
Abstract
N-protected amino acids (Gly, Ala and Phe protected with Boc and Z groups) were reacted with sulfonamide derivatives, leading to the corresponding N-protected amino acid–sulfonamide conjugates. The carbonic anhydrase (CA, EC 4.2.1.1) inhibitory activity of the new compounds was assessed against four human (h) isoforms, hCA I, hCA II, hCA IV and hCA XII. Among them, hCA II, IV and XII are antiglaucoma drug targets, being involved in aqueous humor secretion within the eye. Low nanomolar inhibition was measured against all four isoforms with the 20 reported sulfonamides, but no selective inhibitory profiles, except for some CA XII-selective derivatives, were observed. hCA I, II and XII were generally better inhibited by sulfonamides incorporating longer scaffolds and Gly/Ala, whereas the best hCA IV inhibitors were homosulfanilamide derivatives, incorporating Phe moieties. The amino acid–sulfonamide conjugates show good water solubility and effective hCA II, IV and XII inhibition, and may be considered as interesting candidates for antiglaucoma studies.
Introduction
There are six different genetic families encoding for the metalloprotein carbonic anhydrase (CA, EC 4.2.1.1), a superfamily of metalloenzymes acting as catalysts for the interconversion between CO2 and bicarbonate. These are the α-, β-, γ-, δ-, ζ- and η-CA classessCitation1–11. The catalytic/inhibition mechanisms for the physiologic and other catalyzed by these enzymes are well understood processesCitation12–21. The discovery of new classes of CA inhibitors (CAIs), possessing different inhibition mechanisms compared with the classical inhibitors of the sulfonamide/anion typeCitation1–3,5, has also seen important developments ultimatelyCitation13. At least five different CA inhibition mechanisms were reported so far: (i) the zinc binders are the inhibitors which coordinate to the catalytically crucial Zn(II) ion from the enzyme active site. The metal ion may be in a tetrahedral or trigonal bipyramidal geometries, with the sulfonamides and their isosteres (sulfamides, sulfamates, etc.), most anions, dithiocarbamates and their isosteres, carboxylates and hydroxamates binding in this wayCitation1–3,5,13; (ii) the inhibitors that anchor to the zinc-coordinated water molecule/hydroxide ion, represented by the phenols, some carboxylates, the polyamines, 2-thioxocoumarins, and sulfocoumarinsCitation1–3,13,14; (iii) the inhibitors which occlude the entrance to the active site cavity (coumarins and their isosteres), this binding site coinciding with that where CA activators bindCitation13–20; (iv) the compounds which bind out of the active site cavity (a carboxylic acid derivative was seen to inhibit CA in this manner)Citation21, and (v) compounds for which the inhibition mechanism is not known, among which the secondary/tertiary sulfonamides as well as imatinib/nilotinib are the most investigated examplesCitation13,22–26.
The sulfonamides however remain the main class of CAIs with many clinically used drugs as antiglaucoma agentsCitation27–29, diureticsCitation30, antiobesity drugsCitation31–33, antiepilpeticsCitation34, and more recently agents for the management of hypoxic tumorsCitation35–38, neuropathic painCitation39 and ischaemiaCitation40,Citation41.
Continuing our interest in designing biologically active compounds incorporating N-protected amino acid moieties (the coumarineCitation19 conjugates as CAIs as well as quinine conjugates were recently reportedCitation42), we focused on the synthesis of amino acid–sulfonamide conjugates by using the N-(protected α-aminoacyl)benzotriazole methodologyCitation43 and explore the enzyme inhibitory activities of such compounds against physiologically important isoforms, such as the cytosolic human (h) hCAI and II, as well as the membrane-asssociated/transmembrane hCA IV and XIICitation1–5.
Materials and methods
Chemistry
Anhydrous solvents and all reagents were purchased from Sigma-Aldrich (Milan, Italy), Acros (Milan, Italy) and Merck (Florence, Italy). All reactions involving air- or moisture-sensitive compounds were performed under a nitrogen atmosphere using dried glassware and syringes techniques to transfer solutions. Nuclear magnetic resonance (1H-NMR, 13C-NMR) spectra were recorded using a Bruker Advance III 400 MHz spectrometer in DMSO-d6. Chemical shifts are reported in parts per million (ppm) and the coupling constants (J) are expressed in Hertz (Hz). Splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; m, multiplet; brs, broad singlet; dd, double of doubles. The assignment of exchangeable protons (OH and NH) was confirmed by the addition of D2O. Positive-ion electrospray ionization (ESI) mass spectra were recorded on a double-focusing Finnigan MAT 95 instrument with BE geometry. Analytical thin-layer chromatography (TLC) was carried out on Merck silica gel F-254 plates. All microwave-assisted reactions were carried out in a microwave oven system manufactured by GEM (CEM Corporation, Matthews, NC, USA) under a nitrogen atmosphere. The reaction mixtures were transferred into a 10 mL glass pressure microwave tube equipped with a magnetic stir bar. The tube was closed with a silicon septum and the reaction mixture was subjected to microwave irradiation. Melting points (mp) were measured in open capillary tubes and are uncorrected, using a Gallenkamp MPD350.BM3.5 apparatus. N-protected amino acidsCitation19,43–45; benzyl (2-1H-benzo[d][1,2,3]triazol-1-yl)-2-oxoethyl)carbamate (I), tert-butyl (2-1H-benzo[d][1,2,3]triazol-1-yl)-2-oxoethyl)carbamate (II), (S)-benzyl (1-(1H-benzo[d][1,2,3]triazol-1-yl)-1-oxopropan-2-yl)carbamate (III), (S)-tert-butyl (1-(1H-benzo[d][1,2,3]triazol-1-yl)-1-oxo-3-phenylpropan-2-yl)carbamate (IV) and sulfonamide derivativesCitation46; 2-(4-sulfamoylphenoxy)ethanaminium 2,2,2-trifluoroacetate, 3-(4-sulfamoylphenoxy)propan-1-aminium chloride, 4-(4-sulfamoylphenoxy)butan-1-aminium 2,2,2-trifluoroacetate used as starting materials were prepared according to literature procedure. Compounds 3Citation47, 4Citation48, 5Citation48 and 10Citation47 were found in the literature but their synthesis methods need harsh reaction conditions and cumbersome work-up procedures. Thus, these compounds were synthesized by a new, one-step, easy synthetic method, using N-(protected α-aminoacyl)benzotriazoles in this work.
General procedure for the synthesis of amino acid–sulfonamide conjugates, 1–3
A mixture of equivalent amounts of the appropriate N-protected aminoacylbenzotriazole and 3-aminobenzenesulfonamide was subjected to microwave irradiation (100 W, 70 °C) in anhydrous THF (5 mL) for 30 min. After completion of the reaction, all volatiles were removed by rotavapour and the obtained crude product was crystallized from methanol.
Benzyl (2-oxo-2-((3-sulfamoylphenyl)amino)ethyl)carbamate, 1
White solid (83%); silica gel TLC Rf = 0.16 (MeOH/CH2Cl2 5% v/v); mp 182–183 °C; 1H NMR (DMSO-d6, 400 MHz) 10.32 (s, 1H, NH), 8.21 (s, 1H, Ar-H), 7.79 (d,1H, Ar-H, J = 4.0 Hz), 7.62 (t, 1H, NH, J = 8.0 Hz), 7.56 (s, 2H, NH2), 7.42–7.36 (m, 7H, Ar-H), 5.10 (s, 2H, OCH2Ph), 3.88 (d, 2H, CH2NH, J = 8.0 Hz). 13C NMR (DMSO-d6, 400 MHz) δ 169.3 (COCH2NH), 157.6 (COOCH2Ph), 145.6, 140.1, 137.9, 130.4, 129.3, 128.8, 128.7, 122.9, 121.3, 117.1, 117.4 (Ar-C), 66.5 (OCH2Ph), 45.1 (CH2NH). HRMS m/z for C16H17N3O5S [M + H]+ calcd. 364.1, found 364.0; [M + NH4]+ calcd. 381.1, found 381.1; [M + Na]+ calcd. 386.1, found 386.0; [M − H]− calcd. 362.1, found 362.1; [M + Cl]− calcd. 398.1, found 398.0.
(S)-benzyl (1-oxo-1-((3-sulfamoylphenyl)amino)propan-2-yl)carbamate, 2
White solid (76%); silica gel TLC Rf = 0.20 (MeCOOEt/Hexane 50% v/v); mp 178–179 °C; 1H NMR (DMSO-d6, 400 MHz) 10.32 (s, 1H, NH), 8.24 (s, 1H, Ar-H), 7.79 (d,1H, Ar-H, J = 4.0 Hz), 7.68 (d, 1H, NH, J = 8.0 Hz), 7.57 (s, 2H, NH2), 7.52–7.36 (m, 7H, Ar-H), 5.08 (s, 2H, OCH2Ph), 4.23 (d, 1H, CHNH, J = 8.0 Hz), 1.35 (d, 3H, CH3, J = 8.0 Hz). 13C NMR (DMSO-d6, 400 MHz) δ 172.9 (COCH2NH), 156.7 (COOCH2Ph), 145.5, 140.2, 137.9, 130.3, 129.2, 128.7, 128.6, 123.0, 121.2, 117.2 (Ar-C), 66.4 (OCH2Ph), 51.8 (CHNH), 18.7 (CH3). HRMS m/z for C17H19N3O5S [M + H]+ calcd. 378.1, found 378.0; [M + NH4]+ calcd. 395.1, found 395.1; [M + Na]+ calcd. 400.1, found 400.0; [M − H]− calcd. 376.1, found 376.0; [M + Cl]− calcd. 412.1, found 412.1.
(S)-benzyl (1-oxo-3-phenyl-1-((3-sulfamoylphenyl)amino)propan-2-yl)carbamate, 3
White solid (86%); silica gel TLC Rf = 0.29 (MeOH/CH2Cl2 5% v/v); mp 139–140 °C; 1H NMR (DMSO-d6, 400 MHz) 10.46 (s, 1H, NH), 8.21 (s, 1H, Ar-H), 7.80–7.77 (m, 2H, Ar-H + NH), 7.56 (s, 2H, NH2), 7.40–7.25 (m, 12H, Ar-H), 5.01 (s, 2H, OCH2Ph), 4.48–4.43 (m, 1H, CHNH), 3.11–3.07 (m, 1H, CH2), 2.93–2.87 (m, 1H, CH2). 13C NMR (DMSO-d6, 400 MHz) δ 171.9 (COCH2NH), 156.9 (COOCH2Ph), 145.5, 140.1, 138.6, 137.8, 130.4, 130.1, 129.2, 129.0, 128.7, 128.5, 127.3, 123.1, 121.4, 117.3 (Ar-C), 66.3 (OCH2Ph), 57.9 (CHNH), 38.2 (CH2CH). HRMS m/z for C23H23N3O5S [M + H]+ calcd. 454.1, found 454.1; [M + NH4]+ calcd. 471.1, found 471.1; [M + Na]+ calcd. 476.1, found 476.1; [M − H]− calcd. 452.1, found 452.2; [M + Cl]− calcd. 488.1, found 488.2.
General procedure for the synthesis of amino acid–sulfonamide conjugates, 4–6
A mixture of equivalent amounts of the appropriate N-protected aminoacylbenzotriazole and 4-aminobenzenesulfonamide was subjected to microwave irradiation (100 W, 70 °C) in anhydrous THF (5 mL) for 30 min. After completion of the reaction, all volatiles were removed by rotavapour and the obtained crude product was crystallized from methanol.
Benzyl (2-oxo-2-((4-sulfamoylphenyl)amino)ethyl)carbamate, 4
White solid (84%); silica gel TLC Rf = 0.38 (MeOH/CH2Cl2 5% v/v); mp 186–187 °C; 1H NMR (DMSO-d6, 400 MHz) 10.34 (s, 1H, NH), 7.82–7.78 (m, 4H, Ar-H), 7.61 (t, 1H, NH, J = 8.0 Hz), 7.42–7.37 (m, 5H, Ar-H), 7.27 (s, 2H, NH2), 5.10 (s, 2H, OCH2Ph), 3.88 (d, 2H, CH2NH, J = 8.0 Hz). 13C NMR (DMSO-d6, 400 MHz) δ 169.4 (COCH2NH), 157.5 (COOCH2Ph), 142.7, 139.3, 137.9, 129.2, 128.7, 128.6, 127.6, 119.5 (Ar-C), 66.4 (OCH2Ph), 45.1 (CH2NH). HRMS m/z for C16H17N3O5S [M + H]+ calcd. 364.1, found 364.0; [M + Na]+ calcd. 386.1, found 386.0; [M − H]− calcd. 362.1, found 362.1; [M + Cl]− calcd. 398.1, found 398.0.
(S)-benzyl (1-oxo-1-((4-sulfamoylphenyl)amino)propan-2-yl)carbamate, 5
White solid (77%); silica gel TLC Rf = 0.25 (MeCOOEt/Hexane 50% v/v); mp 240–241 °C; 1H NMR (DMSO-d6, 400 MHz) 10.36 (s, 1H, NH), 7.81 (brs, 4H, Ar-H), 7.70 (d, 1H, NH, J = 8.0 Hz), 7.41–7.40 (m, 5H, Ar-H), 7.29 (s, 2H, NH2), 5.08 (s, 2H, OCH2Ph), 4.26 (t, 1H, CHNH, J = 8.0 Hz), 1.35 (d, 3H, CH3, J = 8.0 Hz). 13C NMR (DMSO-d6, 400 MHz) δ 173.1 (COCH2NH), 156.8 (COOCH2Ph), 152.8, 142.8, 139.4, 137.9, 129.3, 128.7, 128.6, 127.6, 119.7, 113.4 (Ar-C), 66.4 (OCH2Ph), 51.8 (CHNH), 18.8 (CH3). HRMS m/z for C17H19N3O5S [M + H]+ calcd. 378.1, found 378.0; [M + Na]+ calcd. 400.1, found 400.0; [M − H]− calcd. 376.1, found 376.1; [M + Cl]− calcd. 412.1, found 412.1.
(S)-benzyl (1-oxo-4-phenyl-1-((3-sulfamoylphenyl)amino)propan-2-yl)carbamate, 6
White solid (87%); silica gel TLC Rf = 0.32 (MeOH/CH2Cl2 5% v/v); mp 216–217 °C; 1H NMR (DMSO-d6, 400 MHz) 10.47 (s, 1H, NH), 7.84–7.78 (m, 6H, Ar-H + NH), 7.38–7.23 (m, 14H, Ar-H + NH2), 5.02 (s, 2H, OCH2Ph), 4.52–4.47 (m, 1H, CHNH), 3.12–3.07 (m, 1H, CH2), 2.95–2.89 (m, 1H, CH2). 13C NMR (DMSO-d6, 400 MHz) δ 172.0 (COCH2NH), 156.9 (COOCH2Ph), 142.6, 139.5, 138.5, 137.8, 130.1, 129.2, 129.0, 128.7, 128.5, 127.6, 127.3, 119.8 (Ar-C), 66.3 (OCH2Ph), 57.9 (CHNH), 38.3 (CH2CH). HRMS m/z for C23H23N3O5S [M + H]+ calcd. 454.1, found 454.1; [M + Na]+ calcd. 476.1, found 476.1; [M − H]− calcd. 452.1, found 452.2; [M + Cl]− calcd. 488.1, found 488.2.
General procedure for the synthesis of amino acid–sulfonamide conjugates, 7–9
A mixture of equivalent amounts of the appropriate N-protected aminoacyl-benzotriazole and (4-sulfamoylphenyl)methanaminium chloride was subjected to microwave irradiation (100 W, 70 °C) in anhydrous THF (5 mL) for 30 min. After completion of the reaction, all volatiles were removed by rotavapour and the obtained crude product was crystallized from methanol.
Benzyl (2-oxo-2-((4-sulfamoylbenzyl)amino)ethyl)carbamate, 7
White solid (95%); silica gel TLC Rf = 0.20 (MeOH/CH2Cl2 5% v/v); mp 182–183 °C; 1H NMR (DMSO-d6, 400 MHz) 8.51 (t, 1H, NH, J = 4.0 Hz), 7.89 (d, 2H, Ar-H, J = 8.0 Hz), 7.54 (t, 1H, NH, J = 8.0 Hz), 7.47–7.35 (m, 10H, Ar-H + NH2), 5.08 (s, 2H, OCH2Ph), 4.38 (d, 2H, CH2Ph, J = 4.0 Hz), 3.71 (d,2H, CH2NH, J = 8.0 Hz). 13C NMR (DMSO-d6, 400 MHz) δ 170.2 (COCH2NH), 157.4 (COOCH2Ph), 144.4, 143.5, 137.9, 129.2, 128.7, 128.6, 128.4, 126.5 (Ar-C), 66.4 (OCH2Ph), 44.5 (CH2Ph), 42.6 (CH2NH). HRMS m/z for C17H19N3O5S [M + H]+ calcd. 378.1, found 378.0; [M + NH4]+ calcd. 395.1, found 395.1; [M + Na]+ calcd. 400.1, found 400.0; [M + Cl]− calcd. 412.1, found 412.1; [M + HCOO]− calcd. 422.1, found 422.1
(S)-benzyl (1-oxo-1-((4-sulfamoylbenzyl)amino)propan-2-yl)carbamate, 8
White solid (93%); silica gel TLC Rf = 0.36 (MeOH/CH2Cl2 5% v/v); mp 181–182 °C; 1H NMR (DMSO-d6, 400 MHz) 8.55 (t, 1H, NH, J = 8.0 Hz), 7.80 (d, 2H, Ar-H, J = 8.0 Hz), 7.46–7.35 (m, 10H, Ar-H + NH + NH2), 5.07 (s, 2H, CH2Ph), 4.38 (d, 2H, CH2Ph, J = 8.0 Hz), 4.15–4.10 (m, 1H, CH), 1.28 (d, 3H, CH3, J = 4.0 Hz). 13C NMR (DMSO-d6, 400 MHz) δ 173.7 (COCH2NH), 156.7 (COOCH2Ph), 144.6, 143.5, 137.9, 129.3, 128.8, 128.7, 128.2, 126.6 (Ar-C), 66.4 (OCH2Ph), 51.2 (CH2Ph), 42.6 (CH), 19.2 (CH3). HRMS m/z for C18H21N3O5S [M + H]+ calcd. 392.1, found 392.1; [M + NH4]+ calcd. 409.1, found 409.2; [M + Na]+ calcd. 414.1, found 414.1; [M + Cl]− calcd. 426.1, found 426.1; [M + HCOO]− calcd. 436.1, found 436.1.
(S)-benzyl (1-oxo-3-phenyl-1-((4-sulfamoylbenzyl)amino)propan-2-yl)carbamate, 9
White solid (94%); silica gel TLC Rf = 0.29 (MeOH/CH2Cl2 5% v/v); mp 230–231 °C; 1H NMR (DMSO-d6, 400 MHz) 8.68 (t, 1H, NH, J = 8.0 Hz), 7.77 (d, 2H, Ar-H, J = 8.0 Hz), 7.64 (d,1H, NH, J = 8.0 Hz), 7.36–7.30 (m, 14H, Ar-H + NH2), 5.01 (s, 2H, OCH2Ph), 4.39–4.30 (m, 2H, CH2Ph + CH), 3.07–3.03 (m, 1H, CH2CH), 2.88–2.82 (m, 1H, CH2CH). 13C NMR (DMSO-d6, 400 MHz) δ 172.5 (COCH2NH), 156.8 (COOCH2Ph), 144.3, 143.6, 138.9, 137.9, 130.2, 129.2, 129.0, 128.6, 128.4, 128.3, 127.2, 126.5 (Ar-C), 66.2 (OCH2Ph), 57.3 (CH2Ph), 42.7 (CH), 38.4 (CH3). HRMS m/z for C24H25N3O5S [M + H]+ calcd. 368.2, found 468.1; [M + Na]+ calcd. 490.1, found 460.1; [M + Cl]− calcd. 502.1, found 502.2.1; [M + HCOO]− calcd. 512.1, found 512.2.
General procedure for the synthesis of amino acid–sulfonamide conjugates, 10–13
A mixture of equivalent amounts of the appropriate N-protected aminoacylbenzotriazole and 4-(2-aminoethyl)benzenesulfonamide was subjected to microwave irradiation (100 W, 70 °C) in anhydrous THF (5 mL) for 30 min. After completion of the reaction, all volatiles were removed by rotavapour and the obtained crude product was crystallized from methanol.
Benzyl (2-oxo-2-((4-sulfamoylphenethyl)amino)ethyl)carbamate, 10
Beige solid (95%); silica gel TLC Rf = 0.58 (MeOH/CH2Cl2 5% v/v); mp 161–162 °C; 1H NMR (DMSO-d6, 400 MHz) 7.99 (t, 1H, NH, J = 4.0 Hz), 7.78 (d, 2H, Ar-H, J = 8.0 Hz), 7.45–7.40 (m, 8H, Ar-H + NH), 7.33 (s, 2H, NH2), 5.08 (s, 2H, OCH2Ph), 3.61 (d, 2H, CH2Ph, J = 8.0 Hz), 3.33 (t, 2H, CH2CH2NH, J = 8.0 Hz), 2.83 (t, 2H, CH2CH2NH, J = 8.0 Hz). 13C NMR (DMSO-d6, 400 MHz) δ 170.0 (COCH2NH), 157.4 (COOCH2Ph), 144.6, 143.0, 138.0, 130.0, 129.3, 128.7, 128.6, 126.6 (Ar-C), 66.4 (OCH2Ph), 44.5 (CH2NH), 41.1 (CH2CH2Ph), 35.7 (CH2CH2Ph). HRMS m/z for C18H21N3O5S [M + H]+ calcd. 392.1, found 392.1; [M + NH4]+ calcd. 409.2, found 409.1; [M + Na]+ calcd. 414.1, found 414.0; [M + Cl]− calcd. 426.1, found 426.1; [M + HCOO]− calcd. 436.1, found 436.1.
tert-Butyl (2-oxo-2-((4-sulfamoylphenethyl)amino)ethyl)carbamate, 11
White solid (89%); silica gel TLC Rf = 0.60 (MeOH/CH2Cl2 30% v/v); mp 201–202 °C; 1H NMR (DMSO-d6, 400 MHz) 7.89 (t, 1H, NH, J = 4.0 Hz), 7.78 (d, 2H, Ar-H, J = 8.0 Hz), 7.43 (d, 2H, Ar-H, J = 8.0 Hz), 7.32 (s, 2H, NH2), 6.94 (t, 1H, NH, J = 4.0 Hz), 3.52 (d, 2H, CH2NH, J = 4.0 Hz), 3.34 (t, 2H, CH2CH2N, J = 8.0 Hz), 2.82 (t, 2H, NCH2CH2, J = 8.0 Hz), 1.42 (s, 9H, (CH3)3C). 13C NMR (DMSO-d6, 400 MHz) δ 170.2 (COCH2NH), 156.7 (COOCH2Ph), 144.6, 143.0, 130.0, 126.6 (Ar-C), 79.0 ((CH3)3CO, 44.2 (CH2NH), 40.9 (NHCH2CH2), 35.7 (NHCH2CH2), 29.1 ((CH3)3C). HRMS m/z for C15H23N3O5S [M + Na]+ calcd. 380.1, found 380.1; [M + Cl]− calcd. 392.1, found 392.1; [M + HCOO]− calcd. 402.1, found 402.1.
(S)-benzyl (1-oxo-1-((4-sulfamoylphenethyl)amino)propan-2-yl)carbamate, 12
Yellow solid (92%); silica gel TLC Rf = 0.46 (EtOAc/Hexane 30% v/v); mp 130–131 °C; 1H NMR (DMSO-d6, 400 MHz) 7.99 (t, 1H, NH, J = 8.0 Hz), 7.93 (d, 1H, NH, J = 4.0 Hz), 7.81–7.78 (m, 2H, Ar-H), 7.46–7.35 (m, 9H, Ar-H + NH2), 5.08 (s, 2H, OCH2Ph), 4.02 (t, 2H, CH2CH2Ph, J = 8.0 Hz), 3.39–3.31 (m, 1H, CHCH3), 2.82 (t, 2H, PhCH2CH2, J = 8.0 Hz), 1.20 (d, 3H, CH3, J = 8.0 Hz). 13C NMR (DMSO-d6, 400 MHz) δ 173.4 (COCH2NH), 155.7 (COOCH2Ph), 144.6, 143.0, 138.0, 130.1, 130.0, 129.3, 128.7, 126.6, 126.0, 115.9 (Ar-C), 66.3 (OCH2Ph), 51.1 (CHNH), 42.9 (NCH2CH2), 35.7 (CH2CH2Ph), 19.2 (CH3). HRMS m/z for C19H23N3O5S [M + H]+ calcd. 406.1, found 406.1; [M + Na]+ calcd. 428.1, found 428.1; [M + Cl]− calcd. 440.1, found 440.1; [M + HCOO]− calcd. 450.1, found 450.2.
(S)-benzyl (1-oxo-3-phenyl-1-((4-sulfamoylphenethyl)amino)propan-2-yl)carbamate, 13
Beige solid (97%); silica gel TLC Rf = 0.66 (MeOH/CH2Cl2 30% v/v); mp 195–196 °C; 1H NMR (DMSO-d6, 400 MHz) 8.15 (t, 1H, NH, J = 8.0 Hz), 7.77 (d, 2H, ArH, J = 4.0 Hz), 7.75 (d, 1H, NH, J = 4.0 Hz), 7.37–7.24 (m, 14H, Ar-H + NH2), 4.99 (s, 2H, OCH2Ph), 4.23–4.22 (m, 1H, NHCHCH2Ph), 3.36 (t, 2H, PhCH2CH2, J = 8.0 Hz), 3.54 (t, 2H, PhCH2CH2, J = 8.0 Hz), 2.85–2.76 (m, 3H, CH2), 2.97–2.93 (m, 1H, CHCH2), 2.81–2.78 (m, 3H, CHCH2 + PhCH2CH2) . 13C NMR (DMSO-d6, 400 MHz) δ 172.3 (COCH2NH), 156.7 (COOCH2Ph), 144.6, 143.0, 138.0, 139.0, 138.0, 130.1, 129.2, 129.0, 128.6, 128.4, 127.2, 126.6 (Ar-C), 66.2 (OCH2Ph), 57.2 (CHNH), 40.7 (CH2CH2Ph), 38.5 (NCHCH2), 35.7 (CH2CH2Ph). HRMS m/z for C25H27N3O5S [M + H]+ calcd. 482.2, found 482.2; [M + Na]+ calcd. 504.2, found 504.1; [M + Cl]− calcd. 516.1, found 516.2; [M + HCOO]− calcd. 526.2, found 526.3.
Benzyl (2-oxo-2-((2-(4-sulfamoylphenoxy)ethyl)amino)ethyl)carbamate, 14
A mixture of benzyl (2-(1H-benzo[d][1,2,3]triazol-1-yl)-2-oxoethyl)carbamate (0.12 g; 0.39 mmol), 2-(4-sulfamoylphenoxy)ethanaminium 2,2,2-trifluoroacetate (0.13 g; 0.39 mmol) and Et3N (0.06 mL; 0.43 mmol) was subjected to microwave irradiation (100 W, 70 °C) in anhydrous THF (5 mL) for 30 min. After completion of the reaction, all volatiles were removed by rotavapour and the obtained crude product was crystallized from methanol.
White solid (0.14 g, 93%); silica gel TLC Rf = 0.75 (MeOH/CH2Cl2 5% v/v); mp 183–184 °C; 1H NMR (DMSO-d6, 400 MHz) 7.99 (t, 1H, NH, J = 4.0 Hz), 7.78 (d, 2H, Ar-H, J = 8.0 Hz), 7.45–7.40 (m, 8H, Ar-H + NH), 7.33 (s, 2H, NH2), 5.08 (s, 2H, OCH2Ph), 3.61 (d, 2H, CH2Ph, J = 8.0 Hz), 3.33 (t, 2H, CH2CH2NH, J = 8.0 Hz), 2.83 (t, 2H, CH2CH2NH, J = 8.0 Hz). 13C NMR (DMSO-d6, 400 MHz) δ 170.0 (COCH2NH), 157.4 (COOCH2Ph), 144.6, 143.0, 138.0, 130.0, 129.3, 128.7, 128.6, 126.6 (Ar-C), 66.4 (OCH2Ph), 44.5 (CH2NH), 41.1 (CH2CH2Ph), 35.7 (CH2CH2Ph). HRMS m/z for C18H21N3O6S [M + Na]+ calcd. 430.1, found 430.0.
General procedure for the synthesis of amino acid–sulfonamide conjugates, 15 and 16
A mixture of equivalent amounts of the appropriate N-protected aminoacylbenzotriazole, 3-(4-sulfamoylphenoxy)propan-1-aminium chloride and Et3N was subjected to microwave irradiation (100 W, 70 °C) in anhydrous THF (5 mL) for 30 min. After completion of the reaction, all volatiles were removed by rotavapour and the obtained crude product was crystallized from methanol.
Benzyl (2-oxo-2-((3-(4-sulfamoylphenoxy)propyl)amino)ethyl)carbamate, 15
White solid (91%); silica gel TLC Rf = 0.5 (MeOH/CH2Cl2 5% v/v); mp 125–126 °C; 1H NMR (DMSO-d6, 400 MHz) 7.97 (t, 1H, NH, J = 4.0 Hz), 7.77 (d, 2H, Ar-H, J = 8.0 Hz), 7.46–7.35 (m, 6H, Ar-H + NH), 7.22 (s, 2H, NH2), 7.10 (d, 2H, Ar-H, J = 8.0 Hz), 5.06 (s, 2H, OCH2Ph), 4.09 (t, 2H, CH2CH2OPh, J = 8.0 Hz), 3.80 (d, 2H, CH2NH, J = 4.0 Hz), 3.27 (t, 2H, CH2CH2NH, J = 8.0 Hz), 1.91 (t, 2H, CH2CH2CH2, J = 8.0 Hz). 13C NMR (DMSO-d6, 400 MHz) δ 170.0 (COCH2NH), 161.9 (OC6H4), 157.4 (COOCH2Ph), 137.9, 137.0, 129.3, 128.7, 128.6, 128.5, 115.4 (Ar-C), 66.6 (OCH2Ph), 66.4 (CH2CH2O), 44.5 (CH2NH), 36.9 (NHCH2CH2), 29.6 (CH2CH2CH2). HRMS m/z for C19H23N3O6S [M + Na]+ calcd. 444.1, found 444.1; [M + Cl]− calcd. 456.1, found 456.1; [M + HCOO]− calcd. 466.1, found 466.1.
(S)-benzyl (1-oxo-3-phenyl-1-((3-(4-sulfamoylphenoxy)propyl)amino)propan-2-yl)carbamate, 16
White solid (89%); silica gel TLC Rf = 0.45 (MeOH/CH2Cl2 5% v/v); mp 183–184 °C; 1H NMR (DMSO-d6, 400 MHz) 8.11 (t, 1H, NH, J = 4.0 Hz), 7.77 (d, 2H, Ar-H, J = 8.0 Hz), 7.55 (d, 1H, NH, J = 8.0 Hz), 7.39–7.28 (m, 10H, Ar-H), 7.23 (s, 2H, NH2), 7.08 (d, 2H, Ar-H, J = 8.0 Hz), 4.99 (s, 2H, OCH2Ph), 4.22 (m, 1H, CHCH2), 4.01 (t, 2H, CH2CH2O, J = 8.0 Hz), 3.24 (m, 2H, CH2CH2NH), 3.01–2.96 (m, 1H, CHCH2), 2.81–2.78 (m, 1H, CHCH2), 1.86 (m, 2H, CH2CH2CH2). 13C NMR (DMSO-d6, 400 MHz) δ 172.2 (COCH2NH), 161.8 (OC6H4), 156.7 (COOCH2Ph), 138.9, 137.9, 137.0, 130.0, 129.2, 128.9, 128.6, 128.5, 128.0, 127.1, 115.3 (Ar-C), 66.6 (OCH2Ph), 66.1 (CHCH2), 57.2 (CH2CH2O), 38.6 (NHCH2CH2), 36.3 (CHCH2), 29.5 (CH2CH2CH2). HRMS m/z for C26H29N3O6S [M + H]+ calcd. 512.2, found 512.1; [M + Na]+ calcd. 534.2, found 534.1.
General procedure for the synthesis of amino acid–sulfonamide conjugates, 17–20
A mixture of equivalent amounts of the appropriate N-protected aminoacylbenzotriazole, 4-(4-sulfamoylphenoxy)butan-1-aminium 2,2,2-trifluoroacetate and Et3N was subjected to microwave irradiation (100 W, 70 °C) in anhydrous THF (5 mL) for 30 min. After completion of the reaction, all volatiles were removed by rotavapour and the obtained crude product was crystallized from methanol.
Benzyl (2-oxo-2-((4-(4-sulfamoylphenoxy)butyl)amino)ethyl)carbamate, 17
White solid (94%); silica gel TLC Rf = 0.55 (MeOH/CH2Cl2 5% v/v); mp 135–136 °C; 1H NMR (DMSO-d6, 400 MHz) 7.97 (t, 1H, NH, J = 4.0 Hz), 7.78 (d, 2H, Ar-H, J = 8.0 Hz), 7.45 (t, 1H, NH, J = 4.0 Hz), 7.40–7.36 (m, 5H, Ar-H), 7.23 (s, 2H, NH2), 7.11 (d, 2H, Ar-H, J = 8.0 Hz), 5.07 (s, 2H, OCH2Ph), 4.08 (t, 2H, CH2CH2OPh, J = 8.0 Hz), 3.63 (d, 2H, CH2NH, J = 4.0 Hz), 3.16 (q, 2H, CH2CH2NH, J = 8.0 Hz), 1.78–1.74 (m, 2H, CH2CH2O), 1.60–1.57 (m, 2H, NCH2CH2). 13C NMR (DMSO-d6, 400 MHz) δ 169.9 (COCH2NH), 162.0 (OC6H4), 157.4 (COOCH2Ph), 138.0, 137.0, 129.3, 128.7, 128.6, 128.5, 115.4 (Ar-C), 68.6 (OCH2Ph), 66.4 (CH2CH2O), 44.5 (CH2NH), 39.1 (CH2CH2NH), 26.9 (CH2CH2O), 26.6 (NCH2CH2). HRMS m/z for C20H25N3O6S [M + Na]+ calcd. 458.1, found 458.1; [M + Cl]− calcd. 470.1, found 470.1; [M + HCOO]− calcd. 480.1, found 480.1.
tert-butyl (2-oxo-2-((4-(4-sulfamoylphenoxy)butyl)amino)ethyl)carbamate, 18
White solid (88%); silica gel TLC Rf = 0.29 (EtOAc/Hexane 30% v/v); mp 75–76 °C; 1H NMR (DMSO-d6, 400 MHz) 7.80 (t, 1H, NH, J = 4.0 Hz), 7.77 (d, 2H, Ar-H, J = 8.0 Hz), 7.22 (s, 2H, NH2), 7.10 (d, 2H, Ar-H, J = 8.0 Hz), 6.93 (t, 1H, NH, J = 8.0 Hz), 4.08 (t, 2H, CH2CH2OPh, J = 8.0 Hz), 3.53 (d, 2H, CH2NH, J = 8.0 Hz), 3.16 (q, 2H, CH2CH2NH, J = 8.0 Hz), 1.78–1.74 (m, 2H, CH2CH2O), 1.60–1.56 (m, 2H, NCH2CH2), 1.42 (s, 9H, (CH3)3). 13C NMR (DMSO-d6, 400 MHz) δ 170.0 (COCH2NH), 161.9 (OC6H4), 156.8 (COOCH2Ph), 137.0, 128.6, 115.3 (Ar-C), 78.9 (OC(CH3)3), 68.7 (CH2CH2O), 52.2 (CH2NH), 44.2 (CH2CH2NH), 38.9 (CH2CH2O), 29.1 (NCH2CH2), 26.8 (CH3). HRMS m/z for C17H27N3O6S [M+] calcd. 401.2, found 401.2; [M + Na]+ calcd. 424.2, found 424.1; [M + HCOO]− calcd. 446.2, found 446.1.
(S)-benzyl (1-oxo-1-((4-(4-sulfamoylphenoxy)butyl)amino)propan-2-yl)carbamate, 19
Beige solid (93%); silica gel TLC Rf = 0.45 (EtOAc/Hexane 30% v/v); mp 76–77 °C; 1H NMR (DMSO-d6, 400 MHz) 8.29 (t, 1H, NH, J = 8.0 Hz), 7.78 (d, 2H, Ar-H, J = 8.0 Hz), 7.44–7.35 (m, 6H, Ar-H + NH), 7.22 (s, 2H, NH2), 7.10 (d, 2H, Ar-H, J = 8.0 Hz), 5.05 (s, 2H, OCH2Ph), 4.09–3.99 (m, 3H, CH2CH2OPh + CH), 3.16–3.12 (m, 2H, CH2CH2NH), 1.77–1.74 (m, 2H, CH2CH2O), 1.60–1.56 (m, 2H, NCH2CH2), 1.23 (d, 3H, CH3, J = 8.0 Hz). 13C NMR (DMSO-d6, 400 MHz) δ 173.2 (COCH2NH), 161.9 (OC6H4), 156.5 (COOCH2Ph), 138.0, 137.0, 129.2, 128.7, 128.6, 128.5, 115.3 (Ar-C), 68.6 (OCH2Ph), 66.2 (CH2CH2O), 51.1 (CHNH), 39.0 (CH2CH2NH), 26.8 (CH2CH2O), 26.5 (NCH2CH2), 19.3 (CH3). HRMS m/z for C21H27N3O6S [M + H]+ calcd. 450.2 Found 450.1; [M + Na]+ calcd. 472.2, found 472.1.
(S)-benzyl (1-oxo-3-phenyl-1-((4-(4-sulfamoylphenoxy)butyl)amino)propan-2-yl)carbamate, 20
White solid (97%); silica gel TLC Rf = 0.77 (MeOH/CH2Cl2 30% v/v); mp 197–198 °C; 1H NMR (DMSO-d6, 400 MHz) 8.04 (t, 1H, NH, J = 4.0 Hz), 7.78 (d, 2H, Ar-H, J = 8.0 Hz), 7.52(d, 1H, NH, J = 8.0 Hz), 7.37–7.28 (m, 10H, Ar-H), 7.23 (s, 2H, NH2), 7.11 (d, 2H, Ar-H, J = 8.0 Hz), 4.99 (s, 2H, OCH2Ph), 4.27–4.21 (m, 1H, CHCH2), 4.06 (t, 2H, CH2CH2O), 3.19–3.12 (m, 2H, CH2CH2N), 3.01–2.97 (m, 1H, CHCH2Ph), 2.84–2.78 (m, 1H, CHCH2Ph), 1.71 (t, 2H, CH2CH2O, J = 8.0 Hz), 1.55 (t, 2H, NCH2CH2, J = 8.0 Hz). 13C NMR (DMSO-d6, 400 MHz) δ 172.1 (COCH2NH), 162.0 (OC6H4), 156.7 (COOCH2Ph), 139.0, 138.7, 138.0, 137.0, 130.1, 129.0, 128.6, 128.4, 127.2, 115.4 (Ar-C), 68.6 (OCH2Ph), 66.1 (CHNH), 57.2 (CH2CH2O), 39.1 (CH2CH2NH), 26.8 (CH2CH2O), 26.5 (NCH2CH2). HRMS m/z for C27H31N3O6S [M + H]+ calcd. 526.2 Found 526.1; [M + Na]+ calcd. 548.2, found 548.1.
CA inhibition
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activity by using method of KhalifahCitation49. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM HEPES (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7–17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5%–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled–deionized water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were pre-incubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-square methods using PRISM (www.graphpad.com), and non-linear least squares methods, values representing the mean of at least three different determinations, as described earlier by usCitation50–56.
Results and discussion
New N-protected amino acid–sulfanamide conjugates (1–20) were synthesized by the treatment of the appropriate sulfonamide derivatives, incorporating primary amine moieties, with N-protected aminoacylbenzotriazoles under microwave heating at 70 °C for 30 min, with good yields of 83%–97%. The synthetic pathway of sulfonamides 1–20 is summarized in Scheme 1
Scheme 1. Synthesis pathways of the new sulfonamide conjugates of N-protected amino acids.
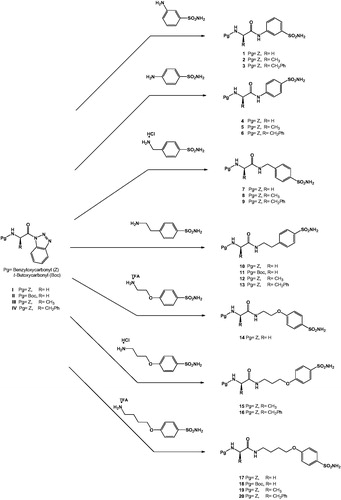
We have included in the study amino-sulfonamides having the NH2 group directly linked to the benzene ring on which the sulfonamide zinc-binding group is found (for example, metanilamide and sulfanilamide) as well as derivatives having various linkers between the benzenesulfonamide fragment and the primary amine group. These were of the alkylene type (methylene, ethylene) or longer ones, of the -(CH2)nO- type, with n varying between 2 and 4 (Scheme 1). The amino acids chosen for derivatization were Gly, Ala and Phe, in order to investigate whether this part of the inhibitor scaffold influences the CA inhibitory properties. Furthermore, the amino group from the amino acid reagent was protected with the benzyloxycarbonyl (Z) or tert-butyloxycarbonyl (Boc) protecting groups, as shown in Scheme 1.
The structures of N-protected amino acid–sulfonamide conjugates (1–20) were elucidated by 1H NMR, 13C NMR and mass spectrometric analyses. All spectral data were in agreement with the proposed structures. The characteristic NH resonances of the sulfonamide part of the conjugates 1–6 and 2–20 were observed between 10.32–10.47 and 7.80–8.68 ppm, respectively. Other amino group resonances for the sulfonamide conjugates 1–20 were observed between 6.93–8.24 ppm. Carbonyl resonances of the amide carbonyl and carbamate carbonyl were observed around 170 and 156 ppm, respectively. The characteristic NH2 resonances for sulfonamide functional group were observed between 7.22–7.57 ppm in the 1H NMR spectrum. All other aliphatic and aromatic protons and carbons were observed in the expected regions (see Materials and Methods).
The hCA I, II, IV and XII enzyme inhibitory activity of the N-protected amino acid–sulfonamide conjugates 1–20 are shown in . We have chosen these isforms as three of them (CA II, IV and XII) are antiglaucoma drug targetsCitation1,27–29 whereas CA I, due to its diffuse distribution in the blood and gastrointestinal tract is one of the main off-targets for such pharmacologic agentsCitation57.
Table 1. hCA I, II, IV and XII inhibition data with amino acid–sulfonamide conjugates 1–20, by a stopped-flow CO2 hydrase assayCitation49.
The following structure–activity relationship (SAR) can be observed from the inhibition data of .
hCA I was potently inhibited by sulfonamides 11, 14, 15, 17–19 (inhibition constants ranging between 5.6 and 24.5 nM), moderately inhibited by 6, 7, 9, 10, 12, 13 and 16 (KIs in the range of 45.9–102.4) and poorly inhibited by the remaining derivatives, which showed KIs in the range of 351.1–1946 nM (). Thus, best inhibition for this isoform was associated with compounds incorporating a long linker between the amino and benzenesulfonamide moieties (14, 15, 17–19), whereas the nature of the protecting group (Pg) seem to be less important for activity (compare the activity of 17–19). The Gly and Ala derivatives were generally much better hCA I inhibitors compared with the corresponding Phe derivatives. Acetazolamide (AZA, 5-acetamido-1,3,4-thiadiazole-2-sulfonamide), a clinically used compound, was a medium potency CAI for this isoform, with an inhibition constant of 250 nM.
Only three derivatives showed highly effective hCA II inhibitory activity, comparable to that of AZA, that is, 17–19 (KIs in the range of 8.6–11.9 nM). Again they incorporate the longest scaffold, being Gly or Ala derivatives with Z- or Boc-protecting groups. A number of other compounds (1, 4–6, and 8–15) were medium potency hCA II inhibitors, with KIs in the range of 24.5–97.2 nM (). They incorporate all the sulfonamide scaffolds employed in this study and this reveals that in this case the nature of the linker and that of the amino acid moiety/protecting groups are the most important factors influencing the CA II inhibitory power. The remaining derivatives (2, 3, 7 and 20) were the least effective inhibitors, with KIs in the range of 139–694 nM.
hCA IV, a membrane-anchored isoformCitation1, was the least sensitive CA among the investigated ones to inhibition by these compounds. A group of derivatives, 1, 4–6, 9, 13 and 17 showed inhibitory activity in the same range or better than AZA, with KIs in the range of 27.9–93.5 nM (KI of AZA is of 74 nM). Interestingly, the most potent CA IV inhibitor, 6, was a Phe derivative (also incorporating Z as protecting group and the homosufanilamide scaffold). Thus, apart 17 which has an elongated molecule, all the best inhibitors of this isoform had a more compact scaffold, being derivatives of metanilamide or homosufanilamide. The remaining derivatives were either inactive (2 and 20 had KIs > 10 μM) or were poorly active CAIs, with KIs in the range of 174.9–1453 nM ().
hCA XII, a transmembrane isoforms with an out of the cell active siteCitation1 was more sensible to inhibition by the reported sulfonamides, which with few exceptions (8, 9, 11 and 18) had KIs < 100 nM. The least active compounds, mentioned above, were the two homosulfanilamide derivatives 8 and 9 incorporating Ala and Phe moieties, and had similar KIs of 101.8–109.0 nM, and two Boc-protected Gly derivatives incorporating longer scaffolds, 11 and 18, which showed KIs of 803.1–951.2 nM. The most effective hCA XII inhibitors (similar to AZA) were 3, 10 and 13, with KIs in the range of 9.5–10.3 nM. They incorporate either metanilamide or 4-aminoethyl-benzenesulfonamide scaffolds, and Gly or Phe moieties. The remaining sulfonamides showed a rather flat SAR, with KIs of 34.7–96.3 nM, proving that a lot of scaffolds and substitution patterns lead to effective (but not highly potent) hCA XII inhibitors.
Some of the new compounds showed rather good selectivity levels for inhibiting hCA XII over the other isoforms (for example, 3, 10, 12 and 13) but generally no highly isoform-selective inhibition profiles were detected for these derivatives ().
Conclusions
A series of N-protected amino acids (Gly, Ala and Phe protected with Z or Boc groups) were reacted with sulfonamide derivatives, leading to the corresponding N-protected amino acid–sulfonamide conjugates in high yields (85%–98%). The carbonic anhydrase (CA, EC 4.2.1.1) inhibitory activity of the new sulfonamides was assessed against human (h) isoforms involved in serious pathologies (glaucoma, cancer, etc.), such as hCA I, hCA II, hCA IV and hCA XII. Among these four isoforms, hCA II, IV and XII are antiglaucoma drug targets, as all of them are involved in aqueous humor secretion within the eye. Low nanomolar inhibition was observed against all these CAs with the 20 prepared sulfonamides, but no selective inhibitory profiles were observed, except for few CA XII-selective derivatives. hCA I, II and XII were generally better inhibited by sulfonamides incorporating longer scaffolds and Gly/Ala, whereas the best hCA IV inhibitors were homosulfanilamide derivatives, incorporating Phe moieties. As the amino acid–sulfonamide conjugates show good water solubility and effective hCA II, IV and XII inhibition, they may be considered as interesting candidates for antiglaucoma studies.
Acknowledgements
We thank İnönü University, Turkey (BAPB), Università degli Studi di Firenze, Italy, and the European Union of the 7th Framework Program Dynano for financial support.
Declaration of interest
The authors declare no conflict of interest.
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77
- Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32
- Supuran CT, Capasso C. The η-class carbonic anhydrases as drug targets for antimalarial agents. Expert Opin Ther Targets 2015;19:551–63
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74
- Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO(2) capture. J Enzyme Inhib Med Chem 2013;28:229–30
- Capasso C, Supuran CT. Sulfa and trimethoprim-like drugs – antimetabolites acting as carbonic anhydrase, dihydropteroate synthase and dihydrofolate reductase inhibitors. J Enzyme Inhib Med Chem 2014;29:379–87
- Supuran CT. Bacterial carbonic anhydrases as drug targets: toward novel antibiotics? Front Pharmacol 2011;2:34
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–89
- Del Prete S, Vullo D, De Luca V, et al. Biochemical characterization of the δ-carbonic anhydrase from the marine diatom Thalassiosira weissflogii, TweCA. J Enzyme Inhib Med Chem 2014;29:906–11
- De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2015. [Epub ahead of print]. DOI: 10.3109/14756366.2015.1122001
- Ferraroni M, Carta F, Scozzafava A, Supuran CT. Thioxocoumarins inhibit carbonic anhydrases through a different mechanism compared to coumarins. J Med Chem 2016;59:462--73
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62
- Touisni N, Maresca A, McDonald PC, et al. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J Med Chem 2011;54:8271–7
- Bonneau A, Maresca A, Winum JY, Supuran CT. Metronidazole-coumarin conjugates and 3-cyano-7-hydroxy-coumarin act as isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2013;28:397–401
- Sharma A, Tiwari M, Supuran CT. Novel coumarins and benzocoumarins acting as isoform-selective inhibitors against the tumor-associated carbonic anhydrase IX. J Enzyme Inhib Med Chem 2014;29:292–6
- Kücükbay FZ, Küçükbay H, Tanc M, Supuran CT. Synthesis and carbonic anhydrase inhibitory properties of amino acid – coumarin/quinolinone conjugates incorporating glycine, alanine and phenylalanine moieties. J Enzyme Inhib Med Chem 2015. [Epub ahead of print]. DOI:10.3109/14756366.2015.1113173
- Isik S, Vullo D, Bozdag M, et al. 7-Amino-3,4-dihydro-1H-quinoline-2-one, a compound similar to thesubstitutedcoumarins, inhibits α-carbonic anhydrases without hydrolysis of the lactam ring. J Enzyme Inhib Med Chem 2015;30:773–7
- D’Ambrosio K, Carradori S, Monti SM, et al. Out of the active site binding pocket for carbonic anhydrase inhibitors. Chem Commun 2015;51:302–5
- Alp C, Ozsoy S, Alp NA, et al. Sulfapyridine-like benzenesulfonamide derivatives as inhibitors of carbonic anhydrase isoenzymes I, II and VI. J Enzyme Inhib Med Chem 2012;27:818–24
- Ekinci D, Fidan I, Durdagi S, et al. Kinetic and in silico analysis of thiazolidin-based inhibitors of α-carbonic anhydrase isoenzymes. J Enzyme Inhib Med Chem 2013;28:370–4
- Abdel-Aziz AA, El-Azab AS, Ekinci D, et al. Investigation of arenesulfonyl-2-imidazolidinones as potent carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:81–4
- Arslan M, Şentürk M, Fidan I, et al. Synthesis of 3,4-dihydroxypyrrolidine-2,5-dione and 3,5-dihydroxybenzoic acid derivatives and evaluation of the carbonic anhydrase I and II inhibition. J Enzyme Inhib Med Chem 2015;30:896–900
- Le Darz A, Mingot A, Bouazza F, et al. Fluorinated pyrrolidines and piperidines containing tertiary benzenesulfonamides: selective carbonic anhydrase II inhibitors. J Enzyme Inhib Med Chem 2015;30:737–45
- Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47
- Carta F, Supuran CT, Scozzafava A. Novel therapies for glaucoma: a patent review 2007–2011. Expert Opin Ther Pat 2012;22:79–88
- Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16
- Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin Ther Pat 2013;23:681–91
- Supuran CT. Carbonic anhydrase inhibitors as emerging drugs for the treatment of obesity. Expert Opin Emerg Drugs 2012;17:11–5
- Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35
- Supuran CT. The safety and clinical efficacy of acetazolamide for the treatment of idiopathic intracranial hypertension. Expert Rev Neurother 2015;15:851–6
- De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810
- Monti SM, Supuran CT, De Simone G. Anticancer carbonic anhydrase inhibitors: a patent review (2008–2013). Expert Opin Ther Pat 2013;23:737–49
- Lou Y, McDonald PC, Oloumi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res 2011;71:3364–76
- Pacchiano F, Carta F, McDonald PC, et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902
- Pettersen EO, Ebbesen P, Gieling RG, et al. Targeting tumour hypoxia to prevent cancer metastasis. From biology, biosensing and technology to drug development: the METOXIA consortium. J Enzyme Inhib Med Chem 2015;30:689–721
- Carta F, Di Cesare Mannelli L, Pinard M, et al. A class of sulfonamide carbonic anhydrase inhibitors with neuropathic pain modulating effects. Bioorg Med Chem 2015;23:1828–40
- Di Cesare Mannelli L, Micheli L, Carta F, et al. Carbonic anhydrase inhibition for the management of cerebral ischemia: in vivo evaluation of sulfonamide and coumarin inhibitors. J Enzyme Inhib Med Chem 2015. [Epub ahead of print]. DOI: 10.3109/14756366.2015.1113407
- Bejaoui M, Panatzi E, De Luca V, et al. Acetazolamide protects steatotic liver grafts against cold ischemia reperfusion injury. J Pharmacol Exp Ther 2015;355:191–8
- Panda SS, Ibrahim MA, Küçükbay H, et al. Synthesis and antimalarial bioassay of quinine peptide conjugates. Chem Biol Drug Des 2013;82:361–6
- Panda SS, Hall CD, Scriven E, Katritzky AR. Aminoacyl benzotriazolides: versatile reagents for the preparation of peptides and their mimetics and conjugates. Aldrichimica Acta 2013;46:43–5
- Khatib ME, Jauregui L, Tala SR, et al. Solution-phase synthesis of chiral O-acyl isodipeptides. Med Chem Comm 2011;2:1087–92
- Ibrahim MA, Panda SS, Oliferenko AA, et al. Macrocyclic peptidomimetics with antimicrobial activity: synthesis, bioassay, and molecular modeling studies. Org Biomol Chem 2015;13:9492–3
- Bozdag M, Pinard M, Carta F, et al. A class of 4-sulfamoylphenyl-ω-aminoalkyl ethers with effective carbonic anhydrase inhibitory action and antiglaucoma effects. J Med Chem 2014;57:9673–86
- Nicolaides E, DeWald H, Lipnik M, et al. Potential antiviral agents. Carbobenzoxy di- and tripeptides active against measles and herpes viruses. J Med Chem 1968;11:74–9
- Trager SF, Blackburn MG. Preparation of 4-(aminoacylamino)benzenesulfonamides useful for treatment of glaucoma. Eur Pat Appl EP399586A119901128; 1990
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73
- Casini A, Scozzafava A, Mincione F, et al. Carbonic anhydrase inhibitors: water-soluble 4-sulfamoylphenylthioureas as topical intraocular pressure-lowering agents with long-lasting effects. J Med Chem 2000;43:4884–92
- Del Prete S, Vullo D, De Luca V, et al. Biochemical characterization of recombinant β-carbonic anhydrase (PgiCAb) identified in the genome of the oral pathogenic bacterium Porphyromonas gingivalis. J Enzyme Inhib Med Chem 2015;30:366–70
- Maresca A, Vullo D, Scozzafava A, Supuran CT. Inhibition of the alpha- and beta-carbonic anhydrases from the gastric pathogen Helycobacter pylori with anions. J Enzyme Inhib Med Chem 2013;28:388–91
- Maresca A, Vullo D, Scozzafava A, et al. Inhibition of the beta-class carbonic anhydrases from Mycobacterium tuberculosis with carboxylic acids. J Enzyme Inhib Med Chem 2013;28:392–6
- Scozzafava A, Passaponti M, Supuran CT, Gulcin I. Carbonic anhydrase inhibitors: guaiacol and catechol derivatives effectively inhibit certain human carbonic anhydrase isoenzymes (hCA I, II, IX and XII). J Enzyme Inhib Med Chem 2015;30:586–91
- De Luca V, Del Prete S, Supuran CT, Capasso C. Protonography, a new technique for the analysis of carbonic anhydrase activity. J Enzyme Inhib Med Chem 2015;30:277–82
- Del Prete S, De Luca V, Vullo D, et al. Biochemical characterization of the γ-carbonic anhydrase from the oral pathogen Porphyromonas gingivalis, PgiCA. J Enzyme Inhib Med Chem 2014;29:532–7
- Durdagi S, Vullo D, Pan P, et al. Protein-protein interactions: inhibition of mammalian carbonic anhydrases I-XV by the murine inhibitor of carbonic anhydrase and other members of the transferrin family. J Med Chem 2012;55:5529–35