoptosis in RKO cells
Enzyme linked immunosorbent apoptosis assay
Compounds 7a and 7b were further investigated for their induction of apoptosis in both FHC and RKO cells using Cell Death Detection ELISAPLUS, which is used for the quantitative in vitro determination of cytoplasmic histone-associated DNA fragments (mono- and oligonucleosomes) in apoptosing cells. The results revealed that 8 μM of 7a and 4 μM of 7b increased apoptosis in RKO cells by approximately 8- and 11-folds, respectively, compared to non-treated control (p < 0.05) (). The results also showed that compounds 7a and 7b induced apoptosis in RKO cells more than FHC cells and this was more pronounced in case of compound 7b.
TUNEL assay. To ascertain induction of apoptosis, the results were confirmed by immune histochemical analysis using TUNEL assay. TUNEL assay involves labeling of the 3′-hydroxyl DNA ends generated during DNA fragmentation by means of rTdT and labeled dUTP. The localized fluorescence of apoptotic cells (labeled-dUTP) in a blue background (DAPI) for nuclei staining or green background (FITC) was visualized using fluorescence microscope. The assay results () revealed the presence of nuclear condensation and TUNEL-positive cells (stained cells).
Impact of compounds 7a and 7b on cell cycle
Annexin V FITC dual staining assay
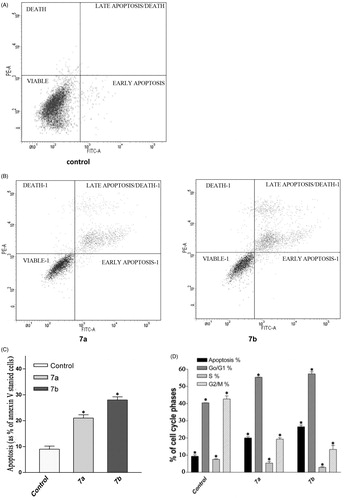
The apoptotic effect of compounds 7a and 7b was finally confirmed by Annexin V. FITC/PI (AV/PI) dual staining assayCitation42. In this assay, RKO cells were treated with 8 μM 7a and 4 μM 7b for 24 h to study the apoptotic effect. Data in showed representative image for untreated group, where the percentage of viable cells was 97%, whereas data in revealed that RKO cells treated with 7a and 7b showed 20% and 28% apoptosis, respectively. reveals that there is a threefold increase in induction of apoptosis in RKO cells treated with 7b compared to control untreated cells under the same experimental conditions. These results suggest that these two compounds induce apoptosis in RKO cells.
Figure 3. Impact of 7a and 7b on cell cycle. (A and B) Flow cytometric analysis of RKO cells stained with Annexin V-FITC and PI. (A) Untreated RKO cells were used as control, (B) RKO cells treated with 8 μM 7a and 4 μM 7b for 24 h. (C) Comparison of 7a and 7b induced apoptosis in RKO colon cancer cells using Annexin V/FITC staining assay. Each bar represents the mean ± SD (n = 3). (D) Percent of distribution of cell cycle phases of RKO colon cancer cells after treatment with 7b for 24 h. (E) A representative image of flow cytometric analysis showing G1 arrest. (F) Total protein was extracted from RKO cells and used for Western blotting analysis to determine the differences in p21 between untreated cells (control; C) and cells treated with 7b for 24 h. β-Actin was used as loading control.
Flow cytometric analysis
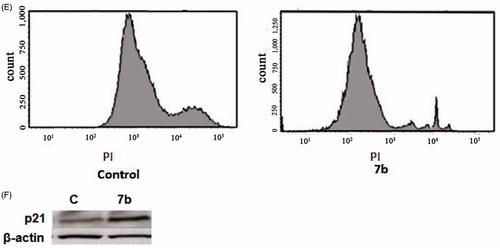
Flow cytometry is regarded as one of the most powerful and specific methods for the integrated study of molecular and morphological events occurring during cell death and cell proliferationCitation43. Generally, anti-cancer drugs inhibit the proliferation of cancer cell either by induction of cell cycle arrest or by apoptosis, or by combination of both these modes. However, by measuring the DNA content, it becomes possible to identify apoptotic cells; to recognize the cell cycle phase specificity and to quantitate apoptosis. Therefore, the effect of 7a and 7b on cell cycle progression was studied by flow cytometry in PI stained cells. The percent of cells in G1, S, G2, M phases and apoptosis were determined after treating cells with 8 and 4 μM of 7a and 7b, respectively, for 24 h (). The results revealed a significant increase in the percent of G1 population (55% and 57.5%, respectively) compared to control group (40%); however, no significant difference was found in the percent of cells in the S phase. Moreover, the apoptotic cells increased to 20% and 26.5% after treating RKO cells with 7a and 7b, respectively, compared to control cells (9%). Thus, it can be concluded that compounds 7a and 7b inhibit the proliferation of RKO cells and the growth inhibition is associated with induction of cycle G1 phase arrest and apoptosis.
Effect of compound 7b on the expression level of signaling proteins
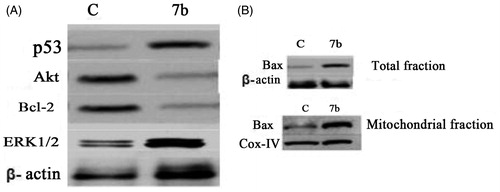
To further investigate the underlying pathways of 7b induced G1 phase arrest and apoptosis, the expression of some apoptotic and survival proteins involved in apoptosis such as p21, p53, Bax and anti-apoptotic proteins such as Bcl-2 in addition to protein kinases, e.g. ERK1/2, AKT was evaluated. p21WAF1 is considered as an important regulator of cell cycleCitation44.To determine whether compound 7b-induced G1 arrest was associated with increased p21/WAF1 level, cells were analyzed for the expression of this protein after treatment with 4 μM of 7b for 24 h and the expression of this protein was quantified using Imagine J software (Scripps, North Torrey Pines, CA). The results showed that, in comparison to the non-treated control, 7b increased p21 level by twofold (). The tumor suppressor protein p53 plays a critical role in inducing cell growth arrest or apoptosis through its transcriptional activityCitation45. Activation of p53 leads to activation of the intrinsic pathway through initiation of the apoptotic cascade that leads to activation of many pro-apoptotic proteins such as Bax and Bid. Bax, a Bcl-2 family protein, induces cell death through disruption of mitochondrial permeability and subsequent release of cytochrome c, where it engages in a cascade of interactions that leads to the execution stage of apoptosis. The obtained data displayed that 7b increased level of p53 by threefold () and increased level of Bax in both mitochondrial and total fractions by four- and threefold, respectively (). It is known that interactions between the pro- and anti-apoptotic members of Bcl-2 family integrate diverse upstream signals to determine the cellular response. Apoptosis occurs through competitive dimerization between the two protein groupsCitation46. Accordingly, an increased ratio of Bax to Bcl-2 leads to programmed cell death. Therefore, we analyzed the level of Bcl-2 protein following treatment with 4 μM 7b for 24 h. The obtained data portrayed that the level of Bcl-2 protein was decreased by 2.5-fold. Finally, we analyzed the influence of 7b on functional status of ERK1/2 and AKT, two pivotal protein kinases involved in mitogenic signal transduction to the cell cycle control system. The results displayed that 7b upregulated ERK1/2 and downregulated Akt by 4.5- and 3-fold, respectively ().
Figure 4. Effect of compound 7b on apoptotic signaling in RKO cells. (A) Cells were treated with 4 μM of compound 7b for 24 h. The cell lysates were collected and the levels of p53, Akt, Bcl-2, ERK1/2 were studied by Western blotting analysis using specific antibodies. β-Actin was used as loading control. C: control (untreated cells). (B) Total protein and mitochondrial fraction were exacted and Western blotting analysis was applied to determine the protein levels of Bax in treated and non-treated cells. β-Actin was used as loading control and IV-Cox was used as internal mitochondrial control.
Compound 7b induced apoptosis through release of cytochrome c and activation of caspases
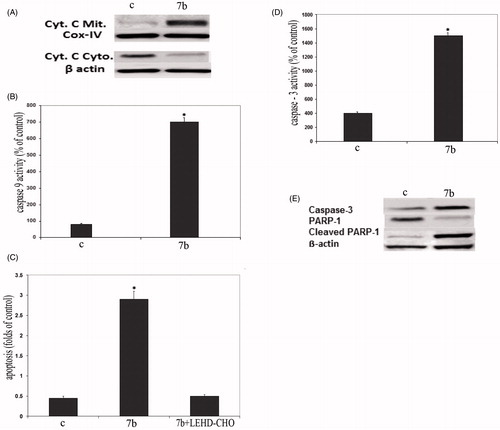
Cytochrome c, a component of the mitochondrial electron transfer chain, is released into the cytosol during the early phases of apoptosisCitation47. The mitochondrial cytochrome c release in the cytosol was determined by immunoblotting of the cell lysate of 7b treated cells. Data from Western blot analysis () showed that the treatment of RKO cells with 4 μM 7b for 24 h increased the expression of cytochrome c in cytoplasm and reduced its expression in mitochondria (). To determine whether caspase-9 and -3 activation is involved in 7b induced apoptosis, caspase-9 and -3 were investigated in RKO cells. The obtained data () revealed that the treatment of cells with 7b increased significantly caspase-9 activity (). Furthermore, pretreatment of RKO cells with the specific caspase-9 inhibitor LEHD-CHO prior to treatment with 7b significantly reduced 7b induced apoptosis (). Additionally, the treatment of cells with 7b increased caspase-3 activity (). Finally, monitoring of the cleavage of PARP-1 poly (ADP-ribose) polymerase, a target of caspase-3, further confirmed the above results. As shown in , cleaved PARP1 (89 kDa fragment) was detected after 24 h exposure to 7b.
Figure 5. Compound 7b induced apoptosis through initiation of the mitochondrial pathway. (A) Western blot analysis was used to determine the release of cytochrome c in the cytosol and mitochondria of 7b-treated RKO cells. β-Actin was used as loading control and IV-Cox was used as internal mitochondrial control. C: control (untreated cells). (B) Effect of 7b on the level of caspase-9 in RKO cells after treatment with 4 μM 7b for 24 h. (C) Effect of 7b and its combination with caspase-9 inhibitor on 7b-induced apoptosis. Cells were preincubated with 20 μM LEHD-CHO for 1 h before the addition of 4 μM of 7b for an additional 24 h. Induction of apoptosis was determined at 405 nm and compared to control (untreated cells). (D) Activation of caspase-3 in RKO cells after treatment with 4 μM 7b for 24 h. (For B, C and D; each data point is the mean of three independent experiments and expressed as M ± SD. The asterisk indicates a significant difference between control (C; untreated cells) and 7b-treated cells; p < 0.05). (E) Western blot analysis was used to determine the expression levels of caspase-3 and cleaved PARP1 after treating cells with 7b for 24 h. β-Actin was used as loading control.
Conclusion
In the present investigation, new 1,5-diphenyl-6-substituted 1H-pyrazolo[3,4-d]pyrimidine-4(5H)-ones were synthesized and screened for their anti-tumor activity in five cancer cell lines.
Results revealed that compounds 7a and 7b were the most active derivatives in this study against RKO colon cancer cells and they showed less cytotoxic effect on normal colon cells (FHC) than colon cancer cells (RKO). The results of apoptosis assays and cell cycle analysis demonstrated that compounds 7a and 7b obviously inhibit the proliferation of RKO cancer cells by inducing apoptosis and arresting the cell cycle at G1 phase. The most active compound 7b increased levels of p53, p21, Bax, ERK1/2 and decreased the level of Bcl-2 and AKT. The regulatory effects of derivative 7b on the Bcl-2 family are correlated with the release of cytochrome c from the mitochondria into the cytoplasm and the activation of caspase-9. Compound 7b also increased caspase-3 and cleaved PARP-1 in RKO colon cancer cells. Finally, it could be concluded that compound 7b was found to induce apoptosis in RKO colon cancer cells via modulating mitochondrial signaling pathway. Accordingly, compound 7b was identified as a new lead compound that merits further optimization and development as an anti-cancer candidate.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the contents and writing the paper. The authors would like to thank the Department of Health Sciences, Qatar University for financial support of the cancer biology part of this work.
References
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics 2014. CA Cancer J Clin 2014;64:9–29
- Haggar FA, Boushey RP. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg 2009;22:191–7
- Borowski E, Bontemps-Gracz MM, Piwkowska A. Strategies for overcoming ABC-transporters-mediated multidrug resistance (MDR) of tumor cells. Acta Biochim Pol 2005;52:609–27
- Meier P, Vousden KH. Lucifer's labyrinth – ten years of path finding in cell death. Mol Cell 2007;28:746–54
- Gupta S, Kass GE, Szegezdi E, Joseph B. The mitochondrial death pathway: a promising therapeutic target in diseases. J Cell Mol Med 2009;13:1004–33
- Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci 2009;122:437–41
- Galluzzi L, Morselli E, Kepp O, et al. Mitochondrial gateways to cancer. Mol Aspects Med 2010;31:1–20
- Portt L, Norman G, Clapp C, et al. Anti-apoptosis and cell survival: a review. Biochim Biophys Acta 2011;1813:238–59
- Rashad AE, Mahmoud AE, Ali MM. Synthesis and anticancer effects of some novel pyrazolo[3,4-d]pyrimidine derivatives by generating reactive oxygen species in human breast adenocarcinoma cells. Eur J Med Chem 2011;46:1019–26
- Ghorab MM, Ragab FA, Alqasoumi SI, et al. Synthesis of some new pyrazolo[3,4-d]pyrimidine derivatives of expected anticancer and radioprotective activity. Eur J Med Chem 2010;45:171–8
- Kumar A, Ahmad I, Chhikara BS, et al. Synthesis of 3-phenylpyrazolopyrimidine-1,2,3-triazole conjugates and evaluation of their Src kinase inhibitory and anticancer activities. Bioorg Med Chem Lett 2011;21:1342–6
- Hassan GS, Kadry HH, Abou-Seri SM, et al. Synthesis and in vitro cytotoxic activity of novel pyrazolo[3,4-d]pyrimidines and related pyrazole hydrazones toward breast adenocarcinoma MCF-7 cell line. Bioorg Med Chem Lett 2011;19:6808–17
- Abd El Hamid MK, Mihovilovic MD, El-Nassan HB. Synthesis of novel pyrazolo[3,4-d]pyrimidine derivatives as potential anti-breast cancer agents. Eur J Med Chem 2012;57:323–8
- Kim DC, Lee YR, Yang B, et al. Synthesis and biological evaluations of pyrazolo[3,4-d]pyrimidines as cyclin-dependent kinase 2 inhibitors. Eur J Med Chem 2003;38:525–32
- Traxler P, Bold G, Frei J, et al. Use of a pharmacophore model for the design of EGFR tyrosine kinase inhibitors: 4-(phenylamino)pyrazolo[3,4-d]pyrimidines. J Med Chem 1997;40:3601–16
- Indovina P, Giorgi F, Rizzo V, et al. New pyrazolo[3,4-d]pyrimidine Src inhibitors induce apoptosis in mesothelioma cell lines through p27 nuclear stabilization. Oncogene 2012;31:929–38
- Carraro F, Naldini A, Pucci A, et al. Pyrazolo[3,4-d]pyrimidines as potent antiproliferative and proapoptotic agents toward A431 and 8701-BC cells in culture via inhibition of c-Src phosphorylation. J Med Chem 2006;49:1549–61
- Santucci MA, Corradi V, Mancini M, et al. C6-unsubstituted pyrazolo[3,4-d]pyrimidines are dual Src/Abl inhibitors effective against imatinib mesylate resistant chronic myeloid leukemia cell lines. Chem Med Chem 2009;4:118–126
- Radi M, Dreassi E, Brullo C, et al. Design, synthesis, biological activity, and ADME properties of pyrazolo[3,4-d]pyrimidines active in hypoxic human leukemia cells: a lead optimization study. . J Med Chem 2011;54:2610–26
- Richard DJ, Verheijen JC, Curran K, et al. Incorporation of water-solubilizing groups in pyrazolopyrimidine mTOR inhibitors: discovery of highly potent and selective analogs with improved human microsomal stability. Bioorg Med Chem Lett 2009;19:6830–5
- Markwalder JA, Arnone MR, Benfield PA, et al. Synthesis and biological evaluation of 1-aryl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-4-one inhibitors of cyclindependent kinases. J Med Chem 2004;47:5894–911
- El-Enany MM, Kamel MM, Khalil OM, El-Nassan HB. Synthesis and antitumor activity of novel 6-aryl and 6-alkylpyrazolo[3,4-d]pyrimidin-4-one derivatives. Eur J Med Chem 2010;45:5286–91
- Rostom SAF, Ashour HMA, Abd ElRazik HA. Synthesis and biological evaluation of some novel polysubstituted pyrimidine derivatives as potential antimicrobial and anticancer agents. Arch Pharm Chem Life Sci 2009;342:299–310
- Ashour HMA, Abdel Wahab AE. Synthesis and biological evaluation of novel pyrazoles and pyrazolo[3,4-d]pyrimidines incorporating a benzenesulfonamide moiety. Arch Pharm Chem Life Sci 2009;342:238–52
- Rostom SAF, Badr MH, Abd ElRazik HA, et al. Synthesis of some pyrazolines and pyrimidines derived from polymethoxy chalcones as anticancer and antimicrobial agents. Arch Pharm Chem Life Sci 2011;344:572–87
- Malki A, Elbayaa RY, Ashour HMA, et al. Novel thiosemicarbazides induced apoptosis in human MCF-7 breast cancer cells via JNK signaling. J Enzyme Inhib Med Chem 2015;30:786–95
- Ibrahim DA, El-Metwally AM. Design, synthesis, and biological evaluation of novel pyrimidine derivatives as CDK2 inhibitors. Eur J Med Chem 2010;45:1158–66
- Zhang H-Z, Crogan-Grundy C, May C, et al. Discovery and structure–activity relationships of (2-(arylthio)benzylideneamino)guanidines as a novel series of potent apoptosis inducers. Bioorg Med Chem Lett 2009;17:2852–8
- Andreani A, Granaiola M, Leoni A, et al. Potential antitumor agents. 37.1 Synthesis and antitumor activity of guanylhydrazones from imidazo[2,1-b]thiazoles and from the new heterocyclic system thiazolo[2′,3′:2,3]imidazo[4,5-c]quinoline. J Med Chem 2005;48:3085–9
- Andreani A, Burnelli S, Granaiola M, et al. New antitumor imidazo[2,1-b]thiazole guanylhydrazones and analogues. J Med Chem 2008;51:809–16
- Zhang H, Drewe J, Tseng B, et al. Discovery and SAR of indole-2-carboxylic acid benzylidenehydrazides as a new series of potent apoptosis inducers using a cell based HTS assay. Bioorg Med Chem 2004;12:3649–55
- Cocco MT, Congiu C, Lilliu V, Onnis V. Synthesis and in vitro antitumoral activity of new hydrazinopyrimidine-5-carbonitrile derivatives. Bioorg Med Chem 2005;14:366–72
- Rollas S, Küçükgüzel G. Biological activities of hydrazone derivatives. Molecules 2007;12:1910–39
- Nasr T, Bondock S, Youns M. Anticancer activity of new coumarin substituted hydrazide-hydrazone derivatives. Eur J Med Chem 2014;76:539–48
- Baviskar AT, Banerjee UC, Gupta M, et al. Synthesis of imine-pyrazolopyrimidinones and their mechanistic interventions on anticancer activity. Bioorg Med Chem 2013;21:5782–93
- Peet NP, Lentz NL, Sunder S, et al. Conformationally restrained, chiral (phenylisopropyl)amino-substituted pyrazolo[3,4-d]pyrimidines and purines with selectivity for adenosine A1 and A2 receptors. J Med Chem 1992;35:3263–9
- Garin J, Loscertales MP, Melendez E, et al. Methyl N-aryldithiocarbamates: useful reagents for the annelation of pyrimidines and 1,3-oxazines to five-membered heterocyclic rings. Heterocycles 1987;26:1303–12
- Shaaban MA, Abou Sier AH, El-Ansary AK, et al. Synthesis and pharmacological activities of certain pyrazolotriazolopyrimidine and pyrazolotriazepine derivatives. Bull Facul Pharm (Cairo Univ) 2001;39:67–77
- Trost BM, Curran DP. Chemoselective oxidation of sulfides to sulfones using potassium hydrogenpersulfate. Tetr Lett 1981;22:1287–90
- Lin CF, Yang JS, Chang CY, et al. Synthesis and anticancer activity of benzyloxybenzaldehyde derivatives against HL-60 cells. Bioorg Med Chem 2005;13:1537–44
- Barboni L, Giarlo G, Ballini R, Fontana G. 14Beta-hydroxy-10-deacetylbaccatin III as a convenient, alternative substrate for the improved synthesis of methoxylated second-generation taxanes. Bioorg Med Chem Lett 2006;16:5389–91
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods 1995;184:39–51
- Shrivastava HY, Ravikumar T, Shanmugasundaram N, et al. Cytotoxicity studies of chromium(III) complexes on human dermal fibroblasts. . Free Radic Biol Med 2005;38:58–69
- Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis – the p53 network. J Cell Sci 2003;116:4077–85
- Taguchi T, Kato Y, Baba Y, et al. Protein levels of p21, p27, cyclin E and Bax predict sensitivity to cisplatin and paclitaxel in head and neck squamous cell carcinomas. Oncol Rep 2004;11: 421–46
- Pellecchia M, Reed JC. Inhibition of anti-apoptotic Bcl-2 family proteins by natural polyphenols: new avenues for cancer chemoprevention and chemotherapy. Curr Pharm Des 2004;10:1387–98
- Liu X, Kim CN, Yang J, et al. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996;86:147–57