
Abstract
Multidrug resistance (MDR) has emerged as the main problem in anti-cancer therapy. Although MDR involves complex factors and processes, the main pivot is the expression of multidrug efflux pumps. P-glycoprotein (P-gp) belongs to the family of adenosine triphosphate (ATP)-binding cassette (ABC) transporters. It functions in cellular detoxification, pumping a wide range of xenobiotic compounds out of the cell. An attractive therapeutic strategy for overcoming MDR is to inhibit the transport function of P-gp and thus, increase intracellular concentration of drugs. Recently, various types of P-gp inhibitors have been found and used in experiments. However, none of them has passed clinical trials due to their high side-effects. Hence, the search for alternatives, such as plant-based P-gp inhibitors have gained attention recently. Therefore, we give an overview of the source, function, structure and mechanism of plant-based P-gp inhibitors and give more attention to cancer-related studies. These products could be the future potential drug candidates for further research as P-gp inhibitors.
Introduction
Some cancer patients can be cured by chemotherapy, immunotherapy and biological-response modifiers, but others respond transiently or do not respond at all to these approachesCitation1–4. Despite recent advances in cancer drug therapy, most cancer patients eventually end up in treatment failureCitation5. Host and tumor-genetic alterations, epigenetic changes and tumor environment, all seem to contribute to the complex process of cancer drug resistance, i.e. the main cause of drug failure is drug resistanceCitation1.
Multidrug resistance (MDR) is described as a phenomenon by which resistance to drugs is accompanied by resistance to other drugs whose structures and mechanisms of action may be completely differentCitation6. In tumorCitation7,Citation8, MDR is usually associated with an adenosine triphosphate (ATP)-dependent decrease in cellular drug accumulation, which is attributed to the over-expression of certain ATP-binding cassette (ABC) transporter proteinsCitation9.
P-glycoprotein (P-gp), the key member of the MDR family proteins, was first characterized in MDR Chinese hamster ovary (CHO) cells by Ling et al.Citation10. It is reported to be the best characterized efflux pump that mediates MDR, and it belongs to the ABC protein superfamilyCitation11. The function of P-gp is to prevent exogenous substances from accumulating in cells by transporting a wide variety of structurally and functionally unrelated compounds from the cell interior into the extracellular space. The energy required for the efflux is supplied by ATP hydrolysisCitation12.
P-glycoprotein inhibitors, the reversal of MDR through direct or indirect interaction with P-gp, are classified into three generations. The classification is based on their specificity, toxicity and affinityCitation13. The first-generation inhibitors have been used in clinics, and exhibited inherent pharmacological activitiesCitation14. These include verapamilCitation15, nifedipinCitation16, cyclosporin ACitation17, ibuprofenCitation18, reserpineCitation19, amiodaroneCitation20 and tamoxifenCitation21 (). However, the usage of these compounds is limited by their toxicity due to the high serum concentrations achieved with the dose required to inhibit P-gp. A greater deal of research by academicians in the direction of decreasing potential toxicity profile and improving specificity resulted in the second- and third-generation inhibitors. Second-generation inhibitors have decreased pharmacological activity but increased affinity than the first-generation inhibitorsCitation22. However, inhibition of two or more ABC transporters leads to complicated drug–drug interactions by this class of compounds. These inhibitors include non-immunosuppresive analogs of cyclosporin A, valspodarCitation23; d-isomer of verapamil, dexverapamilCitation24. Others are MM36Citation25, cinchonineCitation26 and quinine homodimer Q2Citation27 (). In order to avoid the complications associated with second-generation inhibitors, novel third-generation inhibitors were developed. This generation of P-gp inhibitors was shown to be more specific for P-gp; hence, avoiding interference with the pharmacokinetics (bioavailability, excretion, etc) of other drugsCitation13. These P-gp inhibitors include but not limited to tariquidar (XR9576)Citation28, zosuquidar (LY335979)Citation29, elacridar (GF120918)Citation30 and PGP4008Citation31 (). Although proven to be better than second-generation inhibitors, third generation of P-gp inhibitors could not “stand the heat” in clinical studies. For example, the XR9576 was stopped in Phase II trial, because a higher proportion of adverse events were reported in its clinical researchCitation32. This suggests that the search for more effective P-gp inhibitors is undoubtedly needed.
Table 1. Three classifications of P-gp inhibitors.
Plant-based medicines have long been used in the treatment of tumorsCitation33. They are reported to have numerous advantages which include but not limited to low cost, lower toxicity and a wider number of targets. Several plant-based compounds and plant extracts have been reported to inhibit P-gp and modulate MDRCitation34. Based on basic structure, plant-based compounds are classified into flavonoids, alkaloids, coumarins, terpenoids and sterides.
Therefore, in this review, we will discuss these compounds and their sources, functions, structure and mechanism in relation to their P-gp inhibitory activities, giving more attention to cancer-related studies.
P-glycoprotein
Structure, distribution and function
P-glycoprotein consists of 1280 amino acids and has two homologous halves, each containing a transmembrane domain (TMD1 and TMD2), which contain six membrane spanning (TM) segments (TM1–6 on TMD1, and TM7–12 on TMD2), and two nucleotide binding domains, NBD1 and NBD2, which includes an ATP site, located in the cytosol. Also, there are three glycosylation sites at the first extracellular loop in the N terminalCitation35–39 ().
Figure 1. Predicted secondary structure of P-gp and different mechanisms of P-gp inhibition. TMD – transmembrane domain; TM – membrane spanning; NBD – nucleotide binding domains; the ATP sites are rectangles; the three black lines represent glycosylation sites.
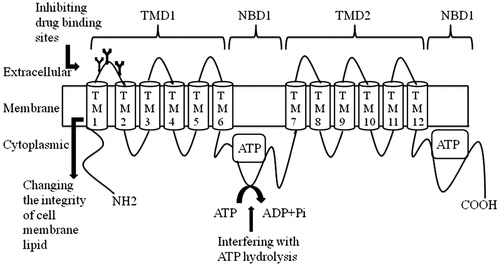
The expression of P-gp was identified in a wide range of non-neoplastic tissues in humans and other mammalian organsCitation40–42. These organs include liverCitation43, pancreasCitation44, kidneyCitation45, intestineCitation46, adrenal glandCitation47 and brainCitation48 ().
Table 2. P-gp expression in different organs.
Due to selective distribution at the port of drug entry and exit, P-gp has been speculated to play a major physiological role in absorption, distribution and excretion of xenobioticsCitation49. P-gp mainly functions as exogenous substances entry barrier and as a vacuum cleaner that expels xenobiotics from the brain, liver, kidney, intestine, etc.
Mechanism of P-glycoprotein efflux
The exact mechanism by which P-gp combines ATP hydrolysis with the movement of xenobiotics across the plasma membrane, to and out of cell and the exact site of substrate interaction with the protein, are not well definedCitation50. However, there are various models which provide clues that can explain the mechanism of P-gp. There are three prevalent models, namely, the pore model, flippase model and hydrophobic vacuum cleaner (HVC) model. The HVC model combines the features of pore and flippase models. In HVC model, P-gp can identify the parcel on the inside of the cell membrane substrate molecule and transfer them through protein channels to the serous lateralCitation51. These models assumed that P-gp substrates divide into the lipid phase prior to interacting with the protein, which would help to explain the P-gp broad substrate specificity.
There are also different models to explain the translocation process of P-gp. But, all models can be summarized into four distinct stages as follows: (i) drugs and one molecule of ATP combine with TMD and NBD dimerization, (ii) reorientation of the TMD from high to low affinity, (iii) hydrolysis of ATP into adenosine diphosphate (ADP) and subsequent production of energy for drugs excretion from the membrane and (iv) termination of the cycle when ADP fell from P-gpCitation52. The next catalytic cycle begins with another molecule of ATP hydrolysis, which provides energy for TMD reset.
Mechanism of P-glycoprotein inhibitors
P-glycoprotein substrates are capable of binding to P-gp specifically or non-specificallyCitation53. Drugs, such as antibiotics, alkaloids, peptides, steroid hormones and local anesthetics are all substrates of P-gp that are widely used in the clinicsCitation54. The physicochemical properties of these substrates are not the same. Most of them are strongly lipophilic, often contain aromatic rings and passively diffuse through cell membraneCitation55. Some P-gp substrates are capable of inhibiting the efflux pump function of P-gp or reversing the MDR of tumor cellsCitation56. The therapeutic use of P-gp inhibitors to improve drug bioavailability in tumor chemotherapy by inhibiting P-gp in the intestine, liver and kidney, has gained considerable interestsCitation49. In fact, since the recognition that P-gp-mediated drug resistance is clinically important, a concerted effort to screen for P-gp inhibitors has extensively dominated researchCitation57. The following sections, throw more light on the mechanisms of P-gp inhibitors.
P-glycoprotein inhibitors can inhibit the function of P-gp through three main mechanisms (): (I) Inhibition using either the competitive, non-competitive or allosterism drug binding sitesCitation56,Citation58. Examples include the drugs such as, competitive inhibitors; vinblastineCitation59, nitrendipineCitation59 and prazosinCitation60; non-competitive inhibitors; amoxapineCitation22 and loxapinCitation22, and allosteric sites; verapamilCitation59. (II) By interfering with ATP hydrolysisCitation61. For example, quercitin, a naturally occurring flavanoid, has been proposed to block P-gp function by interfering with ATPase activityCitation62. (III) By changing the integrity of cell membrane lipids, so that the inhibitors can interfere with membrane fluidity. These types of P-gp inhibitors are pharmaceutical excipients that can change the integrity of cell membrane lipidsCitation63. Examples include poly(ethylene glycol)-300, Tween-80, Cremophor EL, etcCitation63.
Plant-based P-glycoprotein inhibitors
Flavonoids
Flavonoids are a group of natural substances that possess a 2-phenylchromone with substituent groups attached at position 2, 3 or 4, and found in fruits, grains, bark, roots, stems, flowers, tea and wineCitation64,Citation65. Based on different substitution, attached position of ring B and the oxidation status of ring C, flavonoids are divided into many sub-classes: chalcones, flavones, flavonols, flavanones, flavanols, anthocyanins and isoflavonesCitation65. Many plant extracts that are rich in flavonoids have been proved capable of modulation activity and expression of P-gpCitation66.
For instance, Zuccagnia punctata (ZpE) Cav. (Fabaceae), is a monotypic species widely distributed in western Argentina. ZpE and two of its components: 3,7-dihydroxyflavone (DHF) (1) and 2′,4′-dihydroxychalcone (DHC) (2) (, ) have been reported to be modulators of both the expression and activity of P-gp. The rhodamine123 assay, an indicator to show the effects of different treatments on the efflux of P-gp, performed on the human proximal tubule cell line (HK-2) exposed to DHF and DHC shows a significant dose-dependent increase of intracellular fluorescence. Furthermore, the IC50 values for the inhibitory effects on P-gp activity by DHF and DHC were 3.2 and 6.0 μg/mL, respectivelyCitation67.
Figure 2. The structure of flavonoids that can inhibit P-gp. A: the basic flavonoids structure.
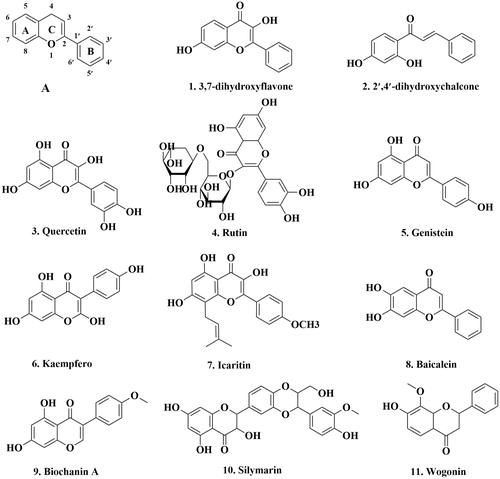
Table 3. An overview of flavonoid inhibitors from in vitro studies.
Another compound, quercetin 3 (, ), isolated from Ginkgo biloba leaves has been shown to strongly inhibit P-gp mediated transport of fluorescent probe Hoechst 33342, a substrate of P-gp, by inhibiting the ATPase activity up to about 33% with 25 μM quercetinCitation75,Citation62. Shapiro and LingCitation62 hypothesized that the effect of quercetin on drug efflux from MDR CHRC5 cells (Chinese hamster ovary cells) has no direct effect on P-gp. Futhermore, Limtrakul et al.Citation68 reported that treatment with 30 μM quercetin was able to significantly decrease P-gp level in MDR human cervical carcinoma cell line (KB-V1).
To add, rutin 4 (, ) was isolated from Ruta graveolens grass naphthaCitation76. Zhang et al.Citation76 reported that rutin decreased the bioavailability of cyclosporine through activation of P-gp. Thus, P-gp is involved in the transmembrane transport and intracellular accumulation of rutin in human colonic cancer cells (Caco-2). Yu and co-researchersCitation69 have found that rutin significantly decreased the accumulation of rhodamine 123 in LS 180, a human colon adenocarcinoma cell line, indicating a possible activation of P-gp.
Additionally, genistein (GNT) 5 (, ) is ingested as a component of vegetables (e.g. soy beans) or as a dietary supplementsCitation77. Castro and AltenbergCitation70 reported that GNT inhibited rhodamine123 efflux in human breast cancer cell lines (MCF-7). GNT also decreased photo-affinity labeling of P-gp with [3H]azidopine, a P-gp substrate, suggesting that GNT could block P-gp-mediated drug efflux by direct interaction with P-gp.
Similarly, kaempfero 6 (, ) from Kaempferia galanga L. root caused a decrease in P-gp levels in human glioblastoma cell line (T98G). A study by Nakatsuma and colleaguesCitation78 revealed that kaempferol reduced P-gp expression and function resulting in the inhibition of P-gp activity. Limtrakul et al.Citation68 also reported that treatment with 30 μM kaempfero was able to significantly decrease the P-gp level KB-V1 cells.
In a similar study, icaritin 7 (, ), a hydrolytic product of icariin, was isolated from Herba epimediu. Icaritin significantly increased the intracellular accumulation of adriamycin (ADR) and decreased the expression of MDR1 gene in HepG2 (a human hepatocellular carcinoma (HCC) cell line)/ADR cell line. Although the HepG2/ADR cell line was developed by treating the cells with ADR only, it was observed that MDR was achieved when assessed with other anti-cancer drugs, such as vincristine (VCR), cisplatin and 5-fluorouracil. Futhermore, the icaritin-mediated reversal of HepG2/ADR cell resistance to ADR was observed at a concentration of 1, 15 and 30 μMCitation71.
In another study, baicalein 8 (, ) was isolated from the roots of Scutellaria. Li et al.Citation72 found that oral administration tamoxifen (10 mg/kg) to rats in the presence of baicalein (0.5, 3, and 10 mg/kg) significantly increased the oral bioavailability of tamoxifen from 47.5 to 89.1%. This was attributed to inhibition of the CYP3A (cytochrome P450, family 3, subfamily A) – mediated metabolism of tamoxifen and inhibition of the P-gp efflux pump in the small intestine.
Last but not the least, biochanin A 9 and silymarin 10 (, ) were isolated from the bark of Aesculus hippocastanum L. and seeds of Milk Thistle, respectively. Zhang and MorrisCitation79,Citation73 reported that at 1.5 h, accumulation of digoxin and vinblastine in Caco-2 significantly increased and the AP-to-BL (from apical side to basolateral side in Caco-2 tranwell assay) transport of digoxin also significantly increased with 50 μM biochanin A or silymarin. This indicates that biochanin A and silymarin can inhibit P-gp-mediated efflux in Caco-2 cells, and may potentially increase the absorption/bioavailability of co-administered drugs that are P-gp substrates.
Lastly, wogonin 11 (, ), was extracted from the roots of Scutellaria baicalensi Georgi. Wogonin was reported to significantly potentiate etoposide-induced apoptosis in human leukemia cell line (HL-60). This effect was attributed to impairment of P-gp function and subsequent increase in cellular content of etoposide in the cells. The potentiation by wogonin is likely to be a specific action for cancer cells, which was also observed by Lee et al.Citation74. Consequently, it is suggested that wogonin may be used to reduce the excretion of the anti-cancer agents via P-gp and enhance their pharmacological actions in cancer cells.
Structural features of flavonoids are found to contribute positively to P-gp inhibition. For instance, it is reported that a hydroxyl group in position 5, double bond between positions 2 and 3, and a methoxyl group in position 3 are required for their P-gp inhibitory activities. Also, the exchange of a 3-methoxy group by an OH group can decrease their activityCitation80. This structural-activity relationship of flavonoids has also been reported by other researchers. For instance, Boumendjel et al.Citation66 have reported that 3-prenylchalcone binds to NBD2 with 20-fold higher affinity compared to the unprenylated chalcone.
As discussed above, flavonoids have significant inhibitory activities on expression of P-gp in various tumor models. While herb–drug interactions may set potential limits flavonoids co-administered with other drugs, opportunities exist for the combination of flavonoids with suitable anti-cancer drugs to improve therapeutic index and bioavailability of such anti-cancer drugs and reduce the dose to minimize side effects associated with anti-cancer drugsCitation81. Also, bioavailability of flavonoids can be influenced by factors, such as pH of the gastrointestinal tractCitation82, low hydrophobicityCitation83 and CYP enzymesCitation84; hence, future scientific research to overcome these challenges are needed to explore the vast pharmacological activities of plant-derived flavonoids.
Alkaloids
Alkaloids are natural nitrogen-containing secondary metabolites found in plants, fungi and bacteriaCitation85. The vast majority of alkaloids are reported to be present in higher plants especially in gymnosperms and angiosperms and a few exist in lower plants. A fundamental characteristic of alkaloids is the possession of nitrogen, which can be derived from amino acids or basic structure of purine or amination of sterides and terpenesCitation85. Many researchers have reported on the modulatory effects of alkaloids on P-gp, and so we discuss these effects as follows.
Glaucine 12 (, ), is an isoquinoline alkaloid, isolated from the stems of Corydalis yanhusuo. Glaucine inhibits P-gp mediated efflux and activates ATPase activities of transporters, indicating that it is a substrate of and can competitively inhibit P-gp. Furthermore, glaucine has been shown to decrease IC50 values of the MCF-7/ADR cells to adriamycin and mitoxantrone in a dose-dependent manner (12.5, 25 and 50 μM)Citation86.
Figure 3. The structure of alkaloids that can inhibit P-gp. B: the basic alkaloids structure.
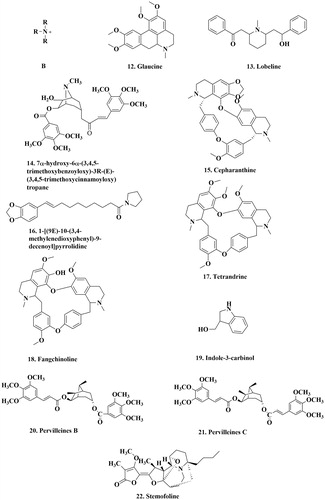
Table 4. An overview of alkaloid inhibitors from in vitro studies.
Lobeline 13 (, ), a piperidine alkaloid from Lobelia inflata, has been reported to specifically inhibit P-gp activity. It reverses doxorubicin resistance in Caco-2 and CEM ADR5000 [a P-gp over-expressing sub-line of CCRF-CEM (the human acute lymphocytic leukemia T lymphocytes)] in vitro at non-toxic concentration, at 10 μMCitation87.
In another study, tropane alkaloid esters were isolated from the stems of Erythroxylum rotundifolium. One of them is 7a-hydroxy-6a-(3,4,5-trimethoxybenzoyloxy)-3R-(E)-(3,4,5-trimethoxycinnamoyloxy) tropane 14 (, ). This compound has been demonstrated to have a significant biological activity on KB-V1 cells incubated in the presence of vinblastine. Thus, tropane esters of this type can reverse the MDR phenotype, presumably via interaction with P-gpCitation88.
Cepharanthine 15 (, ) is a bisbenzylisoquinoline alkaloid extracted from the roots of Stephania cepharantha HayataCitation95. Ikeda et al.Citation89 showed that cepharanthine is an effective agent for the reversal of resistance in human chronic myelogenous leukemia cell line (K562). It has also enhanced the sensitivity to doxorubicin and VCR, and enhanced apoptosis induced by doxorubicin and VCR in K562 cells.
Further, 1-[(9E)-10–(3,4-methylenedioxyphenyl)-9-decenoyl] pyrrolidine 16 (, ) is an aikaloid isolated from Piper boehmeriifoliumCitation90. Wang et al.Citation90 evaluated 25 amide alkaloids from Piper boehmeriifolium and 10 synthetic amide alkaloid derivatives for anti-proliferative activity against eight human tumor cell lines, including P-gp-overexpression of MDR KB (KBvin). They found that 3,4,5-trimethoxy substitution in the phenyl ring of piplartine increased selectivity against KBvin. Furthermore, they demonstrated that Piper methysticum root extracts and its major component, the kavalactones, are potent inhibitors of P-gpCitation96.
To add more, tetrandrine (TET) 17 and fangchinoline (FAN) 18 (, ) are bisbenzylisoquinoline alkaloids derived from the root of Stephania tetrandaCitation91. Choi et al.Citation91 evaluated the MDR-reversal abilities of these two compounds by comparing with verapamil. They found that TET (3.0 μM), FAN (3.0 μM) and verapamil (10.0 μM) reduced the paclitaxel IC50 in HCT15 cells (colorectal cancer cell line, a P-gp over-expression cell line) to about 3100-, 1900- and 410-fold, and enhanced the accumulation rates of rhodamine123 in HCT15 cells (200–250%). Likewise, Sun et al.Citation97 reported that TET and FAN showed a significant synergistic cytotoxic effect in Caco-2 and CEM/ADR5000 cancer cells in combination with doxorubicin. Furthermore, TET and FAN increased intracellular accumulation of rhodamine123 and inhibited its efflux in Caco-2 and CEM/ADR5000 cells. These results suggest that TET and FAN can reverse MDR by increasing the intracellular concentration of anti-cancer drugs, and thus they could serve as lead compounds for developing new drugs to overcome P-gp-mediated drug resistance in clinical cancer therapy.
In other report, indole-3-carbinol (I3C) 19 (, ), an indole type alkaloid, is a glucosinolate present in cruciferous vegetables of the genus BrassicaCitation92. Western blot analysis of vinblastine-resistant human leukemia (K562/R10) cells showed that I3C down-regulated induced levels of P-gp in resistant cells. The quantification of immunocytochemically stained K562/R10 cells showed 24, 48 and 80% decrease in the levels of P-gp by I3C for 24, 48 and 72 h of incubation, respectively. These results indicate that I3C could be used as a novel modulator of P-gp and may be effective as a dietary adjuvant in the treatment of MDR cancersCitation92.
Other compounds, pervilleines B 20 and C 21 (PB and PC) (, ), are two new tropane alkaloid aromatic esters obtained from a chloroform extract of the roots of Erythroxylum pervilleiCitation93. They were unable to inhibit KB-V1 cells growth when vinblastine, PB or PC were administered as single agents, but when co-administered with vinblastine, these compounds showed inhibition of up to 77.7%. Equimolar dose of verapamil when tested was less effectiveCitation98. These suggest that PB and PC are potential inhibitors of P-gp.
Finally on alkaloids, stemona alkaloids were isolated from the roots of Stemona aphylla and Stemona burkillii. There were three isolated alkaloids: stemocurtisine and oxystemokerrine from S. aphylla, and stemofoline 22 (, ) from S. burkilliiCitation98. Chanmahasathien et al.Citation94 found that stemofoline could increase the P-gp substrates in a dose-dependent manner. Additionally, P-gp ATPase activity was stimulated by stemofoline in a concentration-dependent manner. All these indicate that stemofoline may interact directly with P-gp to inhibit its activity.
Basically, there are two features present in alkaloid compounds that inhibit P-gp: (i) basic nitrogen atom and (ii) small lipophilic molecules with two planar aromatic ringsCitation19. Pearce et al.Citation19 have demonstrated that the pendant aromatic ring represented by the benzoyl moiety is an important feature for interaction of reserpine-type inhibitors with P-gp. Also, it is asserted that alkaloid compounds having a pendant benzoyl function under certain conformational constraints most effectively enhance the cytotoxicity of vinblastine and are able to block the binding of 125I-NASV (a P-gp substrate) to P-gp in membranes from CEM/VLB100 (a human MDR leukemia cell line) cells. In line with these reports, Wang et al.Citation90 demonstrated that (i) 3,4,5-trimethoxy phenyl substitution is critical for selectivity against KBvin, and (ii) in arylalkenylacyl amide alkaloids, replacement of an isobutyl amino group with pyrrolidin-1-yl or piperidin-1-yl significantly improved their activity.
Coumarins
Natural coumarins comprise a wide class of compounds found throughout the plant kingdomCitation99. They are found at high levels in some essential oils, particularly cinnamon bark oil, cinnamon leaf oil and cassia leaf oil. Most coumarins occur in higher plants, with the richest sources being the Rutaceae, Araliaceae and UmbelliferaeCitation100. Coumarin is classified as a member of the benzopyrone family of compounds, all of which consist of α-benzopyrone. Based on the position of substitutes and type in α-benzopyrone, coumarins are classified into four main sub-types: the simple coumarins, furanocoumarins, pyranocoumarins and the pyrone-substituted coumarinsCitation101. A number of pharmacological activities of coumarins have been reported along with their P-gp inhibitory activity.
For instance, cnidiadin 23 (, ), a furanocoumarin, was derived from Tordylium apulum (Apiaceae). It was reported that this compound significantly inhibited the extrusion of the rhodamine123 and the radiolabeled anti-cancer agent [3H]-vinblastine out of MDR1-transfected Madin–Darby canine kidney (MDCK-MDR1) cells, via competitive inhibition of P-gp transport activityCitation102. It is notable that the proportion of killed MDCK-MDR1 cells (normal cell line), reached 93%, when vinblastine was used in combination with cnidiadin at 10 μM. However, when co-treatment with cnidiadin (10 μM)/VCR on the KB/VCR cells, the cells resulted from long-term exposure of the parental sensitive KB human oral epidermis carcinoma cell line to VCR. No cell toxicity was detected after exposure to cnidiadin and cnidiadin sensitized KB/VCR cellsCitation103.
Figure 4. The structure of coumarins that can inhibit P-gp. C: the basic coumarins structure.
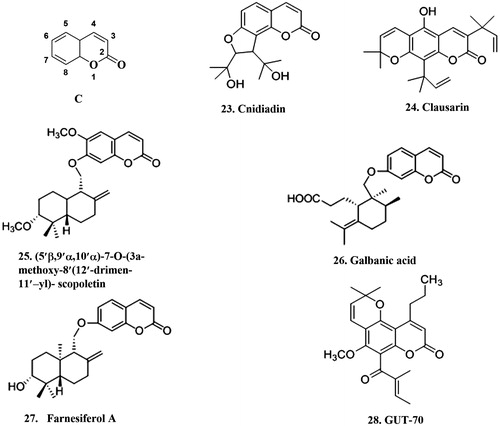
Table 5. An overview of coumarin inhibitors from in vitro studies.
In another study, bioguided fractionation from the roots of Citrus sinensis (Rutaceae) led to the isolation and identification of five coumarins, namely, clausarin, suberosin, poncitrin, xanthyletin and thamnosmoninCitation104. Among these compounds, clausarin 24 (, ) inhibited P-gp-mediated drug efflux in K562/R7 cells, which in daunorubicin accumulation assay showed significant activity of about 45%, compared with cyclosporin A.
A new sesquiterpene-coumarin ether (5′β, 9′α, 10′α)-7-O-(3α-methoxy-8′ (12′)-drimen-11′-yl)- scopoletin 25 (, ) was isolated and characterized from the Me2CO extract of whole dried plant of Euphorbia portlandicaCitation105. Examined for its effects on the reversal of MDR on mouse lymphoma cells (L5178), sesquiterpene-coumarin ether proved to be more active than the verapamil.
Further, two compounds, galbanic acid 26 (from the roots of Ferula szowitsiana) and farnesiferol A 27 (from the roots of Ferula persica) (, ) were assessed on the functionality of drug transporter P-gp using a rhodamine123 efflux assay in MCF7/ADR cellsCitation106. Compared to verapamil, galbanic acid (5, 10 and 25 μg/mL), significantly inhibited P-gp activity. In inhibition of the P-gp transporter, farnesiferol A (0.5 μg/mL) was more potent than verapamil.
Lastly, GUT-70 28 (, ), is a tricyclic coumarin that was extracted from the stem bark of Calophyllum brasiliense. Effects of GUT-70 on P-gp over-expressing MDR K562/D1–9 cells showed that K562/D1–9 cell line was 70 times more resistant to daunorubicin than its parental K562 cell line. The study indicated that GUT-70 may be a useful agent for the treatment of leukemiaCitation107.
Conclusively, it is important to note that, some substituents present on the basic coumarin structure, such as the substituents at position C4 (28), C5 (24), C6–C7 (24, 25 and 28), ether at position C7 (25, 26, 27 and 28) and a [α,β-di(hydroxyisopropyl)dihydrofuran] group at positions C7–C8 (23), exhibit a significant inhibitory effect on P-gpCitation101. For example, Jabeen et al.Citation108 reported that heterocyclic substituents at position C4 or C3–C4 not only enhanced lipophilicity (or log P) but also showed higher inhibitory activity of P-gp. Notably, coumarin itself has low absolute bioavailability in man (< 5%), due to extensive first-pass hepatic conversion of α-benzopyrone to 7-hydroxycoumarin followed by glucuronidationCitation109. It can therefore be proposed that, improving on the bioavailability of coumarins could help enhance their pharmacological activities.
Terpenoids
Terpenoids are a class of isoprenoids from a large and structurally diverse family of natural products. Typical structures contain carbon skeletons represented by (C5) n, and are classified as monoterpenoids (C10), sesquiterpenoids (C15), diterpenoids (C20), sesterterpenoids (C25), triterpenoids (C30), tetraterpenes (C40) and polyterpenesCitation110. Pharmacological activities of terpenoids are extensively researched, particularly, their inhibitory effects on P-gp as discussed below.
(R)-(+)-citronellal 29 (, ) is a component of monoterpenoid found in essential oil from Zanthoxyli fructus and present in some edible plantsCitation117. Abietic acid 30 (, ) is a major component diterpenoid of the rosin fraction of turpentine from pines and other conifersCitation110. Glycyrrhetic acid 31 (, ) is found in Glycyrrhizae radix as a major terpenoid and has been used as an anti-allergic and anti-inflammatory drugCitation118. The inhibitory activities of (R)-(+)-citronellal, abietic acid and glycyrrhetic acid on P-gp-mediated efflux of [3H]digoxin at the screened concentration were greater than 50% on LLC-GA5-COL150 cells, the cells were established by transfection of human MDR1 cDNA encoding P-gp into LLC-PK1 derived from porcine kidney. The IC50 values of (R)-(+)-citronellal (167 ± 8 μM), abietic acid (172 ± 9 μM) and glycyrrhetic acid (80.8 ± 5.0 μM) were lower than that of verapamil (237 ± 13 μM) on LLC-GA5-COL150 cells. In vitro model study on Caco-2 cells, showed that these three compounds increased the AP-to-BL transport and decreased the BL-to-AP transport of [3H]digoxin and efflux ratio. Based on these findings, it was concluded that (R)-(+)-citronellal, abietic acid and glycyrrhetic acid could inhibit P-gp-mediated transportCitation111.
Figure 5. The structure of terpenoids that can inhibit P-gp. D: the basic terpenoids structure.
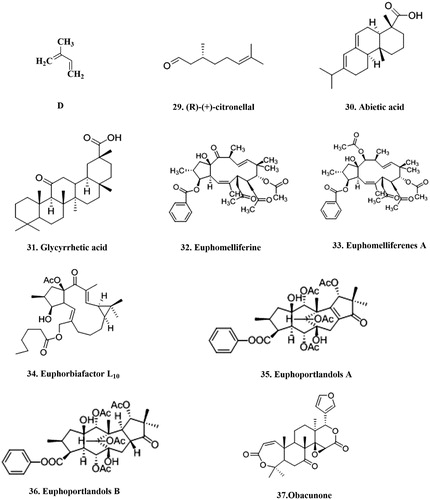
Table 6. An overview of terpenoid inhibitors from in vitro studies.
Furthermore, two new macrocyclic jatrophane diterpenes, namely euphomelliferine 32 () and euphomelliferenes A 33 (, ), were isolated from methanolic extract of Euphorbia melliferaCitation112.The two Jatrophane diterpenes were evaluated for their effects on the reversion of human MDR1 gene-transfected mouse lymphoma cells (L5178Y) and on human colon adenocarcinoma (COLO 320), using the fluorescence activity ratio (FAR, are used to evaluate P-gp modulating potential) values and rhodamine123 exclusion test. The two compounds were able to successfully modulate P-gp efflux activity by reversing the MDR phenotype of MDR cell lines. These results indicate that the two could be promising lead compounds for the development of effective P-gp modulatorsCitation113.
In addition, macrocyclic lathyrane polyester Euphorbia factor L10 34 (, ) was obtained from the seeds of the caper spurge (Euphorbia lathyris) by gravity column chromatography and was further purified by crystallizationCitation114. The interaction of L10 and its acetyl derivative with P-gp was investigated by Appendino et al.Citation114. The detection of dipolar proton–proton correlations between two olefinic protons; the enone proton (H-12) and H-19 and H-8β, and between the oxymethylene protons and H-4 and H-7α showed that L10 assumes a conformation with both the 20-methyl and the 6-oxymethylene syn-oriented on the α-face of the macrocycle. Again, it was shown that L10 could bind to the cytosolic domain of P-gp with high affinity and also strongly inhibits P-gp-mediated daunomycin transport. Interestingly, a 95% increase in intracellular drug accumulation was measured at a 5 μM concentration of L10. Based on these results, it can be assumed that L10 could be a novel chemotype for P-gp inhibitors.
Additionally, two new tetracyclic diterpene polyesters, euphoportlandols A 35 and B 36 (, ), were isolated from an acetone extract of Euphorbia portlandicaCitation115. The evaluation of the inhibition of P-gp-mediated drug efflux from L5178 Y mouse T-lymphoma parental cells was performed by determining the intracellular accumulation of rhodamine 123. At a concentration of 40 μg/mL, both compounds were found to have inhibitory activity on P-gpCitation115.
Lastly, obacunone 37 (, ), is a limonoids isolated from Phellodendron amurenseCitation116. Min et al.Citation116 found that obacunone showed significant P-gp MDR inhibition activity in MES-SA/DX5 (human MDR uterine sarcoma cell line) and HCT15 cells with ED50 value of 0.028 μg/mL and 0.0011 μg/mL, respectively.
It is noteworthy that, two features of terpenoids, the carbonyl groups at the C2α, C3, C8 positions (30, 31 and 35), and a hydrophobic substituent at the C6 position (37), appear to be the key contributors for their P-gp inhibitory activityCitation119.
It is obvious that P-gp inhibitory effects of terpenoids depend on the chemical structure of the compound. For example, it is reported that triterpenes need a particular pi-electron distribution at the C24 and C25 positions and also lipophilic groups for increasing log P. Finally, carbonyl oxygen and dipole moments of terpenes are as well important in binding to membrane lipids and peptides by complex formation via OH, COOH and NH2 groups of the P-gpCitation120.
Sterides
Sterides are generally found in higher plants and have long been used against various diseases, including inflammation and cancerCitation121. The basic structure of sterides contains cyclopentano-perhydrophenanthrene with many substituents, which includes a hydroxy group at the C3 position, methyl at the C10 and C13 positions and different side chains at the C17 positionCitation122. Based on the substituents at C17 position, sterides are classified into cardiac glycosides, steroid saponins, steroid hormones and othersCitation123. Here, in this section, we highlight on plant-derived sterides that possess P-gp modulatory activities.
Paris saponin VII (PSVII) 38 (, ) was isolated from Trillium tschonoskii MaximCitation124. PSVII dose dependently suppressed cell viability, triggered apoptosis and modulated drug resistance of MCF-7/ADR cells. PSVII treatment in MCF-7/ADR cells led to increased TNFR1, TRAIL R1/DR4, TRAIL R2/DR5, FADD expression, and activation of polymerase caspase-8, and 3. In parallel to the alterations, P-gp expression and activity were also reducedCitation124.
Figure 6. The structure of saponins that can inhibit P-gp. E: the basic saponins structure.
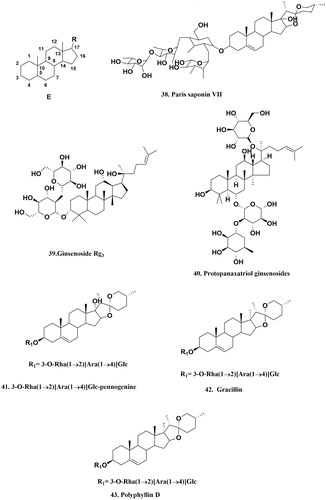
Table 7. An overview of steride inhibitors from in vitro studies.
In a flow cytometric analysis, ginsenoside Rg3 39 (, ), was isolated from Panax ginsengCitation125, promoted accumulation of rhodamine 123 in drug-resistant KBV20C cells in a dose-dependent manner. Additionally, Rg3 inhibited [3H]vinblastine efflux and reversed MDR to doxorubicin, COL, VCR and VP-16 in KBV20C cells. Photo-affinity labeling study with [3H]azidopine revealed that Rg3 competed with [3H]azidopine for binding to P-gp. This research demonstrates that Rg3 can compete with anti-cancer drugs for binding to P-gp thereby blocking drug effluxCitation125.
Moreover, protopanaxatriol ginsenosides (PTG) 40 (, ) is another steride isolated from Panax ginsengCitation126. In a recent study, daunorubicin-resistant acute myelogenous leukemia (AML-2/D100) was shown to increase the expression of P-gp, but treatment with PTG reversed resistance in the AML-2/D100 sub-line in a concentration-dependent manner. The effect of PTG (100 μg/mL) on drug accumulation of daunorubicin in the AML-2/D100 subline was 2-fold higher than that observed in the presence of verapamil (5 μg/mL) and 1.5 times less than cyclosporine A (3 μg/mL). Beyond this, PTG at 200 μg/mL or more completely inhibited the azidopine photo-labeling of P-gp. All these indicate that PTG has a chemosensitizing effect on P-gp-mediated MDR cells by increasing the intracellular accumulation of drugs through direct interaction with P-gp at the azidopine siteCitation126.
In another study, bio-guided fractionation of the roots of Paris polyphylla (Trilliaceae), led to isolation and identification of three saponins 3-O-Rha(1 → 2)[Ara(1 → 4)]Glc-pennogenine 41, gracillin 42 and polyphyllin D 43 (, )Citation127. These three compounds exhibited strong ability to inhibit P-gp-mediated drug efflux in K562/R7 cells, compared to the cyclosporin A, with respective values of 41, 64 and 45%, at 10 μM. On transfected cells study, the results showed that the 3-O-Rha (1 → 2)[Ara(1 → 4)]Glc-pennogenine exhibited the highest ability to modulate P-gp activity (108 and 77% inhibition at 20 and 10 μM, respectively), compared with cyclosporin A. The value of the gracillin and polyphyllin D were about 48 and 32%, 61 and 46% inhibition at 20 and 10 μM, respectivelyCitation127.
Conclusively, it is important to add that, substituents of sterides that mainly exist at the C3 position (38–39, 41–53), at the C16–C17 position (38, 41, 42 and 43) and at the C17 position (39, 40), could be important for their P-gp inhibitory effectsCitation128. Also, the number, the length and the position of side chains, and the type of sugar moiety could have different effects on the P-gp inhibition by sterides. Notably, the hemolytic activity of some steroid saponins (e.g. compounds 41–43) is a major drawback for their clinical development. Fortunately, Wang et al.Citation129 have reported on the structure-activity relationship between steroid saponin and development of hemolysis. They reported that steroid saponin: (i) with the same aglycone and the same length of sugar chain, the sugar linkage determines the hemolytic potency; (ii) with substituents on the hydroxyl groups in the sugar residues could significantly affect their hemolytic activity; (iii) with changes on the aglycone could also change the hemolytic activity. Their report raises hopes that it is feasible to develop steroid saponins into potent P-gp inhibitors with decreased hemolytic effects.
Conclusions and future prospective
Even though MDR involves complex genetic factors, modern scientific research could accelerate its drug discovery process because each factor could provide a novel drug targetCitation5. There have been studies on MDR for the past four decades since the Ling lab used somatic cell genetics to investigate the properties of colchicine resistance in CHO cells and discovered P-gpCitation10. The role of this efflux transporter in drug pharmacokinetics, such as drug absorption and excretion, is increasingly becoming important and well defined. In earlier parts of this review, we have highlighted that P-gp is highly expressed in various tissues, and so it is clear that P-gp inhibition has greater effects on absorption and tissue distribution of drugs.
Many of the plant-based compounds mentioned in this review could provide insights into wide range possibilities of using different methods to develop effective P-gp inhibitors. It is noteworthy that some of the plant-based medicines are reported to be non-specific P-gp inhibitors, which could affect other protein and human enzymatic targets. Therefore, it is important that developing effective P-gp inhibitors in the future should be guided by factors, such as lower side-effect, mechanism and specificity, particularly, blockade in terms of P-gp drug-binding domain inhibitionCitation130. Finally, experimental methods and techniques, such as empirical structure-activity relationships (SAR)Citation120, quantitative structure-activity relationships (QSAR)Citation131, 3-dimensional quantitative structure-activity relationships (3DQSAR)Citation132, pharmacophore studiesCitation133 and cysteine-scanning mutagenesisCitation134 should also be considered as important features to guide researchers to discover highly selective and potent P-gp inhibitors.
Declaration of interest
The authors declare that they have no competing interests.
This work was financially supported by the National Natural Science Foundation of China (No. 81330081, No. 81302845), The Training Program of Academic and Technical Leaders in Universities of Anhui province (No. 34), The Anhui Province Natural Science Foundation in University (No. KJ2013A158) and Grants for Scientific Research of BSKY (No. XJ201211) from Anhui Medical University.
References
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2002;2:48–58
- Romiti A, Cox MC, Sarcina I, et al. Metronomic chemotherapy for cancer treatment: a decade of clinical studies. Cancer Chemother Pharmacol 2013;72:13–33
- Colabufo NA, Pagliarulo V, Berardi F, et al. Bicalutamide failure in prostate cancer treatment: involvement of Multi Drug Resistance proteins. Eur J Pharmacol 2008;601:38–42
- Elkadi OA. MDR-selective microbial-based therapy: a novel approach to cancer treatment. Med Hypotheses 2013;81:207–11
- Huang Y, Sadee W. Drug sensitivity and resistance genes in cancer chemotherapy: a chemogenomics approach. Drug Discov Today 2003;8:356–63
- Montazami N, Aghapour M, Farajnia S, et al. New insights into the mechanisms of multidrug resistance in cancers. Cell Mol Biol (Noisy-le-grand) Mol 2015;61:70–80
- Gatouillat G, Magid AA, Bertin E, et al. Medicarpin and millepurpan, two flavonoids isolated from Medicago sativa, induce apoptosis and overcome multidrug resistance in leukemia P388 cells. Phytomedicine 2015;22:1186–94
- Ji M, Shi Y, Lou H, et al. Overcoming of P-glycoprotein-mediated multidrug resistance in K562/A02 cells using riccardin F and pakyonol, bisbibenzyl derivatives from liverworts. Biosci Trends 2011;5:192–7
- Leslie EM, Deeley RG, Cole SP. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol 2005;204:216–37
- Ling V, Thompson LH. Reduced permeability in CHO cells as a mechanism of resistance to colchicine. J Cell Physiol 1974;83:103–16
- Schinkel AH1, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev 2003;55:3–29
- Gottesman MM, Ling V. The molecular basis of multidrug resistance in cancer: the early years of P-glycoprotein research. FEBS Lett 2006;580:998–1009
- Palmeira A, Sousa E, Vasconcelos MH, et al. Three decades of P-gp inhibitors: skimming through several generations and scaffolds. Curr Med Chem 2012;19:1946–2025
- Sarisozen C, Vural I, Levchenko T, et al. PEG-PE-based micelles co-loaded with paclitaxel and cyclosporine A or loaded with paclitaxel and targeted by anticancer antibody overcome drug resistance in cancer cells. Drug Deliv 2012;19:169–76
- Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res 1981;41:1967–72
- Safa AR. Photoaffinity labeling of the multidrug-resistance-related P-glycoprotein with photoactive analogs of verapamil. Proc Natl Acad Sci USA 1988;85:7187–91
- Shiraga K, Sakaguchi K, Senoh T, et al. Modulation of doxorubicin sensitivity by cyclosporine A in hepatocellular carcinoma cells and their doxorubicin-resistant sublines. J Gastroenterol Hepatol 2001;16:460–6
- Angelini A, Iezzi M, Di Febbo C, et al. Reversal of P-glycoprotein-mediated multidrug resistance in human sarcoma MES-SA/Dx-5 cells by nonsteroidal anti-inflammatory drugs. Oncol Rep 2008;20:731–5
- Pearce HL, Safa AR, Bach NJ, et al. Essential features of the P-glycoprotein pharmacophore as defined by a series of reserpine analogs that modulate multidrug resistance. Proc Natl Acad Sci USA 1989;86:5128–32
- Kalitsky-Szirtes J, Shayeganpour A, Brocks DR, et al. Suppression of drug-metabolizing enzymes and efflux transporters in the intestine of endotoxin-treated rats. Drug Metab Dispos 2004;32:20–7
- Liu ZH, Ma YL, He YP, et al. Tamoxifen reverses the multi-drug-resistance of an established human cholangiocarcinoma cell line in combined chemotherapeutics. Mol Biol Rep 2011;38:1769–75
- Palmeira A, Rodrigues F, Sousa E, et al. New uses for old drugs: pharmacophore-based screening for the discovery of P-glycoprotein inhibitors. Chem Biol Drug Des 2011;78:57–72
- Bark H, Choi CH. PSC833, cyclosporine analogue, downregulates MDR1 expression by activating JNK/c-Jun/AP-1 and suppressing NF-kappaB. Cancer Chemother Pharmacol 2010;65:1131–6
- Bates SF, Chen C, Robey R, et al. Reversal of multidrug resistance: lessons from clinical oncology. Novartis Found Symp 2002;243:83–96; discussion 96–102, 180–5
- Biscardi M, Teodori E, Caporale R, et al. Multidrug reverting activity toward leukemia cells in a group of new verapamil analogues with low cardiovascular activity. Leuk Res 2006;30:1–8
- Lee SY, Rhee YH, Jeong SJ, et al. Hydrocinchonine, cinchonine, and quinidine potentiate paclitaxel-induced cytotoxicity and apoptosis via multidrug resistance reversal in MES-SA/DX5 uterine sarcoma cells. Environ Toxicol 2011;26:424–31
- Pire MM, Emmert D, Hrycyna CA, et al. Inhibition of P-glycoprotein-mediated paclitaxel resistance by reversibly linked quinine homodimers. Mol Pharmacol 2009;75:92–100
- Mistry P, Stewart AJ, Dangerfield W, et al. In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res 2001;61:749–58
- Marcelletti JF, Multani PS, Lancet JE, et al. Leukemic blast and natural killer cell P-glycoprotein function and inhibition in a clinical trial of zosuquidar infusion in acute myeloid leukemia. Leuk Res 2009;33:769–74
- Kuntner C, Bankstahl JP, Bankstahl M, et al. Dose-response assessment of tariquidar and elacridar and regional quantification of P-glycoprotein inhibition at the rat blood-brain barrier using (R)-[(11)C]verapamil PET. Eur J Nucl Med Mol Imaging 2010;37:942–53
- Lee BD, French KJ, Zhuang Y, et al. Development of a syngeneic in vivo tumor model and its use in evaluating a novel P-glycoprotein modulator, PGP-4008. Oncol Res 2003;14:49–60
- Pusztai L, Wagner P, Ibrahim N, et al. Phase II study of tariquidar, a selective P-glycoprotein inhibitor, in patients with chemotherapy-resistant, advanced breast carcinoma. Cancer 2005;104:682–91
- Girdhani S, Bhosle SM, Thulsidas SA, et al. Potential of radiosensitizing agents in cancer chemo-radiotherapy. J Cancer Res Ther 2005;1:129–31
- Abdallah HM, Al-Abd AM, El-Dine RS, et al. P-glycoprotein inhibitors of natural origin as potential tumor chemo-sensitizers: a review. J Adv Res 2015;6:45–62
- Anthony V, Skach WR. Molecular mechanism of P-glycoprotein assembly into cellular membranes. Curr Protein Pept Sci 2002;3:485–501
- Sun J, He ZG, Cheng G, et al. Multidrug resistance P-glycoprotein: crucial significance in drug disposition and interaction. Med Sci Monit 2004;10:RA5–14
- Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, et al. P-glycoprotein: from genomics to mechanism. Oncogene 2003;22:7468–85
- Loo TW, Clarke DM. Recent progress in understanding the mechanism of P-glycoprotein-mediated drug efflux. J Membr Biol 2005;206:173–85
- Ferté J. Analysis of the tangled relationships between P-glycoprotein-mediated multidrug resistance and the lipid phase of the cell membrane. Eur J Biochem 2000;267:277–94
- Ginn PE. Immunohistochemical detection of P-glycoprotein in formalin-fixed and paraffin-embedded normal and neoplastic canine tissues. Vet Pathol 1996;33:533–41
- Borst P, Schinkel AH. P-glycoprotein ABCB1: a major player in drug handling by mammals. J Clin Invest 2013;123:4131–3
- Mealey KL, Fidel J. P-glycoprotein mediated drug interactions in animals and humans with cancer. J Vet Intern Med 2015;29:1–6
- Tucker TG, Milne AM, Fournel-Gigleux S, et al. Absolute immunoquantification of the expression of ABC transporters P-glycoprotein, breast cancer resistance protein and multidrug resistance-associated protein 2 in human liver and duodenum. Biochem Pharmacol 2012;83:279–85
- Tang YZ, Li DQ, Sun FJ, et al. P-glycoprotein regulating biphasic insulin secretion in rat pancreatic beta cells. Chin Med J (Engl) 2009;122:2587–92
- Water FM, Boleij JM, Peters JG, et al. Characterization of P-glycoprotein and multidrug resistance proteins in rat kidney and intestinal cell lines. Eur J Pharm Sci 2007;30:36–44
- Ellis K, Marlin JW, Taylor TA, et al. The effects of human immunodeficiency virus infection on the expression of the drug efflux proteins P-glycoprotein and breast cancer resistance protein in a human intestine model. J Pharm Pharmacol 2015;67:178–88
- Johnson WW. P-glycoprotein-mediated efflux as a major factor in the variance of absorption and distribution of drugs: modulation of chemotherapy resistance. Methods Find Exp Clin Pharmacol 2002;24:501–14
- Huang L, Li X, Roberts J, et al. Differential role of P-glycoprotein and breast cancer resistance protein in drug distribution into brain, CSF and peripheral nerve tissues in rats. Xenobiotica 2015;45:547–55
- Lin JH, Yamazaki M. Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet 2003;42:59–98
- Sauna ZE, Smith MM, Müller M, et al. The mechanism of action of multidrug-resistance-linked P-glycoprotein. J Bioenerg Biomembr 2001;33:481–91
- Sharom FJ. Complex Interplay between the P-Glycoprotein multidrug efflux pump and the membrane: its role in modulating protein function. Front Oncol 2014;4:41
- Callaghan R, Ford RC, Kerr ID. The translocation mechanism of P-glycoprotein. FEBS Lett 2006;580:1056–63
- Orelle C, Gubellini F, Durand A, et al. Conformational change induced by ATP binding in the multidrug ATP-binding cassette transporter BmrA. Biochemistry 2008;47:2404–12
- García-Carrasco M, Mendoza-Pinto C, Macias Díaz S, et al. P-glycoprotein in autoimmune rheumatic diseases. Autoimmun Rev 2015;14:594–600
- Sharom FJ. The P-glycoprotein multidrug transporter. Essays Biochem 2011;50:161–78
- Amin ML. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights 2013;7:27–34
- Varma MV, Ashokraj Y, Dey CS, et al. P-glycoprotein inhibitors and their screening: a perspective from bioavailability enhancement. Pharmacol Res 2003;48:347–59
- Srivalli KMR, Lakshmi PK. Overview of P-glycoprotein inhibitors: a rational outlook. Braz J Pharm Sci 2012;48:353–67
- Pascaud C, Garrigos M, Orlowski S. Multidrug resistance transporter P-glycoprotein has distinct but interacting binding sites for cytotoxic drugs and reversing agents. Biochem J 1998;333:351–8
- Ramachandra M, Ambudkar SV, Gottesman MM, et al. Functional characterization of a glycine 185-to-valine substitution in human P-glycoprotein by using a vaccinia-based transient expression system. Mol Biol Cell 1996;7:1485–98
- Ambudkar SV, Kim IW, Sauna ZE. The power of the pump: mechanisms of action of P-glycoprotein (ABCB1). Eur J Pharm Sci 2006;27:392–400
- Shapiro AB, Ling V. Effect of quercetin on Hoechst 33342 transport by purified and reconstituted P-glycoprotein. Biochem Pharmacol 1997;53:587–96
- Hugger ED, Novak BL, Burton PS, et al. A comparison of commonly used polyethoxylated pharmaceutical excipients on their ability to inhibit P-glycoprotein activity in vitro. J Pharm Sci 2002;91:1991–2002
- Nijveldt RJ, van Nood E, van Hoorn DE, et al. Flavonoids: a review of probable mechanisms of action and potential applications. Am J Clin Nutr 2001;74:418–25
- Narayana KR, Reddy MS, Chaluvadi MR, et al. Bioflavonoids classification, pharmacological, biochemical effects and therapeutic potential. Ind. J Pharmacol 2001;33:2–16
- Boumendjel A, Di Pietro A, Dumontet C, et al. Recent advances in the discovery of flavonoids and analogs with high-affinity binding to P-glycoprotein responsible for cancer cell multidrug resistance. Med Res Rev 2002;22:512–29
- Chieli E, Romiti N, Catiana Zampini I, et al. Effects of Zuccagnia punctata extracts and their flavonoids on the function and expression of ABCB1/P-glycoprotein multidrug transporter. J Ethnopharmacol 2012;144:797–801
- Limtrakul P, Khantamat O, Pintha K. Inhibition of P-glycoprotein function and expression by kaempferol and quercetin. J Chemother 2005;17:86–95
- Yu CP, Wu PP, Hou YC, et al. Quercetin and rutin reduced the bioavailability of cyclosporine from Neoral, an immunosuppressant, through activating P-glycoprotein and CYP 3A4. J Agric Food Chem 2011;59:4644–8
- Castro AF, Altenberg GA. Inhibition of drug transport by genistein in multidrug-resistant cells expressing P-glycoprotein. Biochem Pharmacol 1997;53:89–93
- Sun L, Chen W, Qu L, et al. Icaritin reverses multidrug resistance of HepG2/ADR human hepatoma cells via downregulation of MDR1 and P-glycoprotein expression. Mol Med Rep 2013;8:1883–7
- Li C, Kim M, Choi H, Choi J. Effects of baicalein on the pharmacokinetics of tamoxifen and its main metabolite, 4-hydroxytamoxifen, in rats: possible role of cytochrome P450 3A4 and P-glycoprotein inhibition by baicalein. Arch Pharm Res 2011;34:1965–72
- Zhang S, Morris ME. Effects of the flavonoids biochanin A, morin, phloretin, and silymarin on P-glycoprotein-mediated transport. J Pharmacol Exp Ther 2003;304:1258–67
- Lee E, Enomoto R, Koshiba C, et al. Inhibition of P-glycoprotein by wogonin is involved with the potentiation of etoposide-induced apoptosis in cancer cells. Ann N Y Acad Sci 2009;1171:132–6
- Shapiro AB, Ling V. Reconstitution of drug transport by purified P-glycoprotein. J Biol Chem 1995;270:16167–75
- Zhang X, Song J, Shi X, et al. Absorption and metabolism characteristics of rutin in Caco-2 cells. Scientific World J 2013;10:382350
- Floyd KA, Stella DR, Wang CC, et al. Genistein and genistein-containing dietary supplements accelerate the early stages of cataractogenesis in the male ICR/f rat. Exp Eye Res 2011;92:120–7
- Nakatsuma A, Fukami T, Suzuki T, et al. Effects of kaempferol on the mechanisms of drug resistance in the human glioblastoma cell line T98G. Pharmazie 2010;65:379–83
- Zhang S, Morris ME. Effect of the flavonoids biochanin A and silymarin on the P-glycoprotein-mediated transport of digoxin and vinblastine in human intestinal Caco-2 cells. Pharm Res 2003;20:1184–91
- Pick A, Muller H, Mayer R, et al. Structure-activity relationships of flavonoids as inhibitors of breast cancer resistance protein (BCRP). Bioorg Med Chem 2011;19:2090–102
- Srinivas NR. Recent trends in preclinical drug-drug interaction studies of flavonoids -Review of case studies, issues and perspectives. Phytother Res 2015;29:1679–91
- Ma Y, Zeng M, Sun R, et al. Disposition of flavonoids impacts their efficacy and safety. Curr Drug Metab 2014;15:841–64
- Actis-Goretta L, Lévèques A, Rein M, et al. Intestinal absorption, metabolism, and excretion of (-)-epicatechin in healthy humans assessed by using an intestinal perfusion technique. Am J Clin Nutr 2013;98:924–33
- Yang Z, Kulkarni K, Zhu W, et al. Bioavailability and pharmacokinetics of genistein: mechanistic studies on its ADME. Anticancer Agents Med Chem 2012;12:1264–80
- Qiu S, Sun H, Zhang AH, et al. Natural alkaloids: basic aspects, biological roles, and future perspectives. Chin J Nat Med 2014;12:401–6
- Lei Y, Tan J, Wink M, et al. An isoquinoline alkaloid from the Chinese herbal plant Corydalis yanhusuo W.T. Wang inhibits P-glycoprotein and multidrug resistance-associate protein 1. Food Chem 2013;136:1117–21
- Ma Y, Wink M. Lobeline, a piperidine alkaloid from Lobelia can reverse P-gp dependent multidrug resistance in tumor cells. Phytomedicine 2008;15:754–8
- Chavez D, Cui B, Chai HB, et al. Reversal of multidrug resistance by tropane alkaloids from the stems of Erythroxylum rotundifolium. J Nat Prod 2002;65:606–10
- Ikeda R, Che XF, Yamaguchi T, et al. Cepharanthine potently enhances the sensitivity of anticancer agents in K562 cells. Cancer Sci 2005;96:372–6
- Wang YH, Goto M, Wang LT, et al. Multidrug resistance-selective antiproliferative activity of Piper amide alkaloids and synthetic analogues. Bioorg Med Chem Lett 2014;24:4818–21
- Choi SU, Park SH, Kim KH, et al. The bisbenzylisoquinoline alkaloids, tetrandine and fangchinoline, enhance the cytotoxicity of multidrug resistance-related drugs via modulation of P-glycoprotein. Anticancer Drugs 1998;9:255–61
- Arora A, Seth K, Kalra N, et al. Modulation of P-glycoprotein-mediated multidrug resistance in K562 leukemic cells by indole-3-carbinol. Toxicol Appl Pharmacol 2005;202:237–43
- Mi Q, Cui B, Silva GL, et al. Pervilleines B and C, new tropane alkaloid aromatic esters that reverse the multidrug-resistance in the hollow fiber assay. Cancer Lett 2002;184:13–20
- Chanmahasathien W, Ohnuma S, Ambudkar SV, et al. Biochemical mechanism of modulation of human P-glycoprotein by stemofoline. Planta Med 2011;77:1990–5
- Shiraishi N, Akiyama S, Nakagawa M, et al. Effect of bisbenzylisoquinoline (biscoclaurine) alkaloids on multidrug resistance in KB human cancer cells. Cancer Res 1987;47:2413–16
- Weiss J, Sauer A, Frank A, et al. Extracts and kavalactones of Piper methysticum G. Forst (kava-kava) inhibit P-glycoprotein in vitro. Drug Metab Dispos 2005;33:1580–3
- Sun YF, Wink M. Tetrandrine and fangchinoline, bisbenzylisoquinoline alkaloids from Stephania tetrandra can reverse multidrug resistance by inhibiting P-glycoprotein activity in multidrug resistant human cancer cells. Phytomedicine 2014;21:1110–19
- Chanmahasathien W, Ampasavate C, Greger H, et al. Stemona alkaloids, from traditional Thai medicine, increase chemosensitivity via P-glycoprotein-mediated multidrug resistance. Phytomedicine 2011;18:199–204
- Egan D, O’Kennedy R, Moran E, et al. The pharmacology, metabolism, analysis and applications of coumarin and coumarin-related compounds. Drug Metab Rev 1990;22:503–29
- Lacy A, O'Kennedy R. Studies on coumarins and coumarin-related compounds to determine their therapeutic role in the treatment of cancer. Curr Pharm Des 2004;10:3797–811
- Raad I, Terreux R, Richomme P, et al. Structure-activity relationship of natural and synthetic coumarins inhibiting the multidrug transporter P-glycoprotein. Bioorg Med Chem 2006;14:6979–87
- Barthomeuf C, Demeule M, Grassi J, et al. Conferone from Ferula schtschurowskiana enhances vinblastine cytotoxicity in MDCK-MDR1 cells by competitively inhibiting P-glycoprotein transport. Planta Med 2006;72:634–9
- Barthomeuf C, Grassi J, Demeule M, et al. Inhibition of P-glycoprotein transport function and reversion of MDR1 multidrug resistance by cnidiadin. Cancer Chemother Pharmacol 2005;56:173–81
- Bayet C, Fazio C, Darbour N, et al. Modulation of P-glycoprotein activity by acridones and coumarins from Citrus sinensis. Phytother Res 2007;21:386–90
- Madureira AM, Molnar A, Abreu PM, et al. A new sesquiterpene-coumarin ether and a new abietane diterpene and their effects as inhibitors of P-glycoprotein. Planta Med 2004;70:828–33
- Hanafi-Bojd MY, Iranshahi M, Mosaffa F, et al. Farnesiferol A from Ferula persica and galbanic acid from Ferula szowitsiana inhibit P-glycoprotein-mediated rhodamine efflux in breast cancer cell lines. Planta Med 2011;77:1590–3
- Kimura S, Ito C, Jyoko N, et al. Inhibition of leukemic cell growth by a novel anti-cancer drug (GUT-70) from Calophyllum brasiliense that acts by induction of apoptosis. Int J Cancer 2005;113:158–65
- Jabeen I, Wetwitayaklung P, Chiba P, et al. 2D- and 3D-QSAR studies of a series of benzopyranes and benzopyrano[3,4b][1,4]-oxazines as inhibitors of the multidrug transporter P-glycoprotein. J Comput Aided Mol Des 2013;27:161–71
- Hoult JR, Payá M. Pharmacological and biochemical actions of simple coumarins: natural products with therapeutic potential. Gen Pharmacol 1996;27:713–22
- Dewick PM. The biosynthesis of C5-C25 terpenoid compounds. Nat Prod Rep 2002;19:181–222
- Yoshida N, Koizumi M, Adachi I, et al. Inhibition of P-glycoprotein-mediated transport by terpenoids contained in herbal medicines and natural products. Food Chem Toxicol 2006;44:2033–9
- Appendino G, Jakupovic S, Tron GC, et al. Macrocyclic diterpenoids from Euphorbia semiperfoliata. J Nat Prod 1998;61:749–56
- Valente I, Reis M, Duarte N, et al. Jatrophane diterpenes from Euphorbia mellifera and their activity as P-glycoprotein modulators on multidrug-resistant mouse lymphoma and human colon adenocarcinoma cells. J Nat Prod 2012;75:1915–21
- Appendino G, Della Porta C, Conseil G, et al. A new P-glycoprotein inhibitor from the caper spurge (Euphorbia lathyris). J Nat Prod 2003;66:140–2
- Madureira AM, Gyemant N, Ascenso JR, et al. Euphoportlandols A and B, tetracylic diterpene polyesters from Euphorbia portlandica and their anti-MDR effects in cancer cells. J Nat Prod 2006;69:950–3
- Min YD, Kwon HC, Yang MC, et al. Isolation of limonoids and alkaloids from Phellodendron amurense and their multidrug resistance (MDR) reversal activity. Arch Pharm Res 2007;30:58–63
- Lota ML, de Rocca Serra D, Tomi F, et al. Volatile components of peel and leaf oils of lemon and lime species. J Agric Food Chem 2002;50:796–805
- Kawakami J, Yamamura Y, Santa T, et al. Kinetic analysis of glycyrrhetic acid, an active metabolite of glycyrrhizin, in rats: role of enterohepatic circulation. J Pharm Sci 1993;82:301–5
- Reyes CP, Munoz-Martinez F, Torrecillas IR, et al. Biological evaluation, structure-activity relationships, and three-dimensional quantitative structure-activity relationship studies of dihydro-beta-agarofuran sesquiterpenes as modulators of P-glycoprotein-dependent multidrug resistance. J Med Chem 2007;50:4808–17
- Molnar J, Gyemant N, Tanaka M, et al. Inhibition of multidrug resistance of cancer cells by natural diterpenes, triterpenes and carotenoids. Curr Pharm Des 2006;12:287–311
- Fang S, Hao C, Sun W, et al. Rapid analysis of steroidal saponin mixture using electrospray ionization mass spectrometry combined with sequential tandem mass spectrometry. Rapid Commun Mass Spectrom Commun 1998;12:589–94
- Yang Y, Laval S, Yu B. Chemical synthesis of saponins. Adv Carbohydr Chem Biochem 2014;71:137–226
- D'Uva G, Lauriola M. Towards the emerging crosstalk: ERBB family and steroid hormones. Semin Cell Dev Biol 2015;12:250–5
- Li Y, Fan L, Sun Y, et al. Paris saponin VII from trillium tschonoskii reverses multidrug resistance of adriamycin-resistant MCF-7/ADR cells via P-glycoprotein inhibition and apoptosis augmentation. J Ethnopharmacol 2014;154:728–34
- Kim SW, Kwon HY, Chi DW, et al. Reversal of P-glycoprotein-mediated multidrug resistance by ginsenoside Rg(3). Biochem Pharmacol 2003;65:75–82
- Choi CH, Kang G, Min YD. Reversal of P-glycoprotein-mediated multidrug resistance by protopanaxatriol ginsenosides from Korean red ginseng. Planta Med 2003;69:235–40
- Nguyen VT, Darbour N, Bayet C, et al. Selective modulation of P-glycoprotein activity by steroidal saponines from Paris polyphylla. Fitoterapia 2009;80:39–42
- Sun H, Yang Z, Ye Y. Structure and biological activity of protopanaxatriol-type saponins from the roots of Panax notoginseng. Int Immunopharmacol 2006;6:14–25
- Wang Y, Zhang Y, Zhu Z, et al. Exploration of the correlation between the structure, hemolytic activity, and cytotoxicity of steroid saponins. Bioorg Med Chem 2007;15:2528–32
- Loo TW, Bartlett MC, Clarke DM. Transmembrane segment 7 of human P-glycoprotein forms part of the drug-binding pocket. Biochem J 2006;399:351–9
- Tan W, Mei H, Chao L, et al. Combined QSAR and molecule docking studies on predicting P-glycoprotein inhibitors. J Comput Aided Mol Des 2013;27:1067–73
- Kim KH. 3D-QSAR analysis of 2,4,5- and 2,3,4,5-substituted imidazoles as potent and nontoxic modulators of P-glycoprotein mediated MDR. Bioorg Med Chem 2001;9:1517–23
- Palmeira A, Sousa E, Vasconcelos MH, et al. Structure and ligand-based design of P-glycoprotein inhibitors: a historical perspective. Curr Pharm Des 2012;18:4197–214
- Loo TW, Clarke DM. Identification of residues within the drug-binding domain of the human multidrug resistance P-glycoprotein by cysteine-scanning mutagenesis and reaction with dibromobimane. J Biol Chem 2000;275:39272–8