
Abstract
Platelet aggregation is one of the main events involved in vascular thrombus formation. Recently, N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazides were described as antiplatelet derivatives. In this work, we explore the properties of these antiplatelet agents through a series of pharmacological, biochemical and toxicological studies. The antiplatelet activity of each derivative was confirmed as 3a, 3b and 3 h significantly inhibited human platelet aggregation induced by arachidonic acid, with no detectable effect on clotting factors or healthy erythrocytes. Importantly, mice treated with derivative 3a showed a higher survival rate at an in vivo model of pulmonary thromboembolism with a lower bleeding risk in comparison to aspirin. The in silico studies pointed a series of structural parameters related to thromboxane synthase (TXS) inhibition by 3a, which was confirmed by tracking plasma levels of PGE2 and TXB2 through an in vitro enzyme immunoassay. Derivative 3a showed selective TXS inhibition allied with low bleeding risk and increased animal survival, revealing the derivative as a promising candidate for treatment of cardiovascular diseases.
Introduction
Hemostasis is a physiological process that maintains the blood in a liquid and circulating condition. It forms a hemostatic plug under inflammatory conditions to prevent blood loss, which sustains local perfusion and stimulates the repair processCitation1.
Despite the hemostasis process, pathological disorders may occur, leading to thrombotic diseases (e.g. atherothrombosis and venous thromboembolism)Citation1. According to the World Health Organization (WHO), thrombotic diseases are currently among the major causes of death worldwideCitation2.
The inhibition of platelet aggregation is one of the strategies adopted by physicians through antithrombotic drugs to control the burden and risk of cardiovascular diseases. Thus, the mechanisms underlying platelet aggregation in response to a prothrombotic stimuli (inducers/agonists, receptors and enzymes) are feasible druggable targets. Due to its safety and low cost, the most widespread antiplatelet agent used nowadays is still acetylsalicylic acid (ASA – Aspirin®)Citation1. ASA inhibits cyclooxygenase-1 (COX-1) by acetylating a hydroxyl group of the enzyme active site, preventing the proper binding to arachidonic acid (ARA) and further prostaglandin H2 (PGH2) synthesis. This inhibition impairs the activity of thromboxane synthase (TXS), a P450 epoxygenase, reducing the downstream synthesis of thromboxane A2 (TXA2), a potent platelet activator and inflammatory mediatorCitation1, from PGH2. Ozagrel, also an antiplatelet drug, inhibits TXS through π–cation interactions with the iron atom bound to TXS heme, a mechanism that differs considerably from COX-1 inhibitorsCitation1.
The advance of the antithrombotic therapy with synthetic molecules such as Aspirin®, clopidogrel, triflusal and tirofiban has provided good improvement in the prognostic of patients with acute myocardial infarctionsCitation1. However, considerable limitations still remain on many of these drugs, particularly poor administration routes and adverse effects generally related to: increased bleeding riskCitation3, induced neutropeniaCitation4, aplastic anemiaCitation5, thrombocytopeniaCitation5 and/or drug resistanceCitation6. This context prompts to the rational design and development of new antiplatelet drugs with lower adverse effects and/or better profile to treat these cardiovascular disordersCitation7.
Several families of chemical substances have arisen as potential antiplatelet drug prototypes, including pyrazolopyridinesCitation8. They present a significant therapeutic potential including antimicrobialCitation9, antichagasicCitation10, analgesicCitation11, anti-tumorCitation12 and antithrombotic activitiesCitation13. Previously, N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazide derivatives with antihemostatic profile have been synthesized and their chemical structures were characterized by our group ()Citation8,Citation14. The hit molecules previously found were shown to successfully inhibit platelet aggregation in rabbit platelet-rich plasma (PRP) using ARA as a proaggregant stimuli, highlighting compound 3a, whose potency (IC50=61 μM) was nearly five times better than ASA versus (IC50 =300 μM).
In this work, we proceed in the drug development pipeline of these series by elucidating the pharmacological, biochemical and toxicological features of these most active pyrazolopyridines derivatives (3a, 3b, 3d, 3h and 3j) in human blood using ASA for comparison. The Chemical Abstracts Service (CAS) register numbers of these compounds, previously describedCitation8,Citation14, are 917369-16-5 (3a), 1340493-99-3 (3b), 1340493-48-2 (3d), 917369-21-2 (3h) and 1340494-21-4 (3j). Therefore, we evaluated their inhibition profile on human platelet aggregation against six different inducers (ARA, Collagen, ADP and Epinephrine, thrombin and the thromboxane receptor agonist U-44619). Plasma clotting times and hemocompatibility towards human healthy erythrocytes were also analyzed to evaluate cross-activity and toxicity profile. In silico molecular docking and SAR studies were used as a strategy to evaluate feasible targets for the active compounds, whereas enzyme immune assays were performed to assess the feasible mechanism of action by measuring the production of TXA2 and PGH2 by human platelets. Finally, we tested the in vivo antithrombotic profile of the derivatives using a mice model of pulmonary embolism. The overall bleeding risk of the molecules was also assessed by combining two in vivo tail bleeding assays: total bleeding time and quantitative total hemoglobin loss with Drabkin reagent.
Methods
Human and animal ethics
For in vitro assays, human blood samples were obtained from adult individuals in accordance to the Human Research Ethics Committee (ID number: 140/10) whereas for the in vivo assays, male and females BALB/c mice around 20–25 g (4–5 weeks of age) were provided by NAL-UFF (Núcleo de Animais de Laboratório – Universidade Federal Fluminense). All experiments were approved by CEUA-UFF (Ethics Committee on Animal Use) under protocol 146/13 and were in accordance with COBEA (Brazilian College of Animal Experimentation)Citation15 and CARE (Committee on Animal Research and Ethics; 2012)Citation16.
In vitro assays
Platelet aggregation assays
PRP and platelet-poor plasma (PPP) were prepared by differential centrifugation and the platelet aggregation was monitored by light transmission aggregometryCitation17 using an aggregometer Packs-4® (Helena Laboratories®, Beaumont, TX). The derivatives (100 μM) were pre-incubated in PRP for two minutes before the addition of the inducer (ARA – 500 μM; ADP – 3 μM; epinephrine – 2 μM; collagen – 5 μg/mL; U-44619 – 1 μM and thrombin – 1 UI/mL). Different concentrations of the derivatives were tested to determine the concentration required to inhibit 50% of platelet aggregation (IC50) induced by ARA. The platelet aggregation tests were performed in triplicate and the positive controls were ASA (100 μM – Sigma®, St. Louis, MO); AA-2414 (100 μM – Tocris®) and Argatroban (100 μM – Sigma®); having the vehicle, dimethyl sulfoxide (1% – Sigma®), as a negative control.
Clotting assays
We evaluated the changes in plasma clotting time for all active derivatives (100 μM) using both activated partial thromboplastin time (aPTT) and prothrombin time (PT) assays. Briefly, the citrated plasma samples of donors from HUAP with no disturbances in the hemostatic system and international normalized ratio (INR in the coagulation ≤1.3) were pooled in a total of six donors. Assays were performed in quadruplicate using the coagulation analyzer CoagLab® IV (Beijing Shining Sun Technology Co.®, Beijing, China) according to the manufacturer’s instructions (WAMA Diagnostics®, São Carlos, Brazil) as described by Sathler et al.Citation18. The positive controls were heparin (500 UI – Blausiegel®, Cotia, Brazil) and argatroban (100 μM – Sigma®) using dimethyl sulfoxide (1% – Sigma®) and NaCl (0.15 M) as negative controls.
Hemocompatibility analysis
Blood samples were obtained as described in the platelet aggregation assay and were centrifuged at 2500 rpm for 15 min. The erythrocyte pellet was washed three times with PBS by centrifugation and suspended in the same buffer. Then, 6 μL of the derivatives (100 μM) were added and incubated at 37 °C in water bath for 3, 6, 12 and 24 h. After centrifugation of the samples (2500 rpm for 15 min), the release of hemoglobin was measured using a microplate reader at 540 nm. Complete hemolysis was obtained using 1% of Triton X-100 and assays were performed in triplicates of two independent experimentsCitation19.
Determination of plasma TXB2 levels
Plasma samples pretreated with 100 μM of each derivative were used to determine the concentrations of Thromboxane B2, an index of in vitro COX-1 and TXS activityCitation20. Briefly, 10 μL of ARA (500 μM) was added to PRP samples containing the active derivative (100 μM) under stirring conditions. After five minutes, the suspensions were mixed with 3 μL of indomethacin (10 mM) stop-solution. Platelets were disrupted by sonication for five minutes and the homogenates were centrifuged at 2000 g for 10 min to allow complete release of TXB2. Plasma levels of TXB2 were measured in duplicate by competitive immunoassay using commercially available kits for EIA (TXB2 EIA Kit®, Cayman Chemical Co®, Ann Arbor, MI) according to the manufacturer’s instructions using ozagrel (100 μM – Sigma®) as a positive control.
Determination of plasma PGH2 levels
Plasma samples containing 100 μM of ASA and/or the most active derivative (100 μM) were used to determine the concentrations of prostaglandin H2, an index of in vitro COX-1 activityCitation20. Briefly, 3 μL of ozagrel (100 μM) was added to block TXS activity while COX-1 reaction was triggered with 10 μL of ARA (500 μM) in PRP samples containing each active derivative (100 μM) under stirring conditions. After five minutes, the suspensions were mixed with 3 μL of indomethacin (10 mM) stop-solution. After five minutes of sonication, platelets were disrupted and the purification step was performed according to the manufacturer’s instructions (Cayman Chemical Co®, Ann Arbor, MI). Plasma levels of PGH2 were measured in duplicate by competitive immunoassay using commercially available kits for EIA (PGH2 EIA Kit®, Cayman Chemical Co®, Ann Arbor, MI) according to the manufacturer’s instructions.
In silico assays
Structure–activity relationship studies and molecular docking
Molecular modeling calculations were performed as described elsewhereCitation8,21–24. Briefly, the three-dimensional structure of each derivative was built using SPARTAN (Wavefunction®, Irvine, CA) and the geometry optimization was obtained through the RM1 semi-empirical method followed by an ab initio calculation in quantum level using the 6-31G** basis set to obtain all stereoelectronic properties. The molecular area, volume, HOMO/LUMO energy values and electronic isosurfaces maps were calculated as described by Lourenço et al.Citation23.
Molecular docking studies were performed to identify feasible interactions between the pyrazolopyridine derivatives and COX-1 (PDB code: 1PRH) or a TXS homology modelCitation25, as well as their structural complementarityCitation26.
The three-dimensional structure of each derivative was exported to AutoDock 4.2® with the electrostatic charges calculated by SPARTAN'10 being maintained. Initially, a three-dimensional grid with dimensions of 60 × 60 × 60 Å and spacing of 0.375 Å was positioned in the active site of the target enzyme (COX-1 or TXS). Global surface mapping parameters were calculated using AutoGrid 4® softwareCitation27 and molecular docking was performed using AutoDock 4® softwareCitation27,Citation28. Lamarckian genetic algorithms were used as search parameter to find the best conformation assumed by each docking ligand in a total of 50 energy assessment (generations)Citation27. The enzyme structure was kept rigid and the ligands were kept with maximum allowed flexibility. Finally, the interactions between each enzyme and derivative were analyzed using DeepView/Swiss-PDB Viewer version 4.0® and Pymol (DeLano Scientific®, San Carlos, California).
In vivo assays
Pulmonary thromboembolism – in vivo and histological analysis
After one-hour fasting, each antiplatelet compound (3a, 3b or 3h) was administered orally (10 mM/kg) to BALB/c mice as a suspension in a mixture having 5% of Arabic gum. One hour after compound administration, thrombosis was induced by intravenous injection of thrombin (20 000 UI/kg – SIGMA®) and the surviving rate was determined five minutes after induction. The thrombin concentration used was selected from a concentration/response curve as the dose that leads to 80–90% mortality rate. Animals that did not die within this time were euthanized by anesthesia overdose beside a group of animals pretreated with derivative 3a that survived after thrombin injection (n = 3), which were monitored for five consecutive days. Three groups with three animals were used for statistical analysis of each compound or control. DMSO 0.5% (vehicle, dimethyl sulfoxide 0.5% – SIGMA®) was used as negative control whereas ASA (10 mM/kg – SIGMA®) was used as positive control. Further, necropsy of the lungs was followed by histological processing and HE staining as described elsewhereCitation29.
Tail bleeding assay with Drabkin reagent
Procedures and pretreatment prior to induction were the same as described for our pulmonary thromboembolism model. After complete anesthesia of the animals, a distal 5 mm segment of the tail was amputated 90° vertically to the vessel. After amputation, blood was collected in microtubes containing 1.5 mL of Drabkin reagent (Bioclin®, Quibasa®, Belo Horizonte, Brazil) to allow a quantitative analysis of total hemoglobin loss. At different time points, 100 μL of the solution was transferred to a 96-well plate and absorbance was measured spectrophotometrically using a Microplate spectrophotometer at 420 nm (SpectraMax® 190 –Molecular Devices®, Sunnyvale, CA). Time points were collected each five minutes through a total of 30 min per animal. Hemoglobin concentration per well was obtained through fitting to a standard curve using the equation:
The calibration factor was defined as the ratio between the dose of an hemoglobin standard over the observed absorbance. Titrations of the hemoglobin standard and relative absorbance were carried out for accurate measurement of the calibration factor, as described in manufacturer instructions (Bioclin® Quibasa®). Three groups of three animals were used for the statistical evaluation of each compound and controls. DMSO 0.5% (vehicle dimethyl sulfoxide 0.5% – SIGMA®) was used as negative control, ASA (10 mM/kg – SIGMA®) and Rixaroxaban (RIVA – 1 mM/kg), were used as positive controls. At the end of experiment, blood samples from each specimen were collected and tested in recalcification time assays.
Tail bleeding assay with filter paper
Procedures prior to tail amputation were the same as described in tail bleeding assay with Drabkin reagent. After amputation, the blood leaking from the tail was blotted every 10 s on filter paper until bleeding has stopped. Importantly, the operator was blinded to the compound administrated to the mice to surpass operator bias. Special care was taken not to traumatize the lesion and re-induce bleedingCitation30. Three groups of three animals were included for statistical analysis of each compound and controls. At the end of experiment, the blood of each animal was extracted by cardiac puncture for further evaluation in ex vivo recalcification time assays.
Ex vivo recalcification time
We collected the blood by cardiac puncture from animals used in tail bleeding assays with a disposable syringe containing 1:100 (v/v) of 3.2% sodium citrate solution. Then, the blood was prepared using the same method described in platelet aggregation assays. PRP was analyzed by adding 200 μL of CaCl2 to trigger the reaction performed in quadruplicate using the coagulation analyzer CoagLab® IV (Beijing Shining Sun Technology Co. Ltd.®, Beijing, China) according to Wang et al.Citation31.
Statistical analysis
The statistical analyses were performed with ANOVA followed by the Tukey test, with significant value of p ≤ 0.05 in the SPSS 14.0® for Windows (SPSS Inc., Chicago, IL). All results were expressed as the mean ± standard deviation.
Results
In vitro assays
The antihemostatic profile of the N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazide derivatives (3a, 3b, 3d, 3h and 3j) was initially evaluated in vitro. These antiplatelet assays showed that 3a, 3b and 3h (100 μM) inhibit human platelet aggregation induced by ARA (3a=95.14%±0.29; 3b=93.57%±0.50; 3h=92.15%±0.52) at a similar extent to ASA (96.00% ± 0.29) and better than ozagrel (79.00% ± 0.50) ().
Table 1. Effect of the N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazide derivatives on in vitro platelet aggregation induced by arachidonic acid (500 μM); collagen (5 μg/mL); ADP (3 μM) and epinephrine (2 μM).
Platelet aggregation induced by collagen (5 μg/mL) was highly inhibited by 3b (76.10% ± 8.30) followed by derivatives 3h and 3a (74.60% ± 0.05 and 51.60% ± 3.80, respectively). None of the derivatives showed statistically significant activity against platelet aggregation induced by ADP, epinephrine, U-44619 or thrombin ().
The IC50 values were determined for all derivatives on AA-induced platelet aggregation to compare their potency as inhibitors. The derivative with OH in R2 position (3a) was the most potent of the series (IC50 = 29.6 μM ± 2.7), displaying superior potency to both ASA (IC50 = 40.5 μM ± 4.9) and ozagrel (IC50 = 53.1 μM ± 2.4) ().
Table 2. Comparison of stereoelectronic parameters of the N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazide derivatives and their interaction with feasible targets, cyclooxygenase-1 (COX-1) and thromboxane synthase (TXS), structure–activity relationship of derivatives.
Determination of thromboxane B2 (TXB2) plasma levels through enzyme immunoassays showed that all active molecules (3a, 3b and 3h) successfully reduced TXB2 production in PRP. Similarly to observed results at platelet aggregation assays, the derivative 3a (85.79% ± 8.37) was the most active whereas 3b and 3h presented similar inhibition rates to ASA (75.66% ± 0.49). In addition, the measured plasma levels of PGE2 showed no reduction to negative controls suggesting that derivative 3a is a selective inhibitor of TXS ().
Table 3. Effect of N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazide derivatives on in vitro plasma TXB2 and PGH2 level determination assays.
The evaluation of coagulation time determined by PT () and APTT () assays revealed no statistically significant cross anticoagulant activity for the derivatives compared to heparin and argatroban.
Figure 1. No active inhibition of the coagulation cascade is observed for N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazide derivatives in both extrinsic (A) or intrinsic pathways (B) determined by PT and aPTT assays using human platelet-poor plasma, (C) effect on healthy human erythrocytes. Pyrazolopyridines (100 μM) incubated with suspension of erythrocytes in 37 °C at different times show no significant hemolysis (>10%). Triton X-100® was used as positive control and DMSO 1% as negative control. The assays were performed in triplicate in two independent experiments. * % hemolysis >10%.
![Figure 1. No active inhibition of the coagulation cascade is observed for N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazide derivatives in both extrinsic (A) or intrinsic pathways (B) determined by PT and aPTT assays using human platelet-poor plasma, (C) effect on healthy human erythrocytes. Pyrazolopyridines (100 μM) incubated with suspension of erythrocytes in 37 °C at different times show no significant hemolysis (>10%). Triton X-100® was used as positive control and DMSO 1% as negative control. The assays were performed in triplicate in two independent experiments. * % hemolysis >10%.](/cms/asset/64e31b4b-ebff-479a-a9ef-f6a978e5f8ec/ienz_a_1158712_f0001_b.jpg)
The hemocompatibility profile for the pyrazolopyridines derivatives was determined based on the degree of erythrocyte lysis observed at four different time points (3, 6, 12 and 24 h) of incubation at 37 °C. Accordingly to Fisher et al.Citation32, hemolysis values below 10% are considered non-hemolytic, confirming the acceptable toxicity profile of the derivatives tested ().
In silico assays
The chemical structures of all N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazide derivatives tested in this work are represented in . Furthermore, the electrostatic potential map showed that the derivatives exhibited local charges at R2 position, which may play an important role in the ligand–enzyme interactions. According to our results, 3b and 3h, both with similar potency for antiplatelet activity (IC50 = 58.2 μM ± 1.9 and 59.0 μM ± 2.5, respectively), presented negative charges at R2 due to the cyan and nitro groups, respectively. A different pattern is observed for derivative 3a (IC50 = 29.6 μM ± 2.7), in which the hydroxyl group add both positive and negative local charges due to the partial charges of hydrogen (δ+) and oxygen (δ−). Such electronic feature revealed R2 as an H-bond donor/acceptor group in 3a, differing from H-bond acceptor groups at R2 of 3b and 3h. Interestingly, the derivatives with no significant inhibitory profile (3d and 3j, IC50>100 μM) presented a neutral local charge at this particular region, reinforcing the importance of polar interactions with R2 groups in the ligand–enzyme binding interface ().
Figure 2. (A) Chemical structure and substituents of N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazide derivatives (3a, 3b, 3d, 3h and 3j). (B) Electrostatic potential map of N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazide derivatives. The black square marks the R2 position. Docking complexes are observed between derivatives 3a (C), 3b (D) and 3h (E) with thromboxane synthase. π–cation interactions with the heme iron are shown in purple. Hydrogen bonds are shown as dashed orange lines.
![Figure 2. (A) Chemical structure and substituents of N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazide derivatives (3a, 3b, 3d, 3h and 3j). (B) Electrostatic potential map of N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazide derivatives. The black square marks the R2 position. Docking complexes are observed between derivatives 3a (C), 3b (D) and 3h (E) with thromboxane synthase. π–cation interactions with the heme iron are shown in purple. Hydrogen bonds are shown as dashed orange lines.](/cms/asset/cb8e4071-2df6-4781-8acc-fd78d0ae8da1/ienz_a_1158712_f0002_c.jpg)
The analysis of the docking complex of cyclooxigenase-1 active site with the derivatives revealed a low free-energy binding (BE = −7.25 to −6.07) (). Importantly, these derivatives were kept at the entrance to the active site, which consists of four amphipathic helices and involves ARG120, GLU524 and TYR355 residues, forming the input channel of the enzymeCitation33,Citation34. Differently, ASA (BE = −3.28 kcal/mol), due to its size, enters the active site and interact with SER530, the higher binding energy observed for ASA is due to the high Brownian movement of the molecule relative to its size comparable to the enzyme active site. A feature that is related to the slow mechanism of action and enzyme inhibition were observed in aspirinCitation35.
Docking analysis of the derivatives with TXS active site revealed lower binding energies (BE = −8.14 to −7.08 kcal/mol) than those observed for COX-1 complexes and ozagrel (BE = −5.15 kcal/mol) (). Accordingly to our results, the stability of derivatives 3a, 3b and 3h in the TXS catalytic site occurs due to: (a) strong 1.65 Å hydrogen bonds between ARG371 and the N′-acylhydrazone group; (b) hydrophobic contacts between N′-substituted-phenyl rings in the pyrazolopyridine moiety and the enzyme active site; and (c) feasible cation–π interactions with heme iron (). Interestingly, due to the substitution of a hydroxyl group to the R2 position, 3a was able to establish two hydrogen bonds with ILE181 and ILE183 (), differing from derivatives 3b () and derivative 3h (), which is supportive for its higher inhibition profile among the series, as pointed by the SAR studies.
In vivo and ex vivo assays
The derivatives (3a, 3b and 3h) were tested in an in vivo model of thrombin-induced pulmonary thromboembolism. Reinforcing both in silico and in vitro data, the derivative 3a showed the highest in vivo antithrombotic profile, increasing the survival rate of the treated BALB/c mice up to 80.0% ± 1.0 in comparison to vehicle controls. The profile observed for 3a surpasses aspirin in almost two-fold ASA (50% ± 2.0) and greatly differs from both 3b and 3h (0.0% ± 3.0 and 25.0% ± 2.0) ().
Figure 3. (A) Survival rates of mice treated with pyrazolopyridine derivatives on in vivo pulmonary thromboembolism induced by thrombin. (B) Effects of pyrazolopyridine treatment on total blood loss through tail bleeding assay. (C) Monitoring of total hemoglobin loss from animals treated with each derivative over a course of 30 min. (D) Ex vivo recalcification time of PRP isolated from mice treated with each derivative. All derivatives and ASA were assayed at 10 mM/kg and RIVA (Rivaroxaban) at 1 mM/kg. The assays were performed with three groups of three animals for each compound or control. The results are presented as the mean ± SD. *p <0.05.
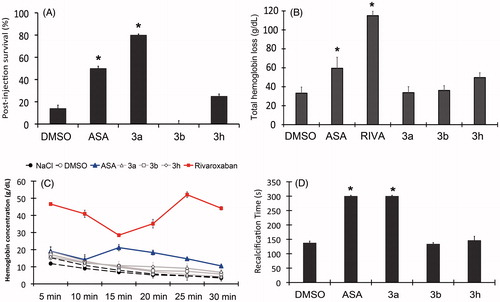
No significant increase on bleeding time was observed for the molecules through tail bleeding assay in comparison to the vehicle control while significant differences were observed for both ASA and Rivaroxaban that produced two- (178.8 s ± 20.9) and three-fold increase (260.0 s ± 13.3) of normal bleeding time (85.3 s ± 16.3), respectively (Figure not shown).
A quantitative evaluation of bleeding through the measurement of total hemoglobin release further confirmed the low bleeding risk associated to the derivatives (). No significant differences were observed between the derivatives and vehicle controls (33.2 g/dL ± 6.4), while significant hemoglobin loss was observed for both ASA and Rivaroxaban (59.5 g/dL ± 11.6 and 115.0 g/dL ± 4.8, respectively), after 30 min of bleeding. This result was reinforced by monitoring the hemoglobin concentration released through the tail amputation over time, where rapid decay of bleeding is observed for all tested derivatives while persistent bleeding is observed for both ASA and Rivaroxaban (at 10 and 15 min, respectively) ().
Although no bleeding risk is associated to the molecules in vivo, the ex vivo evaluation of PRP revealed a significant increase in recalcification time, which was observed for both animals treated with both derivatives 3a and ASA (300 s ± 0.0) (). No statistically significant increase was found in coagulation time for derivatives 3b and 3h, which reinforce their lower antithrombotic profile in vivo.
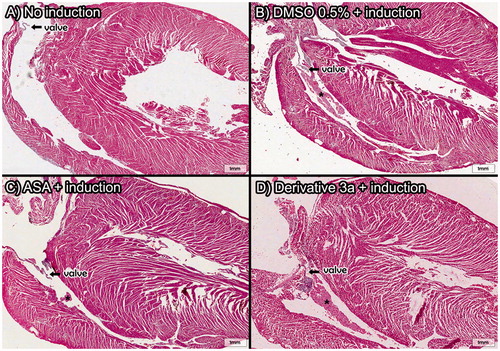
The histopathological evaluation of the lungs from mice treated with 3a showed deposition of thrombi in all thrombin-treated animals without significant differences between treated groups. However, improved blood perfusion and alveoli architecture was observed for derivative 3a in comparison to ASA (). After five days from the induction of thromboembolism, mice treated with 3a show slight sign of distress and complete recovery of alveoli integrity while residual thrombi start to dissemble (). Furthermore, histology of the heart showed recent thrombi located at the right ventricle which connects with the lung via pulmonary artery, which reveals that most of the thrombi observed in the lungs came from heart ().
Figure 4. Histopathology of the lungs isolated from mice subjects included for in vivo pulmonary thromboembolism (HnE staining, 8× magnification; scale bar = 600 μm). Microscopic emboli (*) are seen for all treated groups. (A–D) Intense bronchoconstriction is seen for all treated groups with a slight integrity of alveoli (•) observed for the 3a-treated group, which also displays increased blood perfusion (▪). Only derivative 3a was used because the high survival rate was observed previously (D). All derivatives and ASA were assayed at 10 mM/kg (HnE staining, 8× magnification; scale bar = 600 μm).
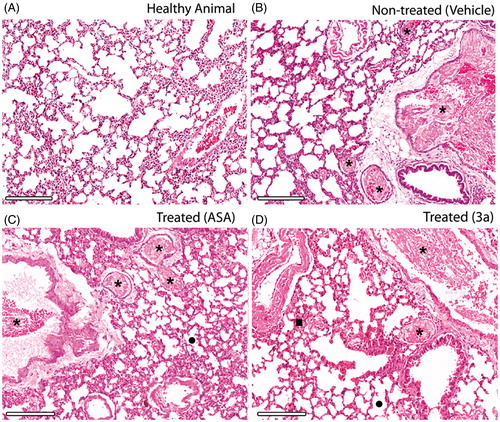
Figure 5. Histopathology of the lungs of thrombin-induced animals at the same day (A) and five days after induction (B). Integrity of alveoli and increased blood perfusion are similar for both, but microscopic emboli (*) after five days from the induction show a residual thrombi starting to disassemble. 3a derivative at 10 mM/kg (HnE staining, 8× magnification; scale bar = 600 μm).
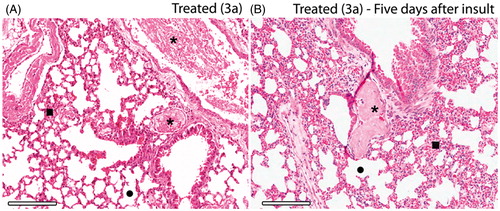
Figure 6. Heart histopathology after in vivo pulmonary thromboembolism model assays induced by thrombin. Thrombi located in the right ventricle reveal the migration of microemboli under formation to the lungs of the animal subjects. Only derivative 3a was used because the high survival rate was observed previously. All derivatives and ASA were assayed at 10 mM/kg (HnE staining, 8× magnification; scale bar = 600 μm) (A) Control group and *=thrombi.
Discussion
In this work we observed that three of the five hit compounds identified as antiplatelet agents against rabbit PRP, were able to selectively inhibit human platelet aggregation induced by ARA (3a, 3b 3h), with comparable potency to ASA. Mild inhibition of collagen-induced aggregation was also observed, as collagen-induced aggregation is partially dependent on TXA2 productionCitation36,Citation37.
According to our data, 3a (IC50 = 29.6 μM ± 2.7) is the most potent derivative of the N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazide series with improved potency in comparison to ASA (IC50 = 40.5 μM ± 4.9) and ozagrel (IC50 = 53.12 μM ± 2.4). Interestingly, both derivatives 3a and ASA presented higher activity against human platelets (IC50 = 29.6 μM ± 2.7 and 40.5 μM ± 4.9, respectively) than their rabbit counterpart (3a – IC50 = 61 μM and ASA – IC50 = 300 μM)Citation8. Since the IC50 is related to the pharmacological potency of a drugCitation38, this suggests a better pharmacological profile for 3a in comparison to ASA and ozagrel in both tested models. Likewise, our pulmonary embolism model successfully reflects the antithrombotic activity of compound 3a, which showed 80% survival in comparison to negative controls, a 30% increase in the survival promoted by ASA.
Peters et al.Citation39 reported that the risk of major bleeding increases in a dose response to ASA, reinforcing the need of finding molecules that require lower doses for maximum response, in addition to a lesser probability of developing adverse effects. Interestingly, none of the derivatives showed increased bleeding risk in all bleeding models tested and no amplification of plasma clotting time was observed for both intrinsic and extrinsic pathways of the coagulation cascade measured with PPP. Although derivative 3a did not alter the coagulation cascade in vitro, it significantly affected the recalcification time of PRP. According to Zhong et al.Citation40, the potency of antiplatelet agents can be reflected into global recalcification time, since platelet α-granules released during activation contain coagulation factors (e.g. FXIII and FV) whereas platelet shape changes upon activation, exposes negatively charged phospholipids, which promotes the further assembly of tenase and protrombinase complexes, leading to a stronger clotting stimuli.
Analogous to the low bleeding risk observed for the compounds, a low toxicity was also observed in vitro towards healthy human erythrocytes regardless of the exposure time, which also relates to the lower concentration of free Hb observed at the in vivo bleeding assays.
Despite the protective role of ASA and 3a against thrombosis and the improved survival rate they promote, no significant difference was detected in the histopathological analysis of the lungs from the animals treated with them. However, animals that survived the induction of pulmonary thromboembolism through prior treatment with derivative 3a, showed reduction of thrombus size and thrombi deposition after five days, indicating that the mechanism of action of this derivative may ease fibrinolysis and clot removal over time, which may be related to the high survival rate observed for this moleculeCitation41,Citation42.
The lack of inhibition observed for U-44619-induced platelet aggregation suggests that the presence of the compounds does not influence the TP receptor activity, prompting that the inhibition of ARA-induced aggregation relies in the inhibition of upstream enzymesCitation43–45. The measurements of TXB2 and PGE2 plasma levels comprise a usual tool to assess the normal function of COX-1 and TXS in PRP samplesCitation46–48. Through the application of this tool, we demonstrate the selectivity of the derivative 3a to inhibit the production of TXB2 but not PGE2, a feature only observed by TXS-selective inhibitorsCitation49,Citation50.
Results from the computational SAR studies reveal that structural substitutions at the R2 position – with the introduction of hydrogen bond donor and acceptor groups – as important to the antithrombotic profile of the active derivatives (3a IC50 = 29.6 μM ± 2.7; 3b IC50 = 58.2 μM ± 1.9; and 3h IC50 = 59.0 μM ± 2.5) in comparison to those substituted with neutral groups (3i and 3j). These results corroborate to the stereoelectronic and pharmacokinetic parameter data previously reported by Geraldo et al. for their activity against rabbit PRPCitation8.
Results from molecular docking support the SAR analysis and in vitro profile all together by revealing an increased number of hydrogen bonds between 3a and isoleucine residues at TXS active site in comparison to other R2-substituted analogs, whose binding modes rely primarily on the establishment of cation–π interactions with the enzyme heme and arginine residues, both of which are also present in 3a interaction panel. The new hydrogen bonds improve stability of 3a over the TXS heme group through the interactions with ARG371 and ARG374, pointed as important anchors sites for TXS inhibitionCitation51,Citation52. Potent inhibitors of TXS display a key cation–π interaction with the iron atom from heme group present in the enzyme catalytic site. This interaction is well-known in literatureCitation53–55 specially for imidazole- or 3-pyridine-based inhibitorsCitation52,Citation56. Accordingly, our results revealed that the phenyl ring in 3a, 3b and 3h derivatives oriented toward the heme group establish a feasible cation–π interaction with the heme iron. This interaction was previously shown to occur between the heme group and aromatic rings lacking nitrogen through the displacement of a conserved water molecule lying close to the heme iron atomCitation57. Likely being the mechanism of action observed by these active compounds, the displacement of this water molecule by aromatic rings was previously reported to reduce the production of TXA2Citation58 which reflects the results observed for 3a in our in vitro EIA.
The inhibition of cyclooxygenases was demonstrated to be an unlikely mechanism, since 3a was not able to inhibit the ex vivo production of PGH2, quantified through EIA. Supported by molecular docking simulations, this work reveals that none of the derivatives were able to bind residues from the COX-1 catalytic site. Although interactions with both ARG120 and TYR355 were observed, the literature reports that such interactions are only significant for molecules capable of occupying the hydrophobic cavity of COX-1, ultimately blocking ARA interaction with TYR385 tyrosyl radicalCitation59 a feature that is absent for all ligand–enzyme complexes with the molecules presented herein, as they are not capable of docking to the vicinity of the active site.
This pharmacological profile suggests an improvement in comparison to current antiplatelet drugs, as the maintenance of endogenous endoperoxides (e.g. PGI2) is not affected by TXS selective inhibitors as it is for inhibitor COX-1Citation42. By keeping PGH levels unaltered through treatment with 3a, the activation of the TP receptor by PGH2 or the production of natural platelet modulators such as PGI2 or prostacyclin is maintained by both platelets, endothelial and inflammatory cells, which lead to improved hemostasisCitation60.
Considering that pulmonary thromboembolism is not only dependent of vessel obstruction but also on bronchoconstriction and vasoconstriction events, the inhibition of prostaglandins production through active COX-1 inhibition decreases lung gas exchange and generates an increase in bleeding timeCitation61–63. TXS inhibitors in other hand, reverse these effects and offer a lower bleeding risk with improved oxygen perfusionCitation3,Citation64 through improved vasodilation and bronchodilation, as well the formation of more soluble thrombiCitation41,Citation42,Citation63. This unique characteristic may be the underlying reason for the improved survival rate observed for derivative 3a in comparison to ASA.
Moreover, ASA is not able to inhibit the formation of thrombi in femoral vein of rats even with complete inhibition of TXA2, different from the profile observed for ozagrel or isbogrel, which showed reduced thrombi size, most likely due to the concurrent maintenance of endogenous PGI2 levelsCitation41. Since an increased TXA2–PGI2 ratio is an important factor to initiate thrombosis, reducing this ratio through selective TXS inhibitors is likely to shift the thrombotic stimuli towards fibrinolysisCitation65. This improved maintenance of hemostasis offered by selective TXS-inhibitors may be the reason why animals treated with 3a have survived thrombin-induced thrombosis and completely recovered from disseminated intravascular coagulation after five consecutive days. This work supports the potential of selective TXS-inhibitors as a promising class of antithrombotic and fosters the development and further characterization of the mechanism underlying the antiplatelet activity of 1H-pyrazolo[3,4-b]pyridine derivatives.
Conclusion
Overall this work evaluated the potential of novel five N′-substituted-phenylmethylene-3-methyl-1,6-diphenyl-1H-pyrazolo[3,4-b]pyridine-4-carbohydrazide derivatives as new antithrombotic agents. Results highlight the antiplatelet activity of the series through selective inhibition of the ARA-induced aggregation of human platelets presenting strong in vitro and in silico evidences of a mechanism able to selectively impair TXB2 formation ex vivo. Active compounds were shown to display equal and superior potency to commercial NSAIDs in vitro, with an enhanced protective role in an animal model of pulmonary thromboembolism. A lack of hemolytic activity of healthy human erythrocytes and a lower bleeding risk observed in two distinct models of tail bleeding in mice support the safety profile of these molecules in comparison to current antithrombotic drugs and prompt the optimization of the series towards the development of novel molecular entities for the treatment of thrombosis. Finally, our data reinforced the potential of pyrazolopyridine 3a for further analysis to explore its mechanism and understand its reduced bleeding time and enhanced survival rate in comparison to ASA.
Declaration of interest
The authors report no declarations of interest. This research was supported by grants and fellowships from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Rio de Janeiro (FAPERJ), CAPES, AGIR-UFF and PROPPI-UFF.
References
- Versteeg HH, Heemskerk JWM, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev 2013;93:327–58
- WHO. World Health Organization [Internet]; 2014. Available from: http://www.who.int/nmh/publications/ncd-status-report-2014/en/ [last accessed 10 Jan 2016]
- Angiolillo DJ, Datto C, Raines S, Yeomans ND. Impact of concomitant low-dose aspirin on the safety and tolerability of naproxen and esomeprazole magnesium delayed-release tablets in patients requiring chronic nonsteroidal anti-inflammatory drug therapy: an analysis from 5 Phase III studies. J Thromb Thrombolysis 2013;38:11–23
- Shah R, Keough LA, Belalcazar-Portacio A, Ramanathan KB. Ticagrelor as an alternative in clopidogrel-associated neutropenia. Platelets 2014;16:1–3
- Kunadian V, Sinclair H, Sutton A, Dangas GD. Aspirin, platelet P2Y12 receptor inhibitors, and other oral antiplatelets: comparative pharmacology and role in elective PCI. Intervent Cardiol Clin 2013;2:527–35
- Choi J-T, Shin K-A, Kim Y-K, Prevalence of aspirin resistance and clinical characteristics in patients with cerebral infarction. Kor Soc Biomed Lab Sci 2013;30:233–8
- Galanaud J-P, Laroche J-P, Righini M. The history and historical treatments of deep vein thrombosis. J Thromb Haemost 2013;11:402–11
- Geraldo RB, Bello ML, Dias LRS, et al. Antiplatelet activity and structure-activity relationship study of pyrazolopyridine derivatives as potential series for treating thrombotic diseases. J Atheroscler Thromb 2010;17:730–9
- Pinheiro LCS, Abreu PA, Afonso IF, et al. Identification of a potential lead structure for designing new antimicrobials to treat infections caused by Staphylococcus epidermidis-resistant strains. Curr Microbiol 2008;57:463–8
- Vera-Divaio MAF, Freitas ACC, Castro HC, et al. Synthesis, antichagasic in vitro evaluation, cytotoxicity assays, molecular modeling and SAR/QSAR studies of a 2-phenyl-3-(1-phenyl-1H-pyrazol-4-yl)-acrylic acid benzylidene-carbohydrazide series. Bioorg Med Chem 2009;17:295–302
- Dias LRS, Alvim MJ, Freitas ACC, et al. Synthesis and analgesic properties of 5-acyl-arylhydrazone 1-H pyrazolo [3,4-b] pyridine derivatives. Pharm Acta Helv 1994;69:163–9
- Mostafa MG, Al-Said MS. Antitumor activity of novel pyridine, thiophene and thiazole derivatives. Arch Pharm Res 2012;35:965–73
- Pinto DJP, Orwat MJ, Koch S, et al. Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4- c] pyridine-3-carboxamide (Apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa. J Med Chem 2007;50:5339–56
- Dias LRS, Santos MB, Albuquerque Sd, et al. Synthesis, in vitro evaluation, and SAR studies of a potential antichagasic 1H-pyrazolo[3,4-b]pyridine series. Bioorg Med Chem 2007;15:211–19
- COBEA. Princípios éticos na experimentação animal [Internet]; 1991 [cited 2013 Jan 1]. Available from: Disponível em <http://www.cobea.org.br> [last accessed 1 Jan 2013]
- CARE. Guidelines for Ethical Conduct in the Care and Use of Animals developed by APA’s [Internet]; 2012 [cited 2013 Jan 1]. Available from: http://www.apa.org/science/anguide.html
- Pierdoná TM, Lima NR, Rodrigues RCM, et al. The Operculina macrocarpa (l.) urb. (jalapa) tincture modulates human blood platelet aggregation. J Ethnopharmacol 2014;151:151–7
- Sathler PC, Lourenço AL, Rodrigues CR, et al. In vitro and in vivo analysis of the antithrombotic and toxicological profile of new antiplatelets N-acylhydrazone derivatives and development of nanosystems: determination of novel NAH derivatives antiplatelet and nanotechnological approach. Thromb Res 2014;134:376–83
- Bauer M, Lautenschlaeger C, Kempe K, et al. Poly(2-ethyl-2-oxazoline) as alternative for the stealth polymer poly(ethylene glycol): comparison of in vitro cytotoxicity and hemocompatibility. Macromol Biosci 2012;12:986–98
- Faye E, Drouet L, De Raucourt E, et al. Absorption and efficacy of acetylsalicylic acid in patients with short bowel syndrome. Ann Pharmacother 2014;48:705–10
- Leal B, Afonso IF, Rodrigues CR, et al. Antibacterial profile against drug-resistant Staphylococcus epidermidis clinical strain and structure–activity relationship studies of 1H-pyrazolo[3,4-b]pyridine and thieno[2,3-b]pyridine derivatives. Bioorg Med Chem 2008;16:8196–204
- Cosconati S, Forli S, Perryman AL, et al. Virtual screening with AutoDock: theory and practice. Expert Opin Drug Discov 2010;5:597–607
- Lourenço AL, Abreu PA, Leal B, et al. Identification of Nor-β-lapachone derivatives as potential antibacterial compounds against Enterococcus faecalis clinical strain. Curr Microbiol 2011;62:684–9
- Santos CBR, Lobato CC, Braga FS, et al. Application of Hartree–Fock method for modeling of bioactive molecules using SAR and QSPR. Comput Mol Biosci 2014;4:1–24
- Sathler PC, Santana M, Lourenço AL, et al. Human thromboxane synthase: comparative modeling and docking evaluation with the competitive inhibitors dazoxiben and ozagrel. J Enzyme Inhib Med Chem 2014;29:527–31
- Meng X-Y, Zhang H-X, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 2011;7:146–57
- Magalhães WS, Corrêa CM, Alencastro RBd, Nagem TJ. Bases moleculares da ação anti-inflamatória dos ácidos oleanólico e ursólico sobre as isoformas da ciclo-oxigenase por docking e dinâmica molecular. Quim Nova 2012;35:241–8
- Bullock CW, Jacob RB, McDougal OM, et al. Dockomatic – automated ligand creation and docking. BMC Res Notes 2010;3:289
- Treuting PM, Dintzis SM. Comparative anatomy and histology: a mouse and human atlas. 1st ed. Amsterdam, Boston: Academic Press; 2011. 474 p
- Liu Y, Jennings NL, Dart AM, Du X-J. Standardizing a simpler, more sensitive and accurate tail bleeding assay in mice. World J Exp Med 2012;2:30–6
- Wang X, Chen X, Xing L, et al. Blood compatibility of a new zwitterionic bare metal stent with hyperbranched polymer brushes. J Mater Chem B 2013;1:5036
- Fischer D, Li Y, Ahlemeyer B, et al. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials 2003;24:1121–31
- Bhattacharyya DK, Lecomte M, Rieke CJ. Involvement of arginine 120, glutamate 524, and tyrosine 355 in the binding of arachidonate and 2-phenylpropionic acid inhibitors to the cyclooxygenase active site of ovine prostaglandin endoperoxide H synthase-1. J Biol Chem 1996;271:2179–84
- Harman CA, Turman MV, Kozak KR, et al. Structural basis of enantioselective inhibition of cyclooxygenase-1 by S-alpha-substituted indomethacin ethanolamides. J Biol Chem 2007;282:28096–105
- Goltsov A, Lebedeva G, Humphery-Smith I, et al. In silico screening of nonsteroidal anti-inflammatory drugs and their combined action on prostaglandin H synthase-1. Pharmaceuticals (Basel) 2010;3:2059–81
- Nakahata N. Thromboxane A2: physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol Ther 2008;118:18–35
- Fintel D. Oral antiplatelet therapy for atherothrombotic disease: overview of current and emerging treatment options. Vasc Health Risk Manage 2012;8:77–89
- Zhang H, Holden-Wiltse J, Wang J, Liang H. A strategy to model nonmonotonic dose-response curve and estimate IC50. PLoS One 2013;8:e69301
- Peters RJG, Mehta SR, Fox KAA, et al. Effects of aspirin dose when used alone or in combination with clopidogrel in patients with acute coronary syndromes observations from the Clopidogrel in Unstable angina to prevent Recurrent Events (CURE) Study. Circulation 2003;108:1682–7
- Zhong S-R, Jin Y, Wu J-B, et al. Characterization and molecular cloning of dabocetin, a potent antiplatelet C-type lectin-like protein from Daboia russellii siamensis venom. Toxicon 2006;47:104–12
- Terashita Z, Imura Y, Kawamura M, et al. Effects of thromboxane A2 synthase inhibitors (CV-4151 and ozagrel), aspirin, and ticlopidine on the thrombosis caused by endothelial cell injury. Thromb Res 1995;77:411–21
- Capra V, Bäck M, Angiolillo DJ, et al. Impact of vascular thromboxane prostanoid receptor activation on hemostasis, thrombosis, oxidative stress, and inflammation. J Thromb Haemost 2014;12:126–37
- Strack AM, Carballo-Jane E, Wang S, et al. Nicotinic acid and DP1 blockade: studies in mouse models of atherosclerosis. J Lipid Res 2013;54:177–88
- Needleman P, Truk J, Jakschik BA, et al. Arachidonic acid metabolism. Annu Rev Biochem 1986;55:69–102
- Santiago E, Contreras C, García-Sacristán A, et al. Signaling pathways involved in the H2O2-induced vasoconstriction of rat coronary arteries. Free Radic Biol Med 2013;60:136–46
- Sadilkova L, Paluch Z, Mottlova J, et al. The purification step is not crucial in EIA measurements of thromboxane B2 and 11-dehydrothromboxane B2 in human plasma. Clin Lab 2012;58:177–83
- Cuendet M, Mesecar AD, DeWitt DL, Pezzuto JM. An ELISA method to measure inhibition of the COX enzymes. Nat Protoc 2006;1:1915–21
- Uno K, Iuchi Y, Fujii J, et al. In vivo study on cross talk between inducible nitric-oxide synthase and cyclooxygenase in rat gastric mucosa: effect of cyclooxygenase activity on nitric oxide production. J Pharmacol Exp Ther 2004;309:995–1002
- Fontana P, Zufferey A, Daali Y, Reny J-L. Antiplatelet therapy: targeting the TxA2 pathway. J Cardiovasc Transl Res 2013;7:29–38
- Cathcart MC, Gately K, Cummins R, et al. Thromboxane synthase expression and correlation with VEGF and angiogenesis in non-small cell lung cancer. Biochim Biophys Acta (BBA) 2014;1842:747–55
- Yanai TK, Mori S. Mechanistic insights into prostanoid transformations catalyzed by cytochrome P450. Prostacyclin and thromboxane biosyntheses. Adv Med Bio [Internet]; 2011 [cited 2013 Sep 18]. Available from: http://smori.sci.ibaraki.ac.jp/978-1-61122-467-2_ch8.pdf
- Chao W-C, Lu J-F, Wang J-S, et al. Probing ligand binding to thromboxane synthase. Biochemistry 2013;52:1113–21
- Ortiz de Montellano PR, Kunze KL, Augusto O. Hemoprotein destruction. Iron-nitrogen shift of a phenyl group in a porphyrin complex. J Am Chem Soc 1982;104:3545–6
- Montellano PRO, de Chan WK, et al. Mechanism-based probes of the topology and function of fatty acid hydroxylases. FASEB J 1992;6:695–9
- Ruan K-H, Milfeld K, Kulmacz RJ, Wu KK. Comparison of the construction of a 3-D model for human thromboxane synthase using P450cam and BM-3 as templates: implications for the substrate binding pocket. Protein Eng 1994;7:1345–51
- Liedtke AJ, Crews BC, Daniel CM, et al. Cyclooxygenase-1-selective inhibitors based on the (E)-2′-des-methyl-sulindac sulfide scaffold. J Med Chem 2012;55:2287–300
- Singh AK, Singh N, Sinha M, et al. Binding modes of aromatic ligands to mammalian heme peroxidases with associated functional implications. J Biol Chem 2009;284:20311–18
- Meunier B, de Visser SP, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem Rev 2004;104:3947–80
- Saxena A, Balaramnavar VM, Hohlfeld T, Saxena AK. Drug/drug interaction of common NSAIDs with antiplatelet effect of aspirin in human platelets. Eur J Pharmacol 2013;721:215–24
- Daham K, James A, Balgoma D, et al. Effects of selective COX-2 inhibition on allergen-induced bronchoconstriction and airway inflammation in asthma. J Allergy Clin Immunol [Internet]; 2014 [cited 2015 Jan 28]. Available from: http://www.sciencedirect.com/science/article/pii/S0091674913018538
- Delcroix M, Melot C, Lejeune P, et al. Cyclooxygenase inhibition aggravates pulmonary hypertension and deteriorates gas exchange in canine pulmonary embolism. Am Rev Respir Dis 1992;145:806–10
- Smulders YM. Pathophysiology and treatment of haemodynamic instability in acute pulmonary embolism: the pivotal role of pulmonary vasoconstriction. Cardiovasc Res 2000;48:23–33
- Petri MH, Tellier C, Michiels C, et al. Effects of the dual TP receptor antagonist and thromboxane synthase inhibitor EV-077 on human endothelial and vascular smooth muscle cells. Biochem Biophys Res Commun 2013;441:393–8
- Bassand J-P. Current antithrombotic agents for acute coronary syndromes: focus on bleeding risk. Int J Cardiol 2013;163:5–18
- Patrono C. Role of clinical pharmacology in the development of antiplatelet drugs. Clin Therapeut 2014;36:2096–111