
Abstract
Transthyretin (TTR), a β-sheet-rich tetrameric protein, in equilibrium with an unstable amyloidogenic monomeric form is responsible for extracellular deposition of amyloid fibrils, is associated with the onset of neurodegenerative diseases, such as senile systemic amyloidosis, familial amyloid polyneuropathy and familial amyloid cardiomyopathy. One of the therapeutic strategies is to use small molecules to stabilize the TTR tetramer and thus curb amyloid fibril formation. Here, we report the synthesis, the in vitro evaluation of several halogen substituted 9-fluorenyl- and di-benzophenon-based ligands and their three-dimensional crystallographic analysis in complex with TTR. The synthesized compounds bind TTR and stabilize the tetramer with different potency. Of these compounds, 2c is the best inhibitor. The dual binding mode prevalent in the absence of substitutions on the fluorenyl ring, is disfavored by (2,7-dichloro-fluoren-9-ylideneaminooxy)-acetic acid (1b), (2,7-dibromo-fluoren-9-ylideneaminooxy)-acetic acid (1c) and (E/Z)-((3,4-dichloro-phenyl)-methyleneaminooxy)-acetic acid (2c), all with halogen substitutions.
Introduction
Transthyretin (TTR), is a 55 kDa plasma homotetrameric β-sheet-rich protein, composed by 127-amino acidsCitation1. The main function of the protein is the transport of thyroxine (T4) and retinol, through its association with retinol binding protein (RBP), in cerebrospinal fluid (CSF) and plasmaCitation2,Citation3. The major sites of protein biosynthesis are the liver and the choroid plexus of the brainCitation4. It is also produced in small amounts in the retinaCitation4 and in human placentaCitation5, so that TTR is present in human plasma and in CSF at different concentrationsCitation6. TTR circulates in plasma as a soluble protein but in some individuals, under unknown conditions it can form amyloid fibrils. In approximately 20% of the population over the age of 70, wild-type TTR becomes amyloidogenic and leads to the onset of senile amyloidosis (SSA)Citation7,Citation8. The amyloidogenic potential of TTR is enhanced by single point mutations that can be associated with TTR hereditary amyloidoses, such as familial amyloid polyneuropathy FAP and familial cardiomyopathy FAC (http://amyloidosismutations.com). Until a few years ago, the only therapy available to prevent the formation of additional amyloid deposits was to remove the source of TTR amyloidogenic variants through transplantation of a liver secreting the less amyloidogenic wild-type TTRCitation9. Unfortunately, this therapeutic approach has no effect on retinal epithelial cells and the choroid plexus, where the mutated TTR continues to be synthesized. A different non-invasive therapy to prevent fibril formation or to destroy already formed fibrils will need to be developed to alleviate unsolved ocular dysfunction and CNS symptomsCitation10–12.
The TTR monomer is constituted by 2 four-stranded antiparallel β-sheets and a short α-helix. A stable dimer is generated through a network of hydrogen-bond interactions between the two-edge β-strands H and F of each monomer. Two dimers associate back to back to build a tetramer traversed by a central cavity in which two molecules of T4 bindCitation2. The T4 hormone binds in two identical funnel-shaped sites located at dimer–dimer interface that is bisected by a two-fold axis. Although the TTR tetramer presents two symmetric binding sites, the binding of T4 in solution shows strong negative cooperativityCitation13,Citation14.
Starting from the knowledge that only 1% of TTR binding sites in CSF and in plasma are occupied by T4Citation15, Kelly and coworkersCitation16,Citation17 proposed a strategy to ameliorate TTR amyloidosis by binding small molecules to the remaining 99% of active sites left unoccupied. Consequently, a large number of TTR ligands have been studied for their ability to bind the T4 binding sites and stabilize the TTR tetramerCitation18. Among these, the non-steroidal anti-inflammatory drug diflunisal and tafamidis (Vindaquel®) were found to be able to stabilize TTR tetramers, reduce fibril formation and slow the neurological impairment due to TTR amyloidosisCitation19–21. Tafamidis was approved for the treatment of familial amyloid polineuropathy (FAP) by the European Medicines Agency in 2011 and by the Japanese Pharmaceuticals and Medical Devices Agency in 2013.
In our previous work, we evaluated fluorenyl-based inhibitors with only one unsubstituted aromatic portion linked through a flexible chain to a carboxylic acidCitation22. The most active compound 7BD shows a heteroatomic (C=NO–CH2–CH2) flexible linker between the fluorenyl and the carboxylic moiety ().
Figure 1. Chemical structures of acetic acid compounds.
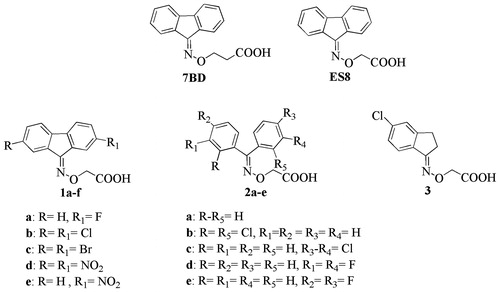
The X-ray structure of the TTR-inhibitor crystal complex shows that the fluorenyl derivative 7BD binds the TTR active sites in both the forward and reverse modes. The same result was obtained for the acetic analog ES8 even though the fluorenyl ring occupies an alternative hydrophobic area in the TTR binding sitesCitation23. The flexibility of the oxime–ether linker enables these small ligands to better tailor their binding along the TTR tunnel.
The acid moieties of 7BD and ES8 can mediate interactions with the key TTR binding site residues: Ser117, Thr119 and Lys15. Based on ES8, a new series of halogen or nitro substituted fluorenyl derivatives 1a–e were synthesized in order to evaluate the influence of such substituents on the positioning of the aromatic moiety and the carboxylic groups within the TTR binding cavity. To achieve a better understanding the spatial relationship between the aromatic and the carboxylic portion, we synthesized a series of benzophenone (2a–e) and indanone (3a) derivatives. Their simplified aromatic portions are less bulky and have different rigidity than the 9-fluorenyl derivatives.
Here we report the synthesis, in vitro evaluation using the turbidimetric UV-Vis assay and three-dimensional crystallographic analysis of TTR complex with the active ligands.
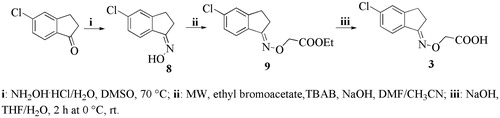
We discuss the finding that the dual binding mode, prevalent in the absence of substitutions on the fluorenyl ring, is replaced by a clear preference for a single orientation in the crystal structures of the halogen substituted analogs, (2,7-dichloro-fluoren-9-ylideneaminooxy)-acetic acid (1c), (2,7-dibromo-fluoren-9-ylideneaminooxy)-acetic acid (1b) and (E/Z)-2-((3,4-dichlorophenyl)(phenyl)methyleneaminooxy)acetic acid (2c) ( and ).
Figure 2. Electron density and binding modes for acetic acid compounds 1b, 1c and 2c. (A) Ligand 1c and its electron density represented as a multi-wire mesh to highlight the electron dense bromine atoms is shown in the context of the tetramer formed by TTR molecule A (left) and B (bottom) and their symmetry related molecules (top). (B) The bromine atoms occupy two of the halogen-specific binding sites with the carboxylate oxygens (O1 and O2) making polar contacts with Ser117 (O2—OG = 2.9 Å; O2—O = 3.1 Å) and Thr119 (O1—OG = 2.4 Å; O2—OG = 2.9 Å). Lys15, shown in the background, does not participate in binding to the carboxylate, characterizing the ligand as reverse mode binder. (C) Ligand 1b also binds to a single site in good electron density. (D) The binding mode is similar but not identical to that of 1c, the differences being characterized by longer H-bond distances with Ser117 (O1—OG = 3.1 Å; O2—O = 3.1 Å) and a single interaction with Thr119 (O1—OG = 2.8 Å). (E) There is good electron density for ligand 2c in both TTR binding sites. The carboxylate points towards Lys15 defining the ligand as a forward mode binder. (F) The salt bridge distances are: O1—NZ = 2.7 Å and O2—NZ = 3.2 Å.
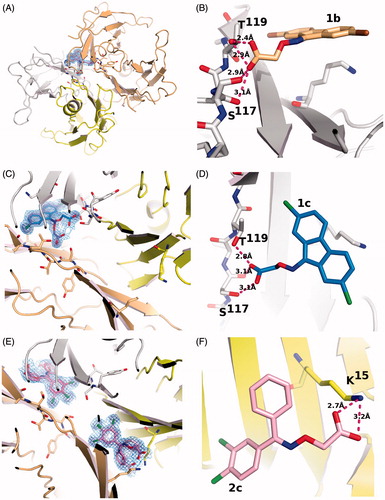
Figure 3. Comparison of ligand binding in the forward and reverse mode. (A) Ligands 1b and 1c show only minor differences in their positioning in the TTR binding site. The differences in radii between the two halides is responsible for the changes. (B) Ligands 1c and 2c show differences in the binding mode, in the halogen pocket that is used for their binding but occupy the same overall volume, when the superposition with its symmetry related molecule is considered. (C) The binding of ligand 1c is better compared to ligand 7BDCitation23, shown in salmon sticks, that to ligands ES8Citation23 shown in panel (D) Since the addition of the halogen atoms precludes a perpendicular binding mode similar to ES8, it must adopt a reverse mode binding similar to 7BD but shifted due to the presence of the halogen atoms. (E) Ligand 2c binds to both TTR binding sites with only minor differences. (F and G) These minor binding differences appear amplified when 2c is compared to ES8 in the two sites. (H) The superimposed ES8 binding to TTR show the same qualitative variation observed for 2c and for 1b relative to 1c where the carboxylate changes its orientation.
Methods
Chemistry
The 9-fluorenylaminooxyacetic (1a–e), the benzophenoneaminooxyacetic (2a–e) and indanoneaminooxyacetic (3) derivatives were prepared as shown in Scheme 1, Scheme 2 and Scheme 3, respectively. The appropriate ketones were treated with hydroxylamine hydrochloride to give the corresponding oximes (4a–e, 6a–e and 8, respectively) which, after reaction with ethyl bromoacetate, yielded the corresponding esters (5a–e, 7a–e and 9, respectively). The esters were hydrolyzed with a stoichiometric amount of 1M NaOH solution to yield the corresponding acids (1a–e, 2a–e and 3, respectively).
Scheme 1. Synthesis of fluorenyl derivatives 1a–e.
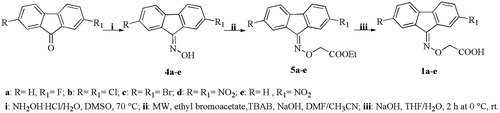
Scheme 2. Synthesis of benzophenone compounds 2a–e.
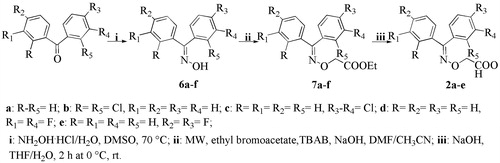
Scheme 3. Synthesis of indanone derivatives 3.
Turbidimetric assay
The percentage of fibril formation was determined by observing the turbidity of wt-TTR at pH 4.4 in absence and in the presence of inhibitory compoundsCitation22. Commercially available Prealbumin from Human Plasma was purchased from Calbiochem (Merck Millipore, Darmstadt, Germany). 7.2 μM wt-TTR in 10 mM phosphate buffer (100 mM KCl, 1.7 mM EDTA, pH 7.6) was preincubated with 2 molar equivalents of aminoxyacetic acid derivatives (1a–e, 2a–e, 3) dissolved in DMSO or for negative and positive controls, DMSO and 2 molar equivalents of diflunisal, respectively. The assay was performed in wells of a 96-well microplate (Corning Incorporated, New York, NY)Citation24. After incubation for 30 min at room temperature, an equal volume of 200 mM acetate buffer, 100 mM KCl, 1.7 mM EDTA, pH 4.2 was added to each well in order to shift the pH to that for optimal fibrillogenesis. After acidification, the 96-well microplate was incubated at 37 °C for 72 h without stirring. After a 3-day incubation, the plate was vortexed in double orbital mode for 15 s, after which the optical density (OD) was measured at 400 nm using a SPECTROstarNano (200–1000 nm) UV/Vis spectrophotometer (Ortenberg, Germany). The assay was repeated in triplicate. The OD ratio was calculated from the OD for each inhibitor and compared to that of a sample prepared in absence of any inhibitors multiplied by 100% to give the percentage of fibril formationCitation25. Before the assay, the solubility of each inhibitor was evaluated by verifying the absence of absorbance at 400 nm prior to mixing with the protein to ensure that the turbidity depended only on TTR amyloid fibril formation.
Crystal preparation and structure determination
Lyophilized human TTR (Calbiochem, Merck Millipore, Darmstadt, Germany) was dissolved, 1 mg in 100 mL 0.02% (w/v) NaN3, and dialyzed against 0.1M NaCl, 50 mM sodium acetate, pH 5.5, overnight. The (2,7-dibromo-fluoren-9-ylideneaminooxy)-acetic acid (1b), (2,7-dichloro-fluoren-9-ylideneaminooxy)-acetic acid (1c) and (E/Z)-((3,4-dichloro-phenyl)-methyleneaminooxy)-acetic acid (2c) were dissolved at 10 mM concentration in DMSO. The ligand-TTR complexes were prepared from TTR at 5 mg/mL to which the ligand was added in a volumetric ratio of 1:6. This gives a molar ratio of 30 ligands per protein monomer and a ligand concentration of 1.3 mM. The crystals were grown from a 1 μL TTR-ligand solution and 1 μL precipitant from the reservoir (500 μL), by sitting drop vapor diffusion using CrysChem plates (Corning Incorporated, New York, NY) that were equilibrated in a cooled incubator at 20 °C. The reservoir solutions were mixed using various combinations of the commercial working solutions (WS) from the Stura Footprint Screen (Molecular Dimensions Ltd., Newmarket, UK)Citation26. The reservoir conditions used for growing the crystals analyzed in this study consisted of 50–60% of (PEG_4D: 30% or PEG_4C: 20% PEG-4000, 0.2 M imidazole malate, pH 6.0) with 40–50% of (PEG_6C: 22.5% PEG-10 000, 0.1 M ammonium acetate, pH 4.5) (). The choice of high molecular weight polyethylene glycols (PEG) has been discussed elsewhereCitation27. Whenever crystals did not appear spontaneously after few hours, the drops were streak seededCitation28. In the absence of crystals after overnight storage, a booster solution (50 μL of 5 M NaCl with or without acetic acid) was added to the reservoirCitation27 followed by streak seeding if crystals did not appear within a few hours.
Table 1. Statistics for data collection, processing and refinement on TTR-ligand complexes.
The crystallization follows the strategy of reverse screeningCitation34 making use of different combinations of premixed working solutions to achieve small variation in pH and precipitating power with greater precision than can be achieved by mixing concentrated precipitant and buffer solutions. This strategy has allowed us to obtain in a short time and using only six drops, crystallographic data at high resolution from crystals of TTR in complex with compounds 1b, 1c and 2c. For data collection, the crystals were cryoprotected by quick immersion into a cryoprotectant solution enriched with extra ligand before flash cooling by dipping into liquid nitrogen at 100 K (). The data sets for the ligands-TTR complexes were collected at synchrotron facilities (ESFR beamlines ID23–1 and ID23–2 in Grenoble, France). Data processing was carried out using the automated system available at the ESRF synchrotron facility or on laboratory computers using XDS with the “xdsme” script (https://github.com/legrandp/xdsme) to optimize data quality. Unfortunately, the initial data set collected on ID23–1 for the 2c complex could not be refined with good statistics, probably because of superposition of diffraction from microcrystals of the poorly solubilized ligand. It was recollected on beamline Proxima-2ACitation35 at the Soleil storage ring in St. Aubin, France. To recollect the data, crystals that had dried out after 18-month storage were re-hydrated by the addition of 300 μL of water to the sitting drop reservoir. The well was then resealed and the crystals allowed to rehydrate over four hours. The re-hydrated crystals were transferred to a cryoprotectant solution as described in and flash cooled in liquid nitrogen. To avoid the problem encountered previously, the ligand-solubilizing cryoprotectant solution SM1Citation29 was used instead of CM29 (). The data were collected over 348.2 μm of a long crystal with 1° angular range for a total of 300 images using the helical scan method in which the crystal is translated in conjunction with each rotation. The final dataset at 1.5 Å could be refined with good statistics (). The structures were solved by rigid body refinement using REFMAC5Citation36 starting with a TTR model (PDB entry 4PM1Citation27 without inhibitor. The electron density maps were viewed in COOTCitation37 and the inhibitor built using the monomer library sketcher from CCP4 program suiteCitation38 was placed in the difference electron density. The structure was subjected to at least three cycles of rebuilding and refinement with REFMAC5Citation36 and phenix.refineCitation33.
Results
Biological assay
The percentage of TTR fibril formation (FF) was evaluated by a doublet concentration of synthesized compounds 1a–e, 2a–e and 3 to determine their potential as inhibitors ().
Table 2. In vitro acid-mediated wt-TTR (7.2 μM) percentage of fibril formation for compounds 1a–e, 2a–f and 3.
The 7.2 μM, the concentration used in the tests represents twice the TTR concentration in plasma (3.6 μM). Diflunisal (9% FF) was used as the reference drug (positive control) against which the ability of the newly synthesized compounds to reduce fibril formation was compared. The negative control, TTR without inhibitor under acid conditions (pH 4.2) is used to determine the 100% FF mark.
Among the 11 compounds tested, three molecules showed better than 50% FF inhibition (1b–c and 2c). The fluorenyl analogs 2,7-dibromo and 2,7-dichloro substituted compounds (1b and 1c) effectively reduced fibril formation to 34% and 37% FF, respectively. The 2-fluoro substituted fluorenyl derivative (1a) was found to be a less effective WT-TTR aggregation inhibitor. The 2-nitro (1e) substituted compound showed poor inhibitory activity and the 2,7-di-nitro substitution (1d) abolished all activity. Among the dibenzophenone derivatives the ((3,4-dichloro-phenyl)-phenyl-methyleneaminooxy)-acetic acid (2c) inhibited TTR fibril formation (only 15% FF) with an effectiveness comparable with diflunisal, while the 2-chloro substituted compound 2b showed a lower inhibition potency. Decreased inhibitory activity was recorded for 2a (unsubstituted) and 2d (3,3′-fluoro substitution) and falls to zero for 2e (4,4′-fluoro substitution). Similarly the indanone-based compound 3 was completely devoid of any activity.
Crystallization and structure determination
The crystals of the TTR complexes with 1b, 1c and 2c belong to the orthorhombic space group P21212 with cell parameters similar to each other () and comparable to other TTR depositions in the PDB database. The diffraction limit for the three structures ranges from 1.5 to 1.33 Å with relative good electron density for the ligands positioned along the two-fold axis of the tetramer () which, since subject to the symmetric crystallographic operation that builds the tetramer, must have occupancies that cannot exceed 0.5. The disordered portions of the protein are limited to the region 99–104 as is commonly the case in many TTR structures. The re-hydrated crystal at 1.5 Å diffracted to a lower resolution than those with 1b or 1c but better than the previous 2c data set collected to only 1.8 Å resolution. Of the seven re-hydrated crystals, three gave data better than 1.8 Å and only one was severely disordered and showed strong anisotropy. It can be noted that the b cell parameter for the re-hydrated dataset is 1 Å smaller than for the other two crystals, this cannot be attributed without ambiguity to the dehydration/re-hydration process. The solubilizing SM1 cryomix was used for this crystal () and the identical change was also observed when the solubilizing SM5 cryomix was used with curcumin complexed TTR crystalsCitation27, while the use of CM29 () gave the identical cell parameters as those for 1b and 1c cryoprotected with CM29.
Crystallographic analysis
The structures show important differences in terms of ligand binding without majors changes in the protein structure. There is electron density for only one of the two TTR molecules in the asymmetric unit for the structures obtained with inhibitors 1b and 1c. The TTR molecules with or without ligand show only minor differences and double conformations for some of the side chains involved in ligand binding. The binding occurs in either the reverse or forward mode depending on the compound, but not both as previously observed with fluorenone derivatives without substitutions (Citation23). The binding mode might be determined by the halogen atoms that occupy the classic halogen binding pockets and by the carboxylic groups that point alternatively towards the inner (Ser117–Thr119) and the outer part of the binding pocket (Lys15). In the reverse mode, the carboxylic moiety directed towards the inner binding pocket in the forward mode it points towards the lysine (). While compound 2c binds in both TTR binding sites in the forward binding mode, 1b and 1c bind select the reverse binding mode, as can be seen with the ligands superimposed in .
Experimental
Chemistry
Melting points were determined on a Köfler (Arthur H. Thomas Co., Philadelphia, PA) hot-stage apparatus and are uncorrected. Mass spectrometry data were collected using a Hewlett Packard 5988A spectrophotometer (Palo Alto, CA), by direct introduction at a nominal electron energy of 70 eV and a source temperature of 350 °C. 1H-NMR and 13C-NMR spectra were determined with a Bruker UltrashieldTM 400 MHz (Fällander, Switzerland) or Varian Gemini 200 MHz (Mountain View, CA) spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) downfield using tetramethylsilane as the internal standard. Coupling constants J are reported in Hertz; 13C-NMR spectra were fully decoupled. The following abbreviations are used: singlet (s), doublet (d), triplet (t), broad (br) and multiplet (m). Microwave assisted (MW) reactions were run in a Discover LabMate Microwave Synthesizer (CEM, Matthews, NC). Reactions were monitored by thin-layer chromatography (TLC) on Merck (Darmstadt, Germany) 0.2 mm pre-coated silica gel aluminum sheets (60 F-254) and visualized under a UV light (254 nm). Evaporation was performed in vacuo with a rotating evaporator (Naples, Italy). Sodium sulfate was used as the drying agent. Analytical reagents, starting materials and solvents were purchased from Sigma Aldrich (St. Louis, MO).
General procedure for the synthesis of the fluoren-9-one oximes (4a–e), benzophenone oximes (6a–e) and 5-chloro-1-indanone oxime (8)
A solution of appropriate carbonyl derivatives variously substituted (0.925 mmol) in DMSO (6 mL) was added drop-wise to hydroxylamine hydrochloride (3.70 mmol) previously solubilized in H2O. The reaction mixture was stirred at room temperature and monitored by TLC (n-hexane/AcOEt 8:2). The resulting solution was dissolved in H2O/ice to obtain a precipitate. The crude solid was collected by filtration, washed with H2O and filtered under vacuum to give the oxime (4a–e, 6a–e and 8).
(E)-2-fluoro-fluoren-9-one oxime (4a)
The title compound was prepared from 2-fluoro-9-fluorenone following the general procedure. The crude product was triturated with EtOH/H2O to give 4a as a yellow solid (88% yield); m.p.: 181–182 °C; 1H-NMR (200 MHz, DMSO-d6) δ: 11.85 (s, 1H); 8.33–8.03 (m, 2H); 7.65–7.00 (m, 5H).
2,7-Dichloro-fluoren-9-one oxime (4b)
The title compound was prepared from 2,7-dichloro-9-fluorenone following the general procedure. The crude product was triturated with EtOH/H2O to give 1b as a yellow solid (91% yield); m.p.: 262–263 °C; 1H-NMR (200 MHz, DMSO-d6) δ: 12.18 (s, 1H); 8.19 (s, 2H); 7.47–7.17 (m, 4H).
2,7-Dibromo-fluoren-9-one-oxime (4c)
The title compound was prepared from 2,7-dibromo-9-fluorenone following the general procedure. The crude product was triturated with MeOH to give 4c as a yellow solid (89% yield); m.p.: 265–266 °C (lit. 243 °CCitation39); 1H-NMR (200 MHz, DMSO-d6) δ: 8.45 (s, 2H); 7.94–7.64 (m, 4H).
2,7-Dinitro-fluoren-9-one-oxime (4d)
The title compound was prepared from 2,7-dinitro-9-fluorenone following the general procedure. The crude product was triturated with EtOH/H2O to give 4d as a yellow solid (89% yield); m.p.: 297–298 °C (lit. 288–289 °CCitation40); 1H-NMR (200 MHz, DMSO-d6) δ: 9.03 (s, 2H); 8.49–8.33 (m, 4H).
(E/Z)-2-Nitro-fluoren-9-one-oxime (4e)
The title compound was prepared from 2,7-dinitro-9-fluorenone following the general procedure. The crude product was triturated with EtOH/H2O to give 4e as a yellow solid (92% yield) in mixture (isomer E/Z ∼50:50), which was not separated; m.p.: 279–280 °C (lit. 262.5–263 °CCitation41). 1H-NMR (200 MHz, DMSO-d6) δ: 9.05 (s, 1H);8.99 (s, 1H), 8.43–8.14 (m, 8H), 7.77–7.59 (m, 4H).
Benzophenone oxime (6a)
The title compound was prepared from benzophenone following the general procedure. The crude product was recrystallized with n-hexane to give 6a as a white solid (70% yield); m.p.: 142–145 °C (lit. 141–142 °CCitation42); 1H-NMR (200 MHz, CDCl3) δ: 11.33 (s, 1H); 7.49–7.42 (m, 5H); 7.41–7.34 (m, 3H); 7.29–7.26 (m, 2H).
Bis-(2-chloro-phenyl)-methanone oxime (6b)
The title compound was prepared from bis-(2-chloro-phenyl)-methanone following the general procedure. The crude product was recrystallized with n-hexane to give 6b as a white solid (48% yield); m.p.: 135–137 °C; 1H-NMR (200 MHz, CDCl3) δ: 8.42 (s, 1H); 7.52–7.28 (m, 8H).
(E/Z)-(3,4-Dichloro-phenyl)-phenyl-methanone oxime (6c)
The title compound was prepared from (3,4-dichloro-phenyl)-phenyl-methanone following the general procedure. The crude product was recrystallized with n-hexane to give 6c as a white solid in mixture (isomer E/Z ∼50:50), which was not separated (46% yield). 1H-NMR (400 MHz, CDCl3) δ: 7.92 (s, 1H); 7.90 (s, 1H), 7.77–7.75 (m, 4H), 7.64–7.49 (m, 10H).
Bis-(3-fluoro-phenyl)-methanone oxime (6d)
The title compound was prepared from bis-(3-fluoro-phenyl)-phenyl-methanone following the general procedure. The crude product was recrystallized with n-hexane to give 6d as a white solid (40% yield); m.p.: 66–67 °C; 1H-NMR (200 MHz, CDCl3) δ: 7.74 (s, 1H); 7.48–7.07 (m, 8H).
Bis-(4-chloro-phenyl)-methanone oxime (6e)
The title compound was prepared from bis-(4-chloro-phenyl)-methanone following the general procedure. The crude product was recrystallized with n-hexane to give 6e as a white solid (66% yield); m.p.: 65–66 °C (lit. 68–69 °CCitation43); 1H-NMR (200 MHz, CDCl3) δ: 8.05 (s, 1H); 7.49–7.00 (m, 8H).
Bis-(4-fluoro-phenyl)-methanone oxime (6f)
The title compound was prepared from bis-(4-fluoro-phenyl)-methanone following the general procedure. The crude product was recrystallized with n-hexane to give 6f as a white solid (52% yield); m.p.: 58–60 °C (lit. 137–138 °CCitation44); 1H-NMR (200 MHz, CDCl3) δ: 8.05 (s, 1H); 7.49–7.00 (m, 8H).
(E)-5-Chloro-indan-1-one oxime (8)
The title compound was prepared from 5-chloro-indan-1-one following the general procedure. The crude product was recrystallized with n-hexane to give 8 as a white solid (58% yield); m.p.: 144–145 °C (lit.144 °CCitation45); 1H-NMR (200 MHz, CDCl3) δ: 8.00 (s, 1H), 7.62–7.28 (m, 3H,); 3.05–3.02 (m, 4H).
General procedure for the synthesis of the fluoren-9-one ethyl esters (5a–e), biphenyl ethyl esters (7a–e) and (5-chloro-indan-1-ylideneaminooxy)-acetic acid ethyl ester (9)
The esters derivatives (5a–e, 7a–e, 9) were synthesized following the general procedure as reported in literature. To a suspension of the appropriate oximes (4a–e, 6a–e, 8) (0.617 mmol) in DMF (0.4 mL) and CH3CN (2 mL) was added of K2CO3 (1.74 mmol), tetrabutylammonium bromide (0.053 mmol) and ethyl bromoacetate (0.694 mmol). The suspension was submitted to microwave irradiation at a power of 80 W, tmax 70 °C, 100 psi and monitored by TLC. The reaction mixture was poured into ice, extracted with Et2O and washed with an aqueous NaOH 1N solution. Organic layer dried and evaporated under vacuum gave exclusively the crude ethyl ester (5a–e, 7a–e, 9), which were used for the successive reaction without purification.
(E)-2-(2-Fluoro-fluoren-9-ylideneaminooxy)-acetic acid ethyl ester (5a)
The title compound was prepared from (E)-2-fluoro-fluoren-9-one oxime (4a) following the general procedure. Yellow oil (64% yield). 1H-NMR (200 MHz, CDCl3) δ: 8.37–7.02 (m, 7H); 4.95 (s, 2H); 4.30 (q, 2H, J = 7.2 Hz); 1.33 (t, 3H, J = 7.2 Hz).
2–(2,7-Dichloro-fluoren-9-ylideneaminooxy)-acetic acid ethyl ester (5b)
The title compound was prepared from 2,7-dichloro-fluoren-9-one oxime (5b) following the general procedure. Yellow oil (57% yield). 1H-NMR (200 MHz, CDCl3) δ: 8.32–7.27 (m, 1H); 7.99–7.91 (m, 2H); 7.68–7.53 (m, 3H); 4.94 (s, 2H); 4.29 (q, 2H, J = 7.2 Hz); 1.32 (t, 3H, J = 7.2 Hz).
2-(2,7-Dibromo-fluoren-9-ylideneaminooxy)-acetic acid ethyl ester (5c)
The title compound was prepared from 2,7-dibromo-fluoren-9-one oxime (5c) following the general procedure. Yellow oil (76% yield). 1H-NMR (200 MHz, DMSO-d6) δ: 8.41–8.40 (m, 2H); 7.94–7.72 (m, 4H); 5.09 (s, 2H); 4.20 (q, 2H, J = 7.2 Hz); 1.24 (t, 3H, J = 7.2 Hz).
2-(2,7-Dinitro-fluoren-9-ylideneaminooxy)-acetic acid ethyl ester (5d)
The title compound was prepared from 2,7-dinitro-fluoren-9-one oxime (5d) following the general procedure. Clear oil (61% yield). 1H-NMR (200 MHz, DMSO-d6) δ: 8.97–8.96 (m, 2H); 8.56–8.30 (m, 4H); 5.23 (s, 2H); 4.24 (q, 2H, J = 7.2 Hz); 1.27 (t, 3H, J = 7.2 Hz).
(E/Z)-2-(2-Nitro-fluoren-9-ylideneaminooxy)-acetic acid ethyl ester (5e)
The title compound was prepared from 2-nitro-fluoren-9-one oxime (5e) following the general procedure. The crude product was obtained as a mixture (isomer E/Z ∼ 54:46), which was not separated. Clear oil (66% yield). 1H-NMR (200 MHz, DMSO-d6) δ: 8.99 (s, 1H); 8.98 (s, 1H); 8.44–8.04 (m, 8H); 7.73–7.48 (m, 4H); 5.15 (s, 2H); 5.12 (s, 2H); 4.23 (q, 2H, J = 7.2 Hz); 1.24 (t, 3H, J = 7.2 Hz).
2-(Diphenylmethylenaminooxy)-acetic acid ethyl ester (7a)
The title compound was prepared from diphenyl-methanone oxime (6a) following the general procedure. Clear oil (70% yield). 1H-NMR (200 MHz, CDCl3) δ: 7.31–7.08 (m, 10H); 4.25 (s, 2H); 4.21 (q, 2H, J = 7.2 Hz); 1.30 (t, 3H, J = 7.2 Hz).
2-(Bis-(2-chloro-phenyl)-methyleneaminooxy)-acetic acid ethyl ester (7b)
The title compound was prepared from bis-(2-chloro-phenyl)-methanone oxime (7b) following the general procedure. Clear oil (75% yield). 1H-NMR (200 MHz, CDCl3) δ: 7.28–7.55 (m, 8H); 4.76 (s, 2H); 4.25 (q, 2H, J = 6.8 Hz); 1.31 (t, 3H, J = 6.8 Hz).
(E/Z)-2-((3,4-Dichloro-phenyl)-phenyl-methyleneaminooxy)-acetic acid ethyl ester (7c)
The title compound was prepared from (3,4-dichloro-phenyl)-phenyl methanone oxime (7c) following the general procedure. The crude product was obtained as a mixture (isomer E/Z ∼50:50). Clear oil (81% yield). 1H-NMR (200 MHz, CDCl3) δ: 7.28–7.62 (m, 16H); 4.74 (s, 2H), 4.73 (s, 2H); 4.26 (q, 4H, J = 7.2 Hz); 1.32 (t, 6H, J = 7.2 Hz).
2-(Bis-(3-fluoro-phenyl)-methyleneaminooxy)-acetic acid ethyl ester (7d)
The title compound was prepared from bis-(3-fluoro-phenyl)-methanone oxime (7d) following the general procedure. Clear oil (73% yield). 1H-NMR (200 MHz, CDCl3) δ: 7.56–7.08 (m, 8H); 4.72 (s, 2H); 4.24 (q, 2H, J = 7 Hz); 1.30 (t, 3H, J = 7 Hz).
2-(Bis-(4-fluoro-phenyl)-methyleneaminooxy)-acetic acid ethyl ester (7e)
The title compound was prepared from bis-(4-fluoro-phenyl)-methanone oxime (7e) following the general procedure. Yellow oil (73% yield). 1H-NMR (200 MHz, CDCl3) δ: 7.54–6.99 (m, 8H); 4.72 (s, 2H); 4.24 (q, 2H, J = 7.2 Hz); 1.32 (t, 3H, J = 7.2 Hz).
(E)-2-(5-Chloro-indan-1-ylideneaminooxy)-acetic acid ethyl ester (9)
The title compound was prepared from 5-Chloro-indan-1-one oxime (8) following the general procedure. Violet oil (68% yield). 1H-NMR (200 MHz, CDCl3) δ: 7.21–7.62 (m, 3H); 4.71 (s, 2H); 4.25 (q, 2H, J = 7 Hz); 3.03 (m, 4H); 1.31 (t, 3H, J = 7 Hz).
General procedure for the synthesis of the 9-fluorenone acetic acid (1a–e), biphenyl acetic acid (2a–e), (5-chloro-indan-1-ylideneaminooxy)-acetic acid (3)
A solution of ethyl ester (1a–e, 2a–e, 3) (0.387 mmol) in H2O (2.22 mL) and THF (4.44 mL) at 0 °C was added dropwise an aqueous NaOH 1N (1.11 mL) solution. The mixture was stirred for 2 h at 0 °C and then at room temperature until the ester was completely hydrolyzed into acid. The reaction was monitored by TLC (n-hexane/AcOEt 8:2). After solvent evaporation, the resultant solution was dissolved in H2O and washed with Et2O. The water solution acidified with 10% HCl at pH 4, extracted with CHCl3, dried and evaporated, gave the crude acetic acids (1a–e, 2a–e, 3).
(E)-2-(2-fluoro-fluoren-9-ylideneaminooxy)-acetic acid (1a)
The title compound was prepared from ethyl ester-(E)-(2-fluoro-fluoren-9-ylideneaminooxy)-acetic acid (5a) following the general procedure. The crude product was recrystallized with n-hexane to give as a yellow solid (64% yield); m.p.: 234–235 °C; 1H-NMR (200 MHz, DMSO-d6) δ: 8.92 (d, 1H); 7.92–7.69 (m, 4H); 7.84–7.94 (m, 2H); 4.97 (s, 2H); 13C NMR (400 MHz, DMSO-d6) δ: 170.8, 152.0, 139.5, 136.6, 134.5, 132.4, 131.4, 130.0, 129.6, 128.5, 122.8, 122.5, 121.9, 118.8, 72.3. MS: 271 (M+, 67), 214 (24), 197 (100), 188 (23), 168 (11), 74 (5).
2-(2,7-Dichloro-fluoren-9-ylideneaminooxy)-acetic acid (1b)
The title compound was prepared from ethyl ester-(2,7-dichloro-fluoren-9-ylideneaminooxy)-acetic acid (5b) following the general procedure. The crude product was recrystallized with n-hexane to give 1b as a yellow solid (50% yield); m.p.: 247–248 °C; 1H-NMR (200 MHz, DMSO-d6) δ: 8.27–8.26 (d, 1H); 7.98–7.91 (m, 2H); 7.68–7.53 (m, 3H); 5.00 (s, 2H); 13C NMR (400 MHz, DMSO-d6) δ: 170.8, 150.7, 139.3, 138.4, 136.3, 134.9, 133.8, 131.9, 131.2, 124.5, 123.4, 123.3, 122.1, 122.0, 72.6. MS: 321 (M+, 56), 264 (30), 247 (100), 211 (77), 177 (22), 83 (78).
2–(2,7-Dibromo-fluoren-9-ylideneaminooxy)-acetic acid (1c)
The title compound was prepared from ethyl ester-(2,7-dibromo-fluoren-9-ylideneaminooxy)-acetic acid (5c) following the general procedure. The crude product was recrystallized with n-hexane to give 1c as a yellow solid (75% yield); m.p.: 243–244 °C; 1H-NMR (200 MHz, DMSO-d6) δ: 8.41 (m, 1H); 7.95–7.68 (m, 5H); 5.01 (s, 2H); 13C NMR (400 MHz, DMSO-d6) δ: 171.1, 150.9, 140.3, 139.4, 136.4, 135.0, 134.2, 133.9, 132.2, 124.6, 123.9, 123.9, 122.8, 122.1,72.9. MS: 411 (M+, 69), 354 (20), 337 (100), 257 (41), 194 (15), 177 (17), 83 (17).
2-(2,7-Dinitro-fluoren-9-ylideneaminooxy)-acetic acid (1d)
The title compound was prepared from ethyl ester-(2,7-dinitro-fluoren-9-ylideneaminooxy)-acetic acid (5d) following the general procedure. The crude product was recrystallized with n-hexane to give 1d as a yellow solid (50% yield); m.p.: 277–278 °C; 1H-NMR (200 MHz, DMSO-d6) δ: 8.99 (m, 1H); 8.52–8.36 (m, 5H); 5.13 (s, 2H); 13C NMR (400 MHz, DMSO-d6) δ: 170.1, 150.7, 140.3, 138.3, 136.1, 134.5, 134.1, 133.9, 133.2, 124.7, 124.2, 124.0, 122.9, 122.5, 72.4. MS: 344 (M+, 4), 269 (100), 255 (15), 239 (73), 197 (40), 164 (65), 83 (78), 75 (13).
(E/Z)-2-(2-nitro-fluoren-9-ylideneaminooxy)-acetic acid (1e)
The title compound was prepared from ethyl ester-(E/Z)-(2-nitro-fluoren-9-ylideneaminooxy)-acetic acid (5e) following the general procedure. The crude product was recrystallized with n-hexane to give 1e as a yellow solid (50% yield) as a mixture (isomer E/Z ∼ 50:50), which was not separated. 1H-NMR (200 MHz, DMSO-d6) δ: 9.00 (s, 1H); 8.99 (s, 1H); 8.50–8.05 (m, 8H); 7.79–7.45 (m, 4H); 5.06 (s, 2H); 5.04 (s, 2H); 13C NMR (400 MHz, DMSO-d6) δ: 170.8, 151.1, 150.9, 147.72, 147.65, 146.87, 145.75, 139.0, 138.1, 135. 8, 135.48, 132.6, 131.5, 131.0, 130.9, 130.6, 129.9, 129.6, 127.7, 126.3, 123.9, 122.8, 112.78, 122.2, 121.9, 121.7, 116.4, 72.7. MS: 289 (M+, 22), 248 (24), 224 (70), 194 (61), 165 (87), 137 (25), 83 (100), 74 (16).
2-(Diphenylmethylenaminooxy)-acetic acid (2a)
The title compound was prepared from ethyl ester-(benzhydrylideneaminooxy)-acetic acid (7a) following the general procedure. The crude product was recrystallized with n-hexane to give 2a as a white solid (50% yield); m.p.: 109–110 °C; 1H-NMR (200 MHz, CDCl3) δ: 7.50–7.27 (m, 8H); 4.08 (s, 2H); 13C NMR (400 MHz, CDCl3) δ: 176.2, 164.3, 153.6, 131.3, 138.8, 129.3, 128.6, 128.4, 127.8, 77.5. MS: 255 (M+, 17), 216 (100), 201 (20), 122 (25), 113 (10), 95 (10), 75 (65).
2-(Bis-(2-chloro-phenyl)-methyleneaminooxy)-acetic acid (2b)
The title compound was prepared from ethyl ester-(bis-(2-chloro-phenyl)-methyleneaminooxy)-acetic acid (7b) following the general procedure. The crude product was recrystallized with n-hexane to give 2b as a white solid (50% yield); m.p.: 113–115 °C, 1H-NMR (200 MHz, CDCl3) δ: 7.50–7.27 (m, 8H); 4.08 (s, 2H); 13C NMR (400 MHz, CDCl3) δ: 173.1, 122.91, 133.9, 133.6, 132.8, 132.8, 132.1, 130.8, 130.8, 130.7, 130.6, 129.8, 126.8, 126.6, 70.9. MS: 323 (M+,1), 288 (2), 248 (13), 214 (34), 177 (4), 149 (4), 75 (100), 56 (25).
(E/Z)-2-((3,4-Dichloro-phenyl)(phenyl)methyleneaminooxy)-acetic acid (2c)
The title compound was prepared from ethyl ester-((3,4-dichloro-phenyl)-(phenyl) methyleneaminooxy)-acetic acid (7c) following the general procedure. The crude product was recrystallized with n-hexane to give 2c as a yellow solid (76% yield) in a mixture (isomer E/Z ∼ 55:45); 1H-NMR (200 MHz, CDCl3) δ: 7.61–7.27 (m, 8H); 4.79 (s, 2H), 4.78 (s, 2H). 13C NMR (400 MHz, CDCl3) δ: 174.9, 156.9, 156.7, 135.7, 134.8, 134.0, 133.5, 132.7, 132.6, 132.4, 131.7, 131.3, 130.3, 130.24, 130.2, 129.7, 129.6, 129.1, 128.8, 128.5, 128.4, 128.1, 127.3, 77.3, 77.2. MS: 323 (M+, 16), 248 (49), 214 (100), 177 (15), 164 (13), 127 (7), 105 (15), 74 (25).
2-(Bis(4-fluoro-phenyl)-methyleneaminooxy)-acetic acid (2d)
The title compound was prepared from ethyl ester-(bis(4-fluoro-phenyl)-methyleneaminooxy)-acetic acid (7d) following the general procedure. The crude product was recrystallized with n-hexane to give 2d as a white solid (54% yield); m.p.: 106–108 °C 1H-NMR (200 MHz, CDCl3) δ: 7.47–7.11 (m, 8H); 4.79 (s, 2H); 13C NMR (400 MHz, CDCl3) δ: 174.5, 156.5, 137.3, 134.1, 134.0, 129.9, 129.8, 129.7, 129.4, 124.7, 116.6, 116.3, 114.9, 114.7, 70.6. MS: 27 (11), 216 (100), 201 (12), 122 (9), 113 (9), 95 (12), 75 (38).
2-(Bis-(4-fluoro-phenyl)-methyleneaminooxy)-acetic acid (2e)
The title compound was prepared from ethyl ester-(bis-(4-fluoro-phenyl)-methyleneaminooxy)-acetic acid (7e) following the general procedure. The crude product was recrystallized with n-hexane to give 2e as a white solid (88% yield); m.p.: 115–116 °C. 1H-NMR (200 MHz, CDCl3) δ: 7.50–6.99 (m, 8H); 4.77 (s, 2H); 13C NMR (400 MHz, CDCl3) δ: 174.5, 157.8, 137.2, 136.5, 134.8, 132.3, 131.8, 130.6, 129.5, 127.7, 127.6, 127.5, 70.8. MS: 291 (M+,12), 216 (100), 201 (17), 122 (15), 133 (15), 95 (10), 75 (69).
(E)-2-(5-Chloro-indan-1-ylideneaminooxy)-acetic acid (3)
The title compound was prepared from ethyl ester-(5-Chloro-indan-1-ylideneaminooxy)-acetic acid (9a) following the general procedure. The crude product was recrystallized with n-hexane to give 3a as a white solid (70% yield); m.p.: 140–141 °C; 1H-NMR (200 MHz, CDCl3) δ: 7.58–7.27 (m, 3H); 4.76 (s, 2H); 3.03 (m, 4H); 13C NMR (400 MHz, CDCl3) δ: 175.16; 164.0, 150.3, 136.7, 133.9, 127.6, 125.8, 122.9, 70.4, 28.4, 26.8. MS: 240 (M+, 50), 229 (16), 164 (100), 137 (15), 130 (14), 103 (16), 75 (9).
Discussion
Human Transthyretin tetramer dissociation is the rate-limiting step in TTR amyloidogenic process. One of the therapeutic strategies to ameliorate TTR amyloidoses and slow the progression of the disease is tetramer stabilization by small molecules, such as tafamidis and diflunisal. TTR stabilizing molecules are typically composed of two aromatic portions directly connected or joined together by a linker of variable composition. The unsubstituted fluorenyl compounds 7BD and ES8 previously studied by us presented only one aromatic portion connected to the terminal carboxylic moiety by an propionic or acetic linker, respectively (22, 23). These fluorenyl derivatives (7BD, ES8) are efficient in stabilizing the tetramer by binding the TTR both in forward and in reverse mode with the distinction that the aromatic portion of the acetic derivative ES8 occupies a different hydrophobic pocket with respect to 7BDCitation23. The new fluorenyl analogs, 1b and 1c, that are halogen substituted also show a low percentage of fibril formations (), result supported by the X-ray analysis which show that they interact with the TTR in similar manner of 7BD but only in reverse mode (). The structures of TTR in complexed with 1b and 1c have ligands binding to a single of the two binding sites. Given that in the co-crystallization experiment, the ligand concentration was in the millimolar range with excess ligand to saturate both sites 30 times over, the lack of ligand at the second site is not due to insufficient ligand. This result can be interpreted as another example of negative cooperativity. The evidence for negative cooperativity in ligand binding to TTR is mountingCitation46, although doubts and Skepticism will undoubtedly persist. Negative cooperative behavior, in the case of two binding sites that are physically well separated from each other, must be relayed via induced conformational changes. The existence of asymmetry due to structural fluctuations that affect the affinity of the two sites for various ligands cannot be considered a cooperative behavior. The asymmetry found in TTR has been characterized by only small differences in the conformation of the cavity of the two binding sites. This asymmetry could be derived from crystal packing constraints (). To prove that ligand binding, and not crystal packing influences the structure of TTR, the differences between monomers with and without ligand should be larger that the differences between A and B monomers. The RMSD between the TTR chain A with 1b compared with chain B without ligand is 1.179. Similarly, the same comparison for the 1c complex gives an RMSD of 1.230, while when the two chains complexed to 2c are compared the value is only 0.591 (). When the various monomers with and without bound ligand are compared, a larger RMSD characterizes heterogeneous couples. To understand the mechanism by which negative cooperation acts, a more detailed analysis would be required, possibly with a TTR polymorph without a crystallographic two-fold axisCitation47. This criticism is difficult to dismiss since almost all the liganded structures are derived from a single crystal polymorph with the protein always confined in the same manner. The affinity of ligands appears to be another factor that can determine different ability of TTR ligands to saturate the two T4 binding sites of the tetrameric protein. Experimental factors, such as the poor aqueous solubility of the ligands and the persistent use of high-salt crystallization conditions also influence the crystallographic results. The crystals used in this study were obtained from polyethylene glycol, conditions in which many ligands are more soluble.
Figure 4. Crystal packing and ligand binding. (A) Crystal packing of TTR monomer A with inhibitor 1b bound and, (B) packing of monomer B without a ligand. The asymmetric unit of the P21212 lattice comprises a dimer A–B that forms a tetramer with A–B′ via a crystallographic symmetry operation. Differences in crystal packing distinguish the two monomers, A and B, where monomers A and A′ pack tighter that B–B′. By convention, ligands bind to monomer A while the empty site is assigned to monomer B. The RMSD deviations on Cα between two chains involved in ligand binding; between chains both not involved in ligand binding and between chains with ligand compared to chains without ligand to show the divergence that exists between monomers involved and not involved in ligand binding. (C) The binding of 1b appears to induce a small conformational change in order to better accommodate the bromine atom in the halogen pocket of molecule A. (D) Molecule B (has been changed from green to yellow after superimposition on molecule A) shows that in order for the same ligand to bind to molecule B, similar conformational changes would need to be induced. (E) Without a change, that would facilitate binding to molecule B, positive cooperativity can be excluded. The evidence for conformational changes that would hinder ligand binding at site B are also lacking, since the dimer interface is essentially the same for the two molecules. (F) The differences observed in the loop 94–104 between molecule B and A are common to most TTR-inhibitor complexes, as shown here for ligand 2c.
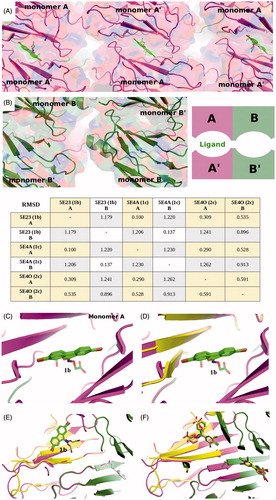
Among the benzophenone (2a–e) and indanone (3a) derivatives in which the aromatic was simplified having less steric hindrance and different rigidity than 9-fluorenyl derivatives, the more potent fibril formation inhibitors was compound 2c. Derivative 2c, like tafamidis, are able to occupy both sites. In those cases, as for ligand 1b, where minor conformational changes are induced on ligand binding () it is difficult to find evidence for the transmission of these changes to the other subunit. The changes observed may be connected to the packing on the molecules in the lattice. We have recently obtained a new crystal formCitation47, without a crystallographic two-fold axis that could be used to re-analyze ligands like 2c that show asymmetric binding. The presence of two tetramers in the asymmetric unit of this new polymorph might be useful to analyze whether conformational changes induced by ligand binding at one site might explain the absence of a ligand at the other site and the apparent negative cooperative behavior of TTR to give structural support to solution studies that show a concerted opening and closing of the dimer–dimer interfaceCitation48, possibly in the form of a see-saw mechanismCitation49.
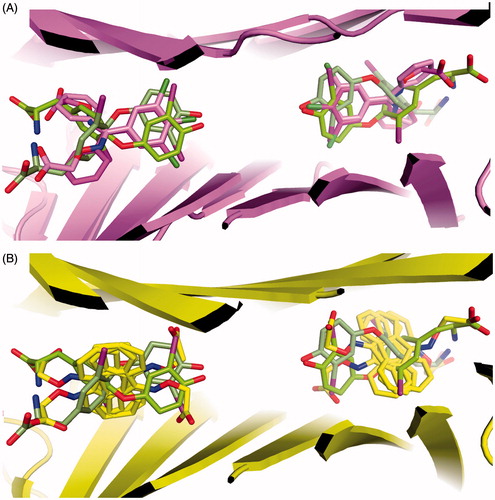
The binding of compound 2c, with a fibril inhibitory activity similar to tafamidis, shows certain features that are reminiscent of the binding of 3,3′-diiodo-l-thyronine () without it occupying of the second halogen pocket used by this ligand. This second pocket is exploited by the fluorenyl moiety of ES8, from our previous study, that has a spacer between the hydrophobic and carboxylate moieties of the same character. This suggests that a combination of the two scaffolds might provide a manner to achieve greater tetramer stabilization.
Figure 5. Superimposition of 2c and ES8 on 3,3′-diiodo-l-thyronine. (A) The halogen binding site exploited by 2b matches with that used by 3,3′-diiodo-l-thyronine. (B) The second halogen binding site used by 3,3′-diiodo-l-thyronine at right angles to the first one superimposes on the fluorenyl ring of ES8.
Conclusion
The X-ray structures of TTR in complex with the non-substituted acetic derivative ES8 showed that a single atom in the linker between the tricyclic ring and acidic moieties can change the positioning of the tricyclic ring that is located in the pocket previously described for 1-amino-5-naphthalene sulfonate (5NS) complex with TTR. In the present work we have explored the effect produced by the decoration of 9-fluorenyl scaffold with halogen groups and their effects on the binding mode. The crystallographic studies have highlighted that in the 9-fluorenyl acetic derivatives of type 1b (PDB:5E23) the presence of halogen atoms on the aromatic moiety repositions the tricyclic ring in the hydrophobic pocket identified for the unsubstituted 9-fluorenyl propionic derivatives, orienting the molecule solely in reverse mode. The X-ray analysis of 2,3-dichlorobenzophenone 2c (PDB: 5E4O) shows a different spatial disposition of the aromatic rings and uni-modal binding opposed to that of halogen substituted 9-fluorenyl derivatives.
The potential of fluorenyl scaffold was revealed by our previous study (23). With the development of these new inhibitors we can now select a specific conformation. There is now an opportunity to improve the potency of fibril inhibitor 2c combining by exploiting the knowledge acquired from these studies to synthesize new inhibitors with improved affinity and without the side effects due to hormonal mimicry or the anti-inflammatory activity typical of NSAIDs.
Declaration of interest
This work was partially supported by Ministero dell’Istruzione, dell’Università e della Ricerca of Italy (PRIN 20109MXHMR_007).
Acknowledgements
The help of the staff at the ESRF and Soleil synchrotrons and the grant of beamtime are acknowledged with gratitude.
References
- Blake CC, Geisow MJ, Oatley SJ, et al. Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J Mol Biol 1978;121:339–56.
- Wojtczak A, Cody V, Luft JR, Pangborn W. Structures of human transthyretin complexed with thyroxine at 2.0 A resolution and 3′,5′-dinitro-N-acetyl-l-thyronine at 2.2 A resolution. Acta Crystallogr D Biol Crystallogr 1996;52:758–65.
- Kanai M, Raz A, Goodman DS. Retinol-binding protein: the transport protein for vitamin A in human plasma. J Clin Invest 1968;47:2025–44.
- Soprano DR, Herbert J, Soprano KJ, et al. Demonstration of transthyretin mRNA in the brain and other extrahepatic tissues in the rat. J Biol Chem 1985;260:11793–8.
- Landers KA, Mortimer RH, Richard K. Transthyretin and the human placenta. Placenta 2013;34:513–17.
- Purdy RH, Woeber KH, Holloway MT, Ingbar SH. Preparation of crystalline thyroxine-binding prealbumin from human plasma. Biochemistry 1965;4:1888–95.
- Buxbaum JN, Tagoe CE. The genetics of the amyloidoses. Annu Rev Med 2000;51:543–69.
- Westermark P, Sletten K, Johansson B, Cornwell GG. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci USA 1990;87:2843–5.
- Pilato E, Dell’Amore A, Botta L, Arpesella G. Combined heart and liver transplantation for familial amyloidotic neuropathy. Eur J Cardiothorac Surg 2007;32:180–2.
- Ohya Y, Okamoto S, Tasaki M, et al. Manifestations of transthyretin-related familial amyloidotic polyneuropathy: long-term follow-up of Japanese patients after liver transplantation. Surg Today 2011;41:1211–18.
- Almeida MR, Cardoso I, Saraiva MJ. In vitro and in vivo effects of genistein on TTR stabilization and aggregation. In: Skinner M, Berk JL, Connors LH, Seldin DC, eds. XIth International symposium on amyloidosis. Boca Raton (FL): CRC Press; 2007:113.
- Connelly S, Choi S, Johnson SM, et al. Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses. Curr Opin Struct Biol 2010;20:54–62.
- Ferguson RN, Edelhoch H, Saroff HA, et al. Negative cooperativity in the binding of thyroxine to human serum prealbumin. Preparation of tritium-labeled 8-anilino-1-naphthalenesulfonic acid. Biochemistry 1975;14:282–9.
- Neumann P, Cody V, Wojtczak A. Structural basis of negative cooperativity in transthyretin. Acta Biochim Pol 2001;48:867–75.
- Herbert J, Wilcox JN, Pham K-TC, et al. Transthyretin: a choroid plexus-specific transport protein in human brain. The 1986 S. Weir Mitchell award. Neurology 1986;36:90011.
- Klabunde T, Petrassi HM, Oza VB, et al. Rational design of potent human transthyretin amyloid disease inhibitors. Nat Struct Biol 2000;7:312–21.
- Ong DST, Kelly JW. Chemical and/or biological therapeutic strategies to ameliorate protein misfolding diseases. Curr Opin Cell Biol 2011;23:231–8.
- Nencetti S, Orlandini E. TTR fibril formation inhibitors: is there a SAR? Curr Med Chem 2012;19:2356–79.
- Coelho T, Maia LF, Da Silva AM, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol 2013;260:2802–14.
- Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013;310:2658–67.
- Obici L, Merlini G. An overview of drugs currently under investigation for the treatment of transthyretin-related hereditary amyloidosis. Expert Opin Investig Drugs 2014;23:1239–51.
- Palaninathan SK, Mohamedmohaideen NN, Orlandini E, et al. Novel transthyretin amyloid fibril formation inhibitors: synthesis, biological evaluation, and X-ray structural analysis. PLoS One 2009;4:e6290.
- Ciccone L, Nencetti S, Rossello A, et al. X-ray crystal structure and activity of fluorenyl-based compounds as transthyretin fibrillogenesis inhibitors. J Enzyme Inhib Med Chem 2015. [Epub ahead of print]. DOI: 10.3109/14756366.2015.1070265.
- Dolado I, Nieto J, Saraiva MJM, et al. Kinetic assay for high-throughput screening of in vitro transthyretin amyloid fibrillogenesis inhibitors. J Comb Chem 2005;7:246–52.
- Oza VB, Smith C, Raman P, et al. Synthesis, structure, and activity of diclofenac analogues as transthyretin amyloid fibril formation inhibitors. J Med Chem 2002;45:321–32.
- Stura EA, Nemerow GR, Wilson IA. Strategies in the crystallization of glycoproteins and protein complexes. J Cryst Growth 1992;122:273–85.
- Ciccone L, Tepshi L, Nencetti S, Stura EA. Transthyretin complexes with curcumin and bromo-estradiol: evaluation of solubilizing multicomponent mixtures. N Biotechnol 2015;32:54–64.
- Stura EA, Wilson IA. Applications of the streak seeding technique in protein crystallization. J Cryst Growth 1991;110:270–82.
- Ciccone L, Vera L, Tepshi L, et al. Multicomponent mixtures for cryoprotection and ligand solubilization. Biotechnol Reports 2015;7:120–7.
- Vera L, Stura EA. Strategies for protein cryocrystallography. Cryst Growth Des 2014;14:427–35.
- Newman J. Novel buffer systems for macromolecular crystallization. Acta Crystallogr D Biol Crystallogr 2004;60:610–12.
- Diederichs K, Karplus PA. Better models by discarding data? Acta Crystallogr D Biol Crystallogr 2013;69:1215–22.
- Adams PD, Afonine PV, Bunkóczi G, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010;66:213–21.
- Stura EA, Satterthwait AC, Calvo JC, et al. Reverse screening. Acta Crystallogr D Biol Crystallogr 1994;50:448–55.
- Duran D, Couster SL, Desjardins K, et al. PROXIMA 2A – a new fully tunable micro-focus beamline for macromolecular crystallography. J Phys Conf Ser 2013;425:012005. doi: http://dx.doi.org/10.1088/17426596/425/1/012005.
- Murshudov GN, Skubák P, Lebedev AA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 2011;67:355–67.
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010;66:486–501.
- Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 2011;67:235–42.
- Holbro T, Tagmann E. Bromofluoranthenes II. Synthesis of 4,11-dibromofluoranthene. Helv Chim Acta 1950;33:2178–85.
- Pan H-L, Fletcher TL. Derivatives of fluorene. XXX. Rearrangement and antitumor activities of some 9-oxofluorene oximes. 6(5H)-Phenanthridinones. J Med Chem 1969;12:822–5.
- Moore FJ, Huntress EH. Unsymmetrical phenanthridones. 11. A new preparative method: 7-nitrophenanthridone by Beckmann rearrangement of 2-nitrofluorenone oxime. J Am Chem Soc 1927;49:2618–24.
- Lachman A. Benzophenone oxime. Org Synth 1930;10:10–11.
- Ulbrich HK, Luxenburger A, Prech P, et al. A novel class of potent nonglycosidic and nonpeptidic pan-selectin inhibitors. J Med Chem 2006;49:5988–99.
- Dunlop RD, Gardner JH. The preparation of 4-fluoro- and 4,4′-difluorobenzophenone. J Am Chem Soc 1933;55:1665–6.
- Novak L, Protiva M. Antihistaminic substances L. Derivatives of N-(1-indanyl)ethylenediamine Coll. Czech Chem Commun 1962;27:2413–18.
- Cianci M, Folli C, Zonta F, et al. Structural evidence for asymmetric ligand binding to transthyretin. Acta Crystallogr D Biol Crystallogr 2015;71:1582–92.
- Polsinelli I, Nencetti S, Shepard W, et al. A new crystal form of human transthyretin obtained with a curcumin derived ligand. J Struct Biol 2016;194:8–17.
- Trivella DBB, Bleicher L, Palmieri LDC, et al. Conformational differences between the wild type and V30M mutant transthyretin modulate its binding to genistein: implications to tetramer stability and ligand-binding. J Struct Biol 2010;170:522–31.
- Lima LMTR, Silva VDA, Palmieri LDC, et al. Identification of a novel ligand binding motif in the transthyretin channel. Bioorg Med Chem 2010;18:100–10.