
Abstract
In the present work, 12 new 2-(5-substituted-benzothiazol-2-ylsulfanyl)-N-(substitutedbenzyl)-N-(4-substitutedphenyl) acetamide derivatives (4a–l) was designed and synthesized. The structures of the synthesized compounds were clarified using Fourier transform infrared spectroscopy (FTIR), proton nuclear magnetic resonance (1H-NMR), carbon-13 nuclear magnetic resonance (13C-NMR) and high-resolution mass spectrometry (HRMS) spectral data. Purity of synthesized compounds was checked by high-performance liquid chromatography (HPLC) analyses and purity ratio was found between 96.5–99.9%. The inhibitory activity of the compounds against MAO-A and MAO-B enzymes was evaluated by using in vitro flurometric method in which kynuramine was used as a substrate. Most of the compounds exhibited more selective inhibitory activity towards monoamine oxidase B (MAO-B) than monoamine oxidase A (MAO-A). Compound 4h was determined as the most potent compound against both enzyme types. The MAO-B enzyme kinetic of the compound 4h was studied and nature of MAO-B inhibition, caused by this compound, was investigated. The graphical analysis of steady-state inhibition data indicated that compound 4h is a mixed type inhibitor. Theoretical calculation of absorption, distribution, metabolism, excretion (ADME) properties for the synthesized compounds was also carried out and observed data supported the potential of compound 4h.
Introduction
In the regulation of different biological processes, the monoamine oxidase (MAO) enzymes play a significant role, thereby crucial targets in drug design for the treatment of psychiatric and neurological disordersCitation1,Citation2. MAOs are in charge of catalysing the oxidative deamination of neurotransmitters and intracellular amines and thus control their concentrations in brain and peripheral tissuesCitation3,Citation4. MAOs include the covalently linked cofactor flavinadenosine-dinucleotide (FAD) and exist in outer mitochondrial membraneCitation5,Citation6. Relying on their affinities to inhibitors and specificities to substrates, two isoforms of MAO, namely MAO-A and MAO-B, have been identified in mammalsCitation7,Citation8. MAO-A predominantly metabolizes serotonin, adrenaline and noradrenaline, while MAO-B utilize phenylethylamine and benzylamine as substrateCitation8,Citation9. On the other hand, tyramine and dopamine were metabolized by both forms of the enzymeCitation10.
By means of their regulatory effect on the metabolism of neurotransmitters, MAOs are increasingly becoming an important area in medicinal chemistry. MAO-A inhibitors are clinically used mostly as antidepressantsCitation11,Citation12, whereas MAO-B inhibitors are generally used in dealing with symptoms associated with Parkinson’s and Alzheimer's diseasesCitation13,Citation14. Iproniazid is the first drug used in the therapy of depressive disorder during 1950sCitation15. First-generation MAO inhibitors, such as iproniazid, imipramine and tranylcipromine, are irreversible and nonselective against MAO isoformsCitation16. Despite their long clinical success, first-generation MAO inhibitors have a number of problems including interactions with other drugs and tyramine, which is a dietary amine and may cause a fatal hypertensive crisisCitation17,Citation18. In view of these adverse actions and restrictions, the clinical use of first generation of MAO inhibitors have been decreased, whereas second-generation MAO inhibitors, such as selegiline and moclobemide, have come into prominence due to lack of above problemsCitation19.
In view of information mentioned previously, researchers have shown an increased interest in developing novel and potent MAO inhibitors. Structural similarity with the current drugs is one of the most used strategy for new MAO inhibitor development. The presence of arylalkylamine or benzylamine functions commonly in many MAO inhibitors may be suggested as a structural requirement for the development of a new class of MAO inhibitors (). Resemblance in the chemical structures of MAO inhibitors and neurotransmitters or intracellular amines also cites to this requirement. Hence, it may conceivably be declared that arylalkylamine or benzylamine moieties contest with endogenic amines. This consideration has been supported by some studies indicating that benzylamine moiety possesses MAO inhibitory activityCitation20–22. In addition to benzylamine, the compounds containing benzothiazole moiety have been reported to inhibit MAO enzymesCitation23. Besides, it has been declared that aromatic nitro compounds have an inhibition potency on MAO enzymesCitation24.
Figure 1. Structure of various MAO inhibitors and synthesized compounds.
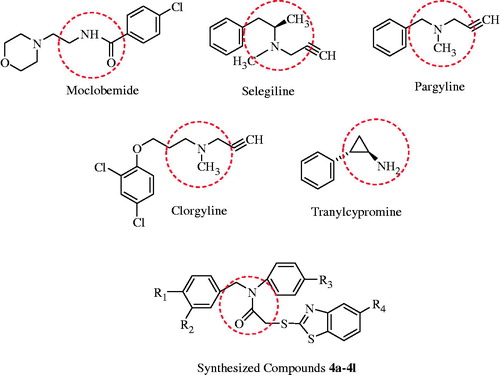
In the light of above information, herein, we report the synthesis and monoamine oxidase inhibitory activity of some novel 2–(5-substituted-benzothiazol-2-ylsulfanyl)-N-(3 or 4-nitro benzyl)-N-(4-substitutedphenyl) acetamide derivatives (4a–l).
Materials and methods
All chemicals were purchased from Sigma-Aldrich Chemicals (Sigma-Aldrich Corp., St. Louis, MO) and Merck Chemicals (Merck KGaA, Darmstadt, Germany). All melting points (m.p.) were determined by MP90 digital melting point apparatus (Mettler Toledo, OH) and were uncorrected. All reactions were monitored by thin-layer chromatography (TLC) using Silica Gel 60 F254 TLC plates (Merck KGaA, Darmstadt, Germany). Spectroscopic data were recorded with the following instruments: IR, Shimadzu Affinity 1S spectrophotometer (Shimadzu, Tokyo, Japan); NMR, Bruker DPX 500 NMR spectrometer (Bruker Bioscience, Billerica, MA), in DMSO-d6, using TMS as internal standard; M + 1 peaks were determined by Shimadzu LC/MS IT-TOF system (Shimadzu, Tokyo, Japan). Purity of synthesized compounds was checked by Shimadzu LC-20A prominence HPLC system, equipped with a Shimadzu DGU-14A degasser, LC-20 A dual piston pump, CTO-10 ASVP column oven and SPD-MI20A PDA detector, reodyne 7725i injection valve and a stainless steel GL Science Inertsil ODS-3 (4.6 × 250 mm) column. Mobile phase was used as solvent A, acetonitrile (95%); solvent B, water (5%) at a flow rate of 0.8 ml/min and a sample injection volume of 20 ml.
General procedure for synthesis of the compounds
Preparation of N-(3 or 4-nitrobenzylidene)-4-substituted anilines (1a–d)
Corresponding nitrobenzaldehyde derivative (30 mmol) appropriate four-substituted aniline (30 mmol) and catalytic amount of glacial acetic acid (0.5 ml) were refluxed in ethanol (100 ml) for 2 h. After completion of reaction, the mixture was cooled, precipitated product was filtered and recrystallized from ethanolCitation25.
Preparation of N-(3 or 4-nitrobenzyl)-4-substituted anilines (2a–d)
The compounds 1a–d (20 mmol) were dissolved in methanol (100 ml). Sodium borohydride (40 mmol, 0.76 g) was divided in to four portion (4 × 0.38 g) and added to the methanolic solution 15 min intervals. After addition of last portion, reaction mixture was allowed to stir for 1 h at room temperature. The excess of solvent was evaporated under reduced pressure, crude product was washed with water, dried and recrystallized from ethanolCitation25.
Preparation of 2-chloro-N-(3- or 4-nitrobenzyl)-N-(4-substitutedphenyl) acetamides (3a–d)
The compounds 2a–d (10 mmol) were dissolved in tetrahydrofuran (50 ml) and triethylamine (11 mmol, 1.55 ml) was added. The mixture was cooled in an ice bath and chloroacetyl chloride (11 mmol, 0.88 ml) was added dropwise with stirring. After addition of chloroacetyl chloride, the reaction mixture was stirred for additional 1 h at room temperature. The solvent was evaporated under reduced pressure, product was washed with water, dried and recrystallized from ethanolCitation26.
Preparation of N-(3- or 4-nitrobenzyl)-N-(4-substitutedphenyl)-2-((5-substitutedbenzo[d]thiazol-2-yl)thio)acetamides (4a–l)
The compounds 3a–d (2 mmol), appropriate 5-substituted benzothiazole-2-thiol (2 mmol) and potassium carbonate (2 mmol, 0.276 g) in acetone (40 ml) was refluxed for 12 h. After TLC screening, the solvent was evaporated under reduced pressure. The product was washed with water, dried and recrystallized from ethanolCitation26.
2-(Benzo[d]thiazol-2-yl-thio)-N-(4-chlorophenyl)-N-(4-nitrobenzyl)acetamide (4a)
Yield 65–67%, m.p. 119–121 °C. HPLC: 99.7% purity. IR νmax (cm−1): 3104.92–3032.06 (aromatic C–H), 2951.49–2928.99 (aliphatic C–H), 1666.50 (C=O amide), 1605.98–1427.32 (C=N, C=C), 839.03 (1,4-disubstitutedbenzene). 1H-NMR (500 Mhz, DMSO-d6, ppm) δ 4.23 (2H, s, CO-CH2), 5.04 (2H, s, N-CH2), 7.38 (1H, t, J = 8.0 Hz, Ar-H), 7.47–7.54 (7H, m, Ar-H), 7.82 (1H, d, J = 8.0 Hz, Ar-H), 8.02 (1H, d, J = 8.0 Hz, Ar-H), 8.11 (2H, d, J = 8.2 Hz, Ar-H). 13C-NMR (125 Mhz, DMSO-d6, ppm) δ 37.08, 52.76, 121.54, 122.37, 123.95, 125.03, 126.82, 129.43, 130.30, 130.50, 133.46, 135.24, 140.66, 145.39, 147.19, 152.84, 166.23, 167.17. HRMS (m/z): [M + H]+ calcd for C22H16ClN3O3S2: 470.0394; found 470.0402.
2-((Benzo[d]thiazol-2-yl-thio)-N-(4-fluorophenyl)-N-(4-nitrobenzyl)acetamide (4b)
Yield 64–65%, m.p. 149–151 °C. HPLC: 98.8% purity. IR νmax (cm −1): 3106.14–3056.02 (aromatic C–H), 2998.51–2939.04 (aliphatic C–H), 1662.64 (C=O amide), 1616.05–1427.98 (C=N, C=C), 856.75 (1,4-disubstitutedbenzene). 1H-NMR (500 Mhz, DMSO-d6, ppm) δ 4.20 (2H, s, CO-CH2), 5.03 (2H, s, N-CH2), 7.32 (2H, m, Ar-H), 7.38 (1H, t, J = 8.0 Hz, Ar-H), 7.47–7.54 (5H, m, Ar-H), 7.82 (1H, d, J = 8.0 Hz, Ar-H), 8.02 (2H, d, J = 8.0 Hz, Ar-H), 8.11 (2H, d, J = 8.4 Hz, Ar-H). 13C-NMR (125 Mhz, DMSO-d6, ppm) δ 37.13, 52.88, 117.14 (d, J = 22.6 Hz), 121.52, 122.37, 123.93, 125.02, 126.81, 129.47, 130.88 (d, J = 8.9 Hz), 135.23, 138.07, 145.45, 147.19, 152.86, 161.93 (d, J = 244.4 Hz), 166.28, 167.27. HRMS (m/z): [M + H]+ calcd for C22H16FN3O3S2: 454.0690; found 454.0693.
2-((Benzo[d]thiazol-2-yl-thio)-N-(4-chlorophenyl)-N-(3-nitrobenzyl)acetamide (4c)
Yield 66–68%, m.p. 116–118 °C. HPLC: 99.8% purity. IR νmax (cm−1): 3116.14–3008.52 (aromatic C–H), 2989.05–2902.29 (aliphatic C–H), 1664.57 (C=O amide), 1620.48–1439.21 (C=N, C=C), 840.96 (1,4-disubstitutedbenzene), 783.10 (1,3-disubstitutedbenzene). 1H-NMR (500 Mhz, DMSO-d6, ppm) δ 4.21 (2H, s, CO-CH2), 5.06 (2H, s, N-CH2), 7.37 (1H, t, J = 8.0 Hz, Ar-H), 7.42–7.47 (3H, m, Ar-H), 7.53–7.57 (3H, m, Ar-H), 7.70 (1H, d, J = 7.0 Hz, Ar-H), 7.77 (1H, d, J = 8.0 Hz, Ar-H), 8.00 (1H, d, J = 8.0 Hz, Ar-H), 8.10–8.12 (2H, m, Ar-H). 13C-NMR (125 Mhz, DMSO-d6, ppm) δ 37.12, 52.43, 121.48, 122.32, 122.81, 123.20, 124.97, 126.80, 130.29, 130.37, 130.60, 133.45, 135.08, 135.20, 139.79, 140.53, 148.31, 152.86, 166.21, 167.21. HRMS (m/z): [M + H]+ calcd for C22H16ClN3O3S2: 470.0394; found 470.0402.
2-((Benzo[d]thiazol-2-yl-thio)-N-(4-fluorophenyl)-N-(3-nitrobenzyl)acetamide (4d)
Yield 68–70%, m.p. 105–106 °C. HPLC: 96.5% purity. IR νmax (cm−1): 3112.49–3028.13 (aromatic C–H), 2997.16–2914.06 (aliphatic C–H), 1660.71 (C=O amide), 1636.62–1429.25 (C=N, C=C), 840.96 (1,4-disubstitutedbenzene), 783.10 (1,3-disubstitutedbenzene). 1H-NMR (500 Mhz, DMSO-d6, ppm) δ 4.19 (2H, s, CO-CH2), 5.04 (2H, s, N-CH2), 7.30–7.38 (3H, m, Ar-H), 7.44–7.48 (3H, m, Ar-H), 7.56 (1H, t, J = 7.9 Hz, Ar-H), 7.69 (1H, d, J = 8.0 Hz, Ar-H), 7.76 (1H, d, J = 8.0 Hz, Ar-H), 8.00 (1H, d, J = 8.0 Hz, Ar-H), 8.09–8.12 (2H, m, Ar-H). 13C-NMR (125 Mhz, DMSO-d6, ppm) δ 37.18, 52.56, 117.12 (d, J = 22.6 Hz), 121.46, 122.30, 122.78, 123.25, 124.95, 126.78, 130.34, 130.98 (d, J = 8.8 Hz), 135.14, 135.19, 137.92, 139.82, 148.30, 152.88, 161.93 (d, J = 244.3 Hz), 166.25, 167.29. HRMS (m/z): [M + H]+ calcd for C22H16FN3O3S2: 454.0690; found 454.0692.
2-((5-Chlorobenzo[d]thiazol-2-yl)thio)-N-(4-chlorophenyl)-N-(4-nitrobenzyl)acetamide (4e)
Yield 63–65%, m.p. 175–177 °C. HPLC: 98.0% purity. IR νmax (cm−1): 3101.59–3009.65 (aromatic C–H), 2994.10–2895.60 (aliphatic C–H), 1664.57 (C=O amide), 1619.91–1425.40 (C=N, C=C), 840.96 (1,4-disubstitutedbenzene). 1H-NMR (500 Mhz, DMSO-d6, ppm) δ 4.22 (2H, s, CO-CH2), 5.04 (2H, s, N-CH2), 7.43–7.55 (7H, m, Ar-H), 7.87 (1H, s, Ar-H), 8.05 (1H, d, J = 8.7 Hz, Ar-H), 8.13 (2H, d, J = 8.2, Ar-H). 13C-NMR (125 Mhz, DMSO-d6, ppm) δ 37.18, 52.72, 121.00, 123.79, 123.95, 125.00, 129.54, 130.35, 130.57, 131.71, 133.49, 134.04, 140.61, 145.37, 147.21, 153.70, 166.97, 169.13. HRMS (m/z): [M + H]+ calcd for C22H15Cl2N3O3S2: 504.0005; found 504.0003.
2-((5-Chlorobenzo[d]thiazol-2-yl)thio)-N-(4-fluorophenyl)-N-(4-nitrobenzyl)acetamide (4f)
Yield 66–68%, m.p. 192–194 °C. HPLC: 98.2% purity. IR νmax (cm−1): 3171.89–3046.19 (aromatic C–H), 2982.18–2893.38 (aliphatic C–H), 1664.57 (C=O amide), 1617.98–1427.32 (C=N, C=C), 842.82 (1,4-disubstitutedbenzene). 1H-NMR (500 Mhz, DMSO-d6, ppm) δ 4.19 (2H, s, CO-CH2), 5.03 (2H, s, N-CH2), 7.32 (2H, t, J = 8.6 Hz, Ar-H), 7.42 (1H, dd, J = 8.5 Hz–1.4 Hz, Ar-H), 7.50–7.54 (4H, m, Ar-H), 7.87 (1H, d, J = 1.4 Hz Ar-H), 8.05 (1H, d, J = 8.5 Hz, Ar-H), 8.13 (2H, d, J = 8.5 Hz, Ar-H). 13C-NMR (125 Mhz, DMSO-d6, ppm) δ 37.27, 52.86, 117.18 (d, J = 22.6 Hz), 120.98, 123.79, 123.93, 124.99, 129.58, 130.91 (d, J = 8.8 Hz), 131.70, 134.03, 138.03 (d, J = 2.9 Hz) 145.44, 147.20, 153.73, 162.01 (d, J = 244.6 Hz), 167.06, 169.21. HRMS (m/z): [M + H]+ calcd for C22H15ClFN3O3S2: 488.0300; found 488.0306.
2-((5-Chlorobenzo[d]thiazol-2-yl)thio)-N-(4-chlorophenyl)-N-(3-nitrobenzyl)acetamide (4 g)
Yield 67–68%, m.p. 164–166 °C. HPLC: 97.1% purity. IR νmax (cm−1): 3104.51–3005.39 (aromatic C–H), 2993.03–2921.66 (aliphatic C–H), 1672.28 (C=O amide), 1629.55–1427.32 (C=N, C=C), 839.03 (1,4-disubstitutedbenzene), 785.03 (1,3-disubstitutedbenzene). 1H-NMR (500 Mhz, DMSO-d6, ppm) δ 4.20 (2H, s, CO-CH2), 5.05 (2H, s, N-CH2), 7.40–7.47 (3H, m, Ar-H), 7.53–7.57 (3H, m, Ar-H), 7.69 (1H, d, J = 7.4 Hz, Ar-H), 7.78 (1H, s, Ar-H), 7.53 (1H, d, J = 8.6 Hz, Ar-H), 8.09–8.13 (2H, m, Ar-H). 13C-NMR (125 Mhz, DMSO-d6, ppm) δ 37.21, 52.44, 120.94, 122.32, 122.81, 123.27, 124.95, 130.33, 130.63, 131.67, 133.50, 134.06, 135.18, 139.77, 140.49, 148.30, 153.71, 167.05, 169.08. HRMS (m/z): [M + H]+ calcd for C22H15Cl2N3O3S2: 504.0005; found 504.0008.
2-((5-Chlorobenzo[d]thiazol-2-yl)thio)-N-(4-fluorophenyl)-N-(3-nitrobenzyl)acetamide (4 h)
Yield 65–67%, m.p. 134–136 °C. HPLC: 99.0% purity. IR νmax (cm−1): 3112.77–3010.59 (aromatic C–H), 2956.40–2891.09 (aliphatic C–H), 1660.71 (C=O amide), 1604.02–1427.32 (C=N, C=C), 842.89 (1,4-disubstitutedbenzene), 785.03 (1,3-disubstitutedbenzene). 1H-NMR (500 Mhz, DMSO-d6, ppm) δ 4.18 (2H, s, CO-CH2), 5.04 (2H, s, N-CH2), 7.32 (2H, t, J = 8.6 Hz, Ar-H), 7.41–7.48 (3H, m, Ar-H), 7.58 (1H, t, J = 7.9 Hz, Ar-H), 7.69 (1H, d, J = 7.5 Hz, Ar-H), 7.79 (1H, s, Ar-H), 8.04 (1H, d, J = 8.6 Hz, Ar-H), 8.09 (1H, s, Ar-H), 8.13 (1H, d, J = 9.1 Hz, Ar-H). 13C-NMR (125 Mhz, DMSO-d6, ppm) δ 37.31, 52.56, 117.16 (d, J = 22.6 Hz), 120.92, 122.36, 122.80, 123.33, 124.95, 130.33, 130.98 (d, J = 8.8 Hz), 131.66, 133.97, 135.25, 137.89 (d, J = 2.3 Hz), 139.82, 148.30, 153.74, 161.92 (d, J = 244.2 Hz), 167.12, 169.17. HRMS (m/z): [M + H]+ calcd for C22H15ClFN3O3S2: 488.0300; found 488.0313.
2-((5-Methoxybenzo[d]thiazol-2-yl)thio)-N-(4-chlorophenyl)-N-(4-nitrobenzyl)acetamide (4i)
Yield 66–68%, m.p. 163–165 °C. HPLC: > 99.9% purity. IR νmax (cm−1): 3097.89–3004.19 (aromatic C–H), 2982.00–2818.65 (aliphatic C–H), 1660.71 (C=O amide), 1633.49–1427.32 (C=N, C=C), 840.96 (1,4-disubstitutedbenzene). 1H-NMR (500 Mhz, DMSO-d6, ppm) δ 3.85 (3H, s, OCH3) 4.22 (2H, s, CO-CH2), 5.05 (2H, s, N-CH2), 7.02 (1H, dd, J = 8.8 Hz–2.0 Hz, Ar-H), 7.33 (1H, d, J = 2.0 Hz, Ar-H), 7.48–7.54 (6H, m, Ar-H) 7.87 (1H, d, J = 8.8 Hz, Ar-H), 8.13 (2H, d, J = 8.3 Hz, Ar-H). 13C-NMR (125 Mhz, DMSO-d6, ppm) δ 37.16, 52.74, 56.00, 105.04, 114.18, 122.62, 123.96, 126.77, 129.48, 130.33, 130.51, 133.47, 140.63, 145.38, 147.19, 154.19, 159.19, 167.14, 167.20. HRMS (m/z): [M + H]+ calcd for C23H18ClN3O4S2: 500.0500; found 500.0503.
2-((5-Methoxybenzo[d]thiazol-2-yl)thio)-N-(4-fluorophenyl)-N-(4-nitrobenzyl)acetamide (4j)
Yield 66–68%, m.p. 175–177 °C. HPLC: > 99.9% purity. IR νmax (cm−1): 3071.46–3029.03 (aromatic C–H), 2919.18–2895.38 (aliphatic C–H), 1662.64 (C=O amide), 1617.98–1419.61 (C=N, C=C), 846.75 (1,4-disubstitutedbenzene). 1H-NMR (500 Mhz, DMSO-d6, ppm) δ 3.85 (3H, s, OCH3), 4.19 (2H, s, CO-CH2), 5.03 (2H, s, N-CH2), 7.02 (1H, dd, J = 8.7 Hz–1.7 Hz, Ar-H), 7.30–7.33 (3H, m, Ar-H), 7.49–7.55 (4H, m, Ar-H), 7.87 (1H, d, J = 8.7 Hz, Ar-H), 8.13 (2H, d, J = 8.3 Hz, Ar-H). 13C-NMR (125 Mhz, DMSO-d6, ppm) δ 37.24, 52.86, 55.99, 105.03, 114.15, 117.16 (d, J = 22.6 Hz), 122.67, 123.94, 126.75, 129.52, 130.90 (d, J = 8.9 Hz), 138.04 (d, J = 2.6 Hz), 145.45, 147.18, 154.21, 159.18, 161.93 (d, J = 244.5 Hz), 167.16, 167.22. HRMS (m/z): [M + H]+ calcd for C23H18FN3O4S2: 484.0796; found 484.0790.
2-((5-Methoxybenzo[d]thiazol-2-yl)thio)-N-(4-chlorophenyl)-N-(3-nitrobenzyl)acetamide (4k)
Yield 62–65%, m.p. 140–143 °C. HPLC: > 99.9% purity. IR νmax (cm−1): 3093.67–3031.09 (aromatic C–H), 2941.18–2891.01 (aliphatic C–H), 1662.64 (C=O amide), 1625.69–1429.25 (C=N, C=C), 840.96 (1,4-disubstitutedbenzene), 783.10 (1,3-disubstitutedbenzene). 1H-NMR (500 Mhz, DMSO-d6, ppm) δ 3.84 (3H, s, OCH3) 4.20 (2H, s, CO-CH2), 5.05 (2H, s, N-CH2), 7.00 (1H, dd, J = 8.8 Hz–1.8 Hz, Ar-H), 7.28 (1H, d, J = 1.8 Hz, Ar-H), 7.45 (2H, d, J = 8.2 Hz, Ar-H), 7.53–7.58 (3H, m, Ar-H) 7.69 (1H, d, J = 8.8 Hz, Ar-H), 7.85 (1H, d, J = 8.8 Hz, Ar-H), 8.10–8.13 (2H, m, Ar-H). 13C-NMR (125 Mhz, DMSO-d6, ppm) δ 37.22, 52.43, 56.00, 104.97, 114.17, 122.56, 122.82, 123.22, 126.71, 130.31, 130.38, 130.61, 133.47, 135.13, 139.77, 140.48, 148.31, 154.21, 159.15, 167.12, 167.20. HRMS (m/z): [M + H]+ calcd for C23H18ClN3O4S2: 500.0500; found 500.0497.
2-((5-Methoxybenzo[d]thiazol-2-yl)thio)-N-(4-fluorophenyl)-N-(3-nitrobenzyl)acetamide (4 l)
Yield 68–70%, m.p. 140–143 °C. HPLC: > 99.9% purity. IR νmax (cm−1): 3108.25–3003.99 (aromatic C–H), 2992.71–2831.04 (aliphatic C–H), 1670.35 (C=O amide), 1619.91–1419.61 (C=N, C=C), 848.68 (1,4-disubstitutedbenzene), 783.10 (1,3-disubstitutedbenzene). 1H-NMR (500 Mhz, DMSO-d6, ppm) δ 3.83 (3H, s, OCH3), 4.18 (2H, s, CO-CH2), 5.04 (2H, s, N-CH2), 7.00 (1H, dd, J = 8.8 Hz–2.0 Hz, Ar-H), 7.28–7.33 (3H, m, Ar-H), 7.46–7.47 (2H, m, Ar-H), 7.58 (1H, t, J = 7.9 Hz, Ar-H), 8.86 (1H, d, J = 8.8 Hz, Ar-H), 8.10–8.13 (2H, m, Ar-H). 13C-NMR (125 Mhz, DMSO-d6, ppm) δ 37.31, 52.55, 55.98, 104.97, 114.13, 117.14 (d, J = 22.5 Hz), 122.54, 122.80, 123.26, 126.70, 130.35, 130.99 (d, J = 8.7 Hz), 135.20, 137.87, 139.80, 148.30, 154.24, 159.15, 161.93 (d, J = 244.5 Hz), 167.18, 167.27. HRMS (m/z): [M + H]+ calcd for C23H18FN3O4S2: 484.0796; found 484.0786.
MAO activity assay
Enzyme activity assay was performed according to the procedure reported by Matsumoto et al.,Citation27 with slight modifications. The entire materials used in enzymatic assay were purchased from Sigma-Aldrich Chemicals (Sigma-Aldrich Corp., St. Louis, MO). All of the pipettings in the assay were performed by Biotek Precision robotic system (BioTek Instruments, Inc., Winooski, VT). In order to prevent light effect on MAO enzymes the assay was performed in 96-well black plates. Phosphate buffer (0.1 M, pH = 7.4) was used for the preparation of stock solutions (5 mg/ml) of both human recombinant MAO-A and MAO-B enzymes. Enzyme stock solutions was further diluted with assay buffer to get a final concentration 0.006 mg/ml for MAO-A and 0.015 mg/ml for MAO-B. Kynuramine was dissolved in sterilized distilled water to get stock solution (25 mM) and then diluted with assay buffer to get a final concentration of 40 μM for MAO-A enzyme and 20 μM for MAO-B enzyme. Synthesized compounds and reference drugs (moclobemide for MAO-A enzyme and selegiline for MAO-B enzyme) were diluted to 10−3 M and 10−4 M concentrations (100 μl/well) using 2% DMSO. The addition of the compounds dilutes was followed by the addition of either MAO-A or MAO-B enzyme (50 μl/well). After 10-min incubation at 37 °C, kynuramine (50 μl/well) was added and enzyme–substrate reaction was initiated. The plate was incubated for 20 min at 37 °C, and then, reaction was terminated by using of 2 N NaOH (75 μl/well). The fluorimetric read from top was performed by BioTek-Synergy H1 multimode microplate reader (BioTek Instruments, Inc., Winooski, VT) at 310/380 nm excitation/emission wavelength pair. The same procedure was followed for further concentrations (10−5–10−9 M, 100 μl/well) of the inhibitors and selected compounds indicating ≥50% inhibition at initial concentrations (10−3 and 10−4 M, 100 μl/well). The IC50 value was calculated from the plots of enzyme activity against concentrations by applying regression analyses on GraphPad Prism Version 5.
Enzyme kinetic studies
The same materials were used in MAO-inhibition assay. The compound 4 h was prepared at IC50 concentration that calculated in enzyme assay and then added to the wells (100 μl/well). Stock solution (25 mM) of kynuramine 0.1 M phosphate buffer was diluted to final concentrations of 40, 20, 10, 5, 2.5 and 1.25 μM and then added to the wells (50 μl/well) that contain the test compound. The monoamine oxidase-B enzyme was added to the plate (50 μl/well) and this step was followed by a 20-min incubation period at 37 °C. The plate was read at 310/340 nm excitation/emission wavelength pairCitation28. Control measurement without inhibitor was also determined simultaneously. The results were analysed as Lineweaver–Burk plots using Microsoft Office Excel 2013.
Theoretical calculation of ADME parameters
In order to evaluate pharmacokinetic profiles of the synthesized compounds, some physicochemical parameters that are used for determination of ADME properties were calculated using the Molinspiration property calculation programCitation29.
Results and discussion
Chemistry
Target molecules (4a–g) were synthesized in four steps as shown in . Initially, N-(3 or 4-nitrobenzylidene)-4-substituted anilines (1a–d) were synthesized via condensation of appropriate benzaldehyde and aniline in ethanol with catalytic amount of glacial acetic acid. Secondly, compounds 1a–d in methanol was reduced to N-(3 or 4-nitrobenzyl)-4-substituted anilines (2a–d) by using NaBH4. In the third step, 2-chloro-N-(3- or 4-nitrobenzyl)-N-(4 substitutedphenyl)acetamides (3a–d) were synthesized by acetylation of the compounds 2a–d in tetrahydrofuran (THF) with chloroacetyl chloride. Finally, bimolecular nucleophilic substitution (SN2) reaction between appropriate 5-substituted benzothiazole-2-thiol derivatives and the compounds 3a–d give the 12 new N-(3- or 4-nitrobenzyl)-N-(4-substitutedphenyl)-2-((5-substitutedbenzo[d]thiazol-2-yl)thio)acetamides (4a–l).
Figure 2. The synthetic pathway of the compounds (4a–l).
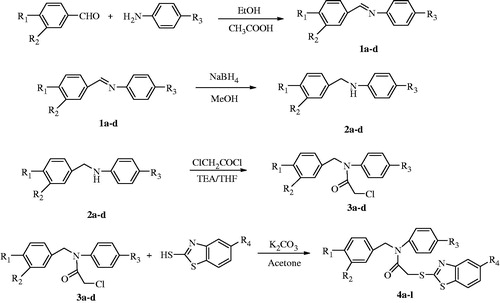
Structures of the obtained compounds were elucidated by spectral data. In the IR spectra, significant stretching bands belonging to C=O were observed between 1670–1660 cm−1. The stretching bands for C=N and C=C were recorded in the region of 1593–1419 cm−1. Stretching bands at 783–786 cm−1 and 839–857 cm−1 were observed for 1,3-disubstituted and 1,4-disubstituted benzenes, respectively. In the 1H-NMR spectra, a singlet peak between 4.18–4.23 ppm was assigned for the protons of acetamide (–NHCOCH2) moiety. A singlet peak at 5.03–5.06 ppm was recorded for methylene (–CH2) protons of benzylamine (–NCH2Ph). The methoxy (–OCH3) protons in compounds 4 g–l were assigned as a singlet at about 3.84 ppm. The protons belonging to nonsubstituted benzothiazole were observed at 7.38–8.05 ppm as two doublets and one triplet. Substituted benzothiazole peaks were determined as two doublet and one doublet of doublet at 7.01–8.05 ppm. Nitrobenzene protons near to nitro group observed at 8.11–8.13 ppm as a doublet or multiplet depending on 1,4 or 1,3-disubstitutednitrobenzene, respectively. Other aromatic protons recorded as multiplet around 7.50 ppm. In the 13C-NMR spectra, peaks between 37.08–37.31 ppm and 52.43–52.88 ppm was observed for the methylene (-CH2) carbons of acetamide (-NHCOCH2) and benzylamine (-NCH2Ph) moieties. The peaks belonging to methoxy (-OCH3) carbons in compounds 4 g–l were observed at about 56.00 ppm. Aromatic carbons were generally observed at 104.97–162.02 ppm. The carbonyl of acetamide and second carbon of benzothiazole were observed between 166.21–167.18 ppm and 169.21–167.17 ppm, respectively. According to HPLC analysis, purity ratio was found between 96.5–99.9%. Peak purity index of all compounds were also checked and no impurity was determined in observed peaks.
Monoamine oxidase inhibitory activity
The synthesized 2–(5-substituted-benzothiazol-2-ylsulfanyl)-N-(substitutedbenzyl)-N-(4-substitutedphenyl)acetamide derivatives (4a–l) were investigated for their MAO-A and MAO-B inhibitory activity by an in vitro flurometric method. The fundamental of the activity determination is based on the ability of both MAO-A and MAO-B enzymes to metabolize non-fluorescent kynuramine, which is a suitable substrate for both isozymes, to the fluorescent product 4-hydroxyquinoline. Both enzymes were pre-incubated with test compounds and the MAO-inhibitory effect of the test compounds was correlated with the amount of 4-hydroxyquinoline formed. The read of the activity was performed by using proper excitation/emission wavelength pairCitation27. DMSO was considered as control (100% activity). Moclobemide and selegiline were used as reference drugs. The inhibitory activity results are listed in and .
Table 1. Inhibitory activity (%) of the compounds against MAO-A enzyme.
Table 2. Inhibitory activity (%) of the compounds against MAO-B enzyme.
In general, it was observed that the synthesized compounds are more potent against MAO-B enzyme when compared with MAO-A enzyme. The compounds 4 h, 4k and 4 l displayed considerable inhibitory activity against MAO-A enzyme, whereas compounds 4c, 4d and 4 h indicated remarkable inhibition profile against MAO-B enzyme. Due to their inhibition potency (>50%) at 10−3 and 10 −4 M, related compounds were tested at further concentrations (10−5–10−9 M) and their IC50 values were calculated ( and ). Compound 4 h was the most active derivative with a 17.00 μM and 2.95 μM IC50 values against MAO-A and MAO-B enzymes, respectively.
The MAO-B enzyme kinetic of the most active compound 4 h was studied. The nature of MAO-B inhibition, caused by this compound, was investigated by the graphical analysis of steady-state inhibition data (). According to reciprocal plots (Lineweaver–Burk plots) compound 4 h was found as a mixed type inhibitor, due to different intercepts on both the y- and x-axes. The values of Km and Vmax were calculated by non-linear regression according to literatureCitation28 and found as 185.19 and 578.57, respectively.
Figure 3. Lineweaver–Burk plots for compound 4 h (IC50 = 2.95 μM). Substrate (kynuramine) concentrations used: 40, 20, 10, 5, 2.5 and 1.25 μM. 1/V: 1/velocity of reaction [1/(nmoles/min/mg protein)], 1/S: 1/substrate concentration (1/μM).
![Figure 3. Lineweaver–Burk plots for compound 4 h (IC50 = 2.95 μM). Substrate (kynuramine) concentrations used: 40, 20, 10, 5, 2.5 and 1.25 μM. 1/V: 1/velocity of reaction [1/(nmoles/min/mg protein)], 1/S: 1/substrate concentration (1/μM).](/cms/asset/916bf22b-5778-4846-8c77-2ff43b486bc0/ienz_a_1161621_f0003_b.jpg)
In the design of the target compounds (4a–l), the substitution pattern was performed on three regions to discuss substitution effect on biological activity. These were 3- or 4-nitrobezyl, 4-chloro or 4-florophenyl and 5-methoxy or 5-chlorobenzothioazole-2-thiol substructures. It was observed that there is no significant relationship between enzyme inhibitory activity and substituent type on benzothiazole and phenyl rings. On the other hand, an important finding was observed with 3- or 4-nitrobezyl fragment. It was determined that 3-nitro substition at this part increases the inhibitory activity significantly against both enzyme types when compared 4-nitro substitution.
Theoretical determination of ADME properties
The theoretical prediction of ADME properties (molecular weight, log p, topological polar surface area (TPSA), number of hydrogen donors and acceptors, volume) of all target compounds (4a–l) was carried out and presented in along with violations of Lipinski’s ruleCitation30,Citation31. This rule suggests that, an orally active drug has no more than one violation. Thus, according to the data in , compounds 4e, 4 g, 4i and 4k does not stick the Lipinski’s rule due to two violations. On the other hand, all calculated physicochemical parameters for the compound 4 h are compatible with the Lipinski’s rule except log p value. Although the log p (6.29) of compound 4 h exceeds Lipinski’s limit it shows that the related compound has a lipophilic character which is suitable to cross central nervous system (CNS). Furthermore, TPSA, described to be a predictive indicator of membrane penetration, is positive (79.03) and as MAO inhibitors have to pass different membranes and reach the CNS, this supports the potential of compound 4 h.
Table 3. Substituent pattern and some physicochemical parameters of the compounds 4a–l used in prediction of ADME profiles.
Declaration of interest
The authors declare no conflicts of interest.
References
- Ramsay RR. Monoamine oxidases: the biochemistry of the proteins as targets in medicinal chemistry and drug discovery. Curr Top Med Chem 2012;12:2189–209.
- Ramsay RR. Inhibitor design for monoamine oxidases. Curr Pharm Des 2013;19:2529–39.
- Khattab SN, Khalil HH, Bekhit AA, et al. Synthesis and preliminary biological evaluation of 1,3,5-triazine amino acid derivatives to study their MAO inhibitors. Molecules 2015;20:15976–88.
- Hadizadeh F, Ghodsi R. Synthesis of novel N-substituted imidazolecarboxylic acid hydrazides as monoamine oxidase inhibitors. Farmaco 2005;60:237–40.
- Passos CDS, Soldi TC, Torres Abib R, et al. Monoamine oxidase inhibition by monoterpene indole alkaloids and fractions obtained from Psychotria suterella and Psychotria laciniata. J Enzyme Inhib Med Chem 2013;28:611–18.
- Khattab SN, Haiba NS, Asal AM, et al. Synthesis and evaluation of quinazoline amino acid derivatives as mono amine oxidase (MAO) inhibitors. Bioorg Med Chem 2015;23:3574–85.
- Bolasco A, Fioravanti R, Carradori S. Recent development of monoamine oxidase inhibitors. Exp Op Ther Patents 2005;15:1763–82.
- Choi JW, Jang BK, Cho NC, et al. Synthesis of a series of unsaturated ketone derivatives as selective and reversible monoamine oxidase inhibitors. Bioorg Med Chem 2015;23:6486–96.
- De Colibus L, Li M, Binda C, et al. Three-dimensional structure of human monoamine oxidase A (MAO A): relation to the structures of rat MAO A and human MAO B. Proc Natl Acad Sci USA 2005;102:12684–9.
- O’Carroll AM, Fowler CJ, Philips JP, et al. The deamination of dopamine by human brain monoamine oxidase. Specificity for the two enzyme forms in seven brain regions. Naunyn-Schmiedebergs Arch Pharmacol 1983;322:198–202.
- Legoabe LJ, Petzer A, Petzer JP. 2-Acetylphenol analogs as potent reversible monoamine oxidase inhibitors. Drug Des Devel Ther 2015;9:3635–44.
- Chirkova ZV, Kabanova MV, Filimonov SI, et al. Inhibition of monoamine oxidase by indole-5,6-dicarbonitrile derivatives. Bioorg Med Chem Lett 2015;25:1206–11.
- Checkoway H, Franklin GM, Costa-Mallen P, et al. A genetic polymorphism of MAO-B modifies the association of cigarette smoking and Parkinson's disease. Neurology 1998;50:1458–61.
- Minders C, Petzer JP, Petzer A, Lourens AC. Monoamine oxidase inhibitory activities of heterocyclic chalcones. Bioorg Med Chem Lett 2015;25:5270–6.
- Ban TA. Pharmacotherapy of depression: a historical analysis. J Neural Transm 2001;108:707–16.
- Nayak BV, Ciftci-Yabanoglu S, Bhakat S, et al. Monoamine oxidase inhibitory activity of 2-aryl-4H-chromen-4-ones. Bioorg Chem 2015;58:72–80.
- Van Dyk AS, Petzer JP, Petzer A, Legoabe LJ. 3-Coumaranone derivatives as inhibitors of monoamine oxidase. Drug Des Devel Ther 2015;9:5479–89.
- Da Prada M, Zürcher G, Wüthrich I, Haefely WE. On tyramine, food, beverages and the reversible MAO inhibitor moclobemide. J Neural Transm Suppl 1988;26:31–56.
- Yildiz O, Karahalil F, Can Z, et al. Total monoamine oxidase (MAO) inhibition by chestnut honey, pollen and propolis. J Enzyme Inhib Med Chem 2014;29:690–4.
- Lu X, Rodríguez M, Gu W, Silverman GB. Inactivation of mitochondrial monoamine oxidase B by methylthio-substituted benzylamines. Bioorg Med Chem 2003;11:4423–30.
- Upadhyay AK, Edmondson DE. Development of spin-labeled pargyline analogues as specific ınhibitors of human monoamine oxidases A and B. Biochemistry 2009;48:3928–35.
- Tripathi RK, Goshain O, Ayyannan SR. Design, synthesis, in vitro mao-B inhibitory evaluation, and computational studies of some 6-nitrobenzothiazole-derived semicarbazones. ChemMedChem 2013;8:462–74.
- Thull U, Carrupt PA, Testa B. Pargyline analogues as potent, non-selective monoamine oxidase ınhibitors. Pharm Pharmacol Commun 1998;4:579–81.
- Kimes AS, Carr DO. Inhibition of rabbit liver monoamine oxidase by nitro aromatic compounds. Biochem Pharmacol 1982;31:2639–42.
- Safa KD, Mardipour JV, Oskoei YM. Synthesis of new imines and amines containing organosilicon groups. J Organomet Chem 2011;696:802–6.
- Turan-Zitouni G, Demirayak Ş, Özdemir A, et al. Synthesis of some 2-[(benzazole-2-yl)thioacetylamino]thiazole derivatives and their antimicrobial activity and toxicity. Eur J Med Chem 2004;39:267–72.
- Matsumoto T, Suzuki O, Furuta T, et al. A sensitive fluorometric assay for serum monoamine oxidase with kynuramine as substrate. Clin Biochem 1985;18:126–9.
- Chaurasiya ND, Ibrahim MA, Muhammad I, et al. Monoamine oxidase inhibitory constituents of propolis: kinetics and mechanism of inhibition of recombinant human MAO-A and MAO-B. Molecules 2014;19:18936–52.
- M Cheminformatics, Bratislava, Slovak Republic Available from: http://www.molinspiration.com/services/properties.html [last accessed Feb 2016].
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001;19:3–26.
- Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem 2000;43:3714–17.