ABSTRACT
Alpha-1 antitrypsin (AAT) deficiency is an established genetic risk factor for pulmonary disease and may lead to severe emphysema. Despite accessible, inexpensive, and straightforward testing procedures, the disorder is still widely undiagnosed due mainly to a lack of awareness among the medical community. AAT deficiency often results in the development of non-specific respiratory symptoms that can be confused with those of other non-hereditary chronic obstructive pulmonary disease or asthma. However, there are published guidelines that provide detailed recommendations on patient testing. Early diagnosis of AAT deficiency is fundamental to improve patient outcomes; it allows preventive measures to be taken, such as smoking cessation, and allows monitoring and initiation of appropriate therapy while lung function is still relatively preserved. Diagnosis should not solely be the domain of the specialist pulmonologist; testing can be easily initiated in the primary care setting. The establishment of process maps and diagnosis algorithms, as suggested in this review, should encourage appropriate suspicion, testing, and follow-up of AAT deficiency in the patient's primary care medical home setting. Primary care physicians have a key role in increasing the awareness, diagnosis, and effective management of this disorder.
INTRODUCTION
Alpha-1 antitrypsin (AAT) deficiency, a classic single-gene disorder, predisposes individuals to the development of lung and liver disease due to insufficient secretion or production of the glycoprotein AAT from the liver. AAT normally protects the lungs against the damaging effects of neutrophil elastase (Citation1), a proteolytic enzyme secreted in response to infection or inflammation. AAT deficiency is an established genetic risk factor for pulmonary disease, with plasma AAT levels of below a protective threshold of 11 μM associated with emphysema (Citation2). Despite AAT deficiency resulting from a single gene mutation, its clinical symptom development can be variable and there is evidence that it may be influenced by other genetic factors, environmental exposure, and interactions between these elements (Citation3).
In chronic diseases such as AAT deficiency, symptoms may only manifest years after the development of the disease. In the absence of symptoms, it is important that clinicians be cognizant of family history and any other disease markers in their usual clinical encounters. Compared with the general population, individuals with AAT deficiency have an accelerated decline in lung function (Citation4), and once symptoms have developed they have an increased risk of morbidity and reduced life expectancy (Citation2). Therefore, timely initiation of appropriate therapy is essential to preserve lung function, as are preventive measures such as smoking cessation and avoiding exposure to second-hand smoke. Primary care physicians can play a pivotal role in improving diagnosis rates and effective clinical management of AAT deficiency. Despite an extensive, and growing, body of scientific literature and a slowly increasing awareness of the condition, AAT deficiency is still widely undiagnosed, and only approximately 5% of individuals receive the correct diagnosis (Citation5). Furthermore, delays of up to 8.3 years between first symptoms and eventual diagnosis of AAT deficiency have been reported (Citation5).
This review analyzes the role of primary care practice in the diagnosis and management of AAT deficiency and suggests practical processes that may facilitate the translation of physician knowledge into effective clinical action. A case study is utilized throughout the article to demonstrate key points in the management of patients with this disorder.
Case study, patient A: History
A 44-year-old male patient visited his primary care physician 2 years previously to discuss becoming a live liver donor for his father. His father had been diagnosed at the age of 72 years with cirrhosis that was attributed to lifestyle, although later AAT testing revealed a PiMZ phenotype (Citation6). No further testing was initiated in other family members at that time. The patient was also under the care of a pulmonary specialist for asthma and reported to his primary care physician that his asthma was not responding to bronchodilators, a finding that his specialist believed to be due to a history of smoking (he smoked socially during college).
IMPEDIMENTS TO IMPROVING DIAGNOSIS AND MANAGEMENT
A number of impediments to the diagnosis and optimal management of AAT deficiency can be identified within the primary care setting (Citation7). These include:
Lack of awareness and knowledge of the condition, which can lead to misdiagnosis or delays in diagnosis.
Insufficient time for detailed patient consultations.
Limited access to supplementary healthcare services such as genetic counseling.
Poor communication between practice personnel, between physicians and patients or their families, and between physicians and specialist consultants.
Provision of serial rather than parallel care, such that activities led by the primary care physician occur in isolation from those led by other healthcare professionals.
Pessimistic physician mindsets, whereby the disease is considered too costly or complicated to treat, or that there are no effective therapies to render it worth trying to treat (“therapeutic nihilism”).
TRANSLATING KNOWLEDGE INTO CLINICAL ACTION
AAT deficiency is a chronic and progressive disease that requires lifelong clinical management. Upon diagnosis of the condition, primary care physicians have a crucial role to play in the delivery of comprehensive and coordinated healthcare for affected individuals. The development of practice recommendations for the identification of individuals with AAT deficiency, and the establishment of a planned-care follow-up model tailored to individual patient needs, may help to surmount some of the obstacles identified above and thereby facilitate early diagnosis and optimize patient care.
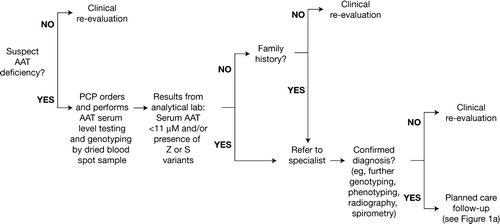
A process map and diagnosis algorithm is suggested here for guidance and to encourage appropriate suspicion, testing, and follow-up in the primary care setting (a and ). The process incorporates many of the defining principles of the patient-centered medical home concept endorsed by various primary care organizations (Citation8). These principles include: an ongoing relationship between a patient and physician from first contact; physician-directed medical practice, whereby the personal physician leads a team of individuals within their practice who take collective responsibility for the patient's care; and co-ordinated or integrated care across various elements of the healthcare system, such as speciality care, hospitals, and family, public, and community-based services. One advantage of following such a process is that it allows physicians to better benchmark their progress in order to measure clinical outcomes within their practice. Benchmarking encourages accurate diagnosis, resulting in better patient outcomes and promotion of clinical excellence.
Figure 1a. AAT deficiency screening, diagnosis, and planned-care follow-up for primary care physicians: Process map. AAT = Alpha-1 antitrypsin; PCP = primary care physician.
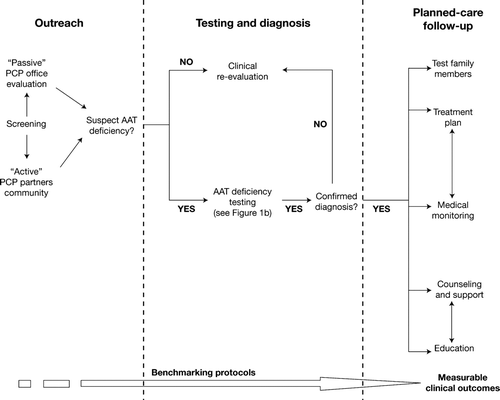
Figure 1b. AAT deficiency screening, diagnosis, and planning-case follow-up for primary care physicians: Diagnosis algorithm. AAT = Alpha-1 antitrypsin.
Screening and outreach
Identification of individuals with AAT deficiency can be achieved in one of two ways: (Citation1) large-scale screening of the general population and (Citation2) targeted screening of “at-risk” individuals. Currently, there is no screening for AAT deficiency in the general population and its practicality and effectiveness (e.g., screening of neonates) is questionable. However, evidence does support the targeted screening of “at-risk” populations (Citation9–12), and the process map suggested here offers two potential methods (): “active” screening, where the physician's practice works in partnership with the community and “passive” screening, which takes place within the physician's office.
Active screening may involve participation of the primary care practice in employer-based screening programs or health fairs that screen at-risk individuals. In addition, at-risk individuals could be reached through smoking cessation clinics. Screening of the families of affected individuals is also an important way to identify at-risk patients and this could be achieved through the healthcare system and various patient support groups.
Passive screening relies upon the physician taking an appropriate patient history and being alert to risk factors in patients presenting at their practice as detailed in published guidelines from the American Thoracic Society (ATS) and European Respiratory Society (ERS) (Citation2). When AAT deficiency is suspected, initial assessments may include a detailed physical examination, liver function tests, pulmonary function tests, and radiography, followed by the initiation of appropriate testing (serum AAT levels and genotyping) (). Other means of identification of at-risk individuals in the physician's office include the development and introduction of a patient self-administered questionnaire, which could increase disease awareness and lead to earlier symptom recognition. A similar questionnaire was recently introduced for patient self-assessment of chronic obstructive pulmonary disease (COPD) (the COPD Population Screener) and was validated as accurately classifying physician-reported COPD (Citation13). There is also preliminary evidence that the inclusion of prompts within the results of pulmonary function tests demonstrating airflow obstruction may increase the rate of testing for AAT deficiency (Citation14).
Clinical features of AAT deficiency
AAT deficiency often results in the insidious development of several non-specific symptoms that can be easily confused with those of other non-hereditary COPD or asthma (Citation15). These include: shortness of breath after activity, decreased exercise tolerance, wheezing (in the presence and/or absence of a respiratory tract infection), cough, high levels of sputum production, frequent infections of the lower respiratory tract, and a history of suspected allergies and/or asthma. Given the general lack of awareness of AAT deficiency and its association with pulmonary disease, it is likely that many physicians will stop their investigations at, for example, a positive diagnosis of asthma or emphysema in affected individuals. However, the appearance of early-onset emphysema regardless of smoking history, emphysema in a non-smoker at any age, a family history of pulmonary disease, or unexplained liver disease (e.g., jaundice or, more seriously, cirrhosis) should immediately alert the physician to the possibility of AAT deficiency ().
Table 1. Recognition of AAT deficiency and recommendations for testing
Testing for AAT deficiency
Guidelines issued by the ATS and ERS provide detailed recommendations on the patients who should be tested () (Citation2). Prior to testing, it is recommended that patients be informed about the implications of a positive diagnosis with respect to the emotional burden and employment and insurance discrimination, even though the 2008 Genetic Information Nondiscrimination Act (GINA) has gone someway to prohibit the improper use of genetic information in these settings. Under the Act, health plans and health insurers cannot deny coverage or charge higher premiums based on a genetic predisposition. Employers are also prohibited from using genetic information as a factor in the decision to hire, promote, or terminate the employment of an individual.
Testing for AAT deficiency is accessible, straightforward, and inexpensive and can be easily performed in a primary care setting. The identification of individuals with AAT deficiency can be determined by the assessment of serum AAT levels and genotyping for the detection of generic variants that are associated with the condition. Perhaps the easiest method for AAT deficiency testing utilizes dried blood spots collected on filter paper which are then submitted by regular mail to a research laboratory for analysis. This circumvents the need to draw intravenous blood and transport biohazardous specimens for testing. These test kits are provided free of charge and contain detailed instructions for their use. If a patient is found to have serum AAT levels below 11 μM and/or Z or S alleles, then it is likely that he/she has AAT deficiency. At this point, the patient should be referred to a respiratory specialist in order for confirmatory tests and phenotyping to be carried out ().
Case study, patient A: AAT deficiency testing
The patient's primary care physician had a good understanding of guidelines issued by the ATS and ERS with respect to AAT deficiency. These recommend AAT testing in individuals with a family history and in individuals with asthma not completely reversible with bronchodilators. In this case, the physician suspected AAT deficiency and initiated testing. Following AAT testing consisting of a serum level and genotyping, the patient was found to have a PiZZ phenotype, and lung function tests revealed a forced expiratory volume in 1 second (FEV1) of 38% predicted. Earlier tests, 4 years previously and under a different physician, had shown an FEV1 of 61% predicted, but these findings had not been investigated further. As a result of earlier misdiagnosis, the patient had suffered a substantial delay before appropriate treatment/management could be initiated. Current guidelines from the ATS/ERS do not recommend the use of augmentation therapy for AAT deficiency in individuals with an FEV1 ≥ 60% predicted, as in this case. However, the supportive evidence for this guidance does not come from randomized controlled trials but rather from underpowered observational studies. As such, physicians are best advised to exercise their clinical judgement when deciding upon the best course of treatment for their patients.
Planned-care follow-up
Planned-care follow-up of individuals with diagnosed AAT deficiency incorporates several different activities and medical functions all co-ordinated at the practice level and aimed at providing consistent and coherent care. These activities include testing of family members for AAT deficiency by both serum AAT levels and genotyping. Testing for AAT levels alone in this setting may not detect individuals who are heterozygous for an abnormal AAT gene, and thus family members might mistakenly believe they are not at risk of passing on an abnormal gene to their offspring. Other activities may include the development of a treatment plan; medical monitoring; genetic counseling and counseling from family support groups, social support groups, and patient support groups; and ongoing education through literature sources and patient support groups. In addition, patient self-management education is a cornerstone of the chronic care model. Treatment comprises lifestyle changes such as preventive therapy that may importantly include smoking cessation management (if necessary) and vaccination against pneumococcal and influenza viruses, hepatitis A, and hepatitis B, limited alcohol intake, and the avoidance of liver and respiratory toxins. Medical monitoring may take the form of routine follow-up visits, treatment adherence monitoring, regular spirometry, regular medical history, physical examination, and liver and pulmonary function tests.
Treatment
Even when there is awareness of AAT deficiency, physicians may hesitate to diagnose the disorder because it is perceived to be a complicated and costly disease to manage. The concept of “therapeutic nihilism” (Citation7) may also be a consideration in AAT deficiency. This occurs when a physician deems there are no effective treatments for the condition and thus no benefit in identifying affected individuals—an attitude that can significantly contribute to poor diagnosis rates. In reality, there are a number of effective treatments for the disorder (). Recommended therapy for patients with COPD unrelated to AAT deficiency is also applicable to those patients with AAT deficiency and includes inhaled bronchodilators and corticosteroids (Citation2). However, the major focus of therapy is on the correction of the deficiency so that further lung deterioration may be prevented. Several observational studies have demonstrated that intravenous augmentation therapy with a purified AAT preparation increases levels of AAT in the lung and slows the rate of lung function decline (Citation16-19). On the basis of these results, the ATS/ERS guidelines recommend the use of augmentation therapy in individuals with moderate airflow obstruction (FEV1 35–60% predicted) (Citation2). Two randomized, controlled trials, including a recent report, have demonstrated a trend toward a reduced loss of lung tissue as measured by computed tomography (CT) scanning in patients treated with augmentation therapy (Citation20, 21). Furthermore, an integrated analysis of these two trials has shown that augmentation therapy significantly reduces the decline in lung density in patients with AAT deficiency (Citation22).
Table 2. Medical treatments for AAT deficiency (2)
Case study, patient A: Follow-up and outcomes
The primary care physician consequently recommeded AAT testing for other family members and the patient's mother and brother were found to have an MZ and MM phenotype, respectively. Affected family members were offered genetic and social counseling and support, which is being led by the primary care practice working alongside other healthcare professionals. Scheduled medical monitoring was initiated by the primary care physician, and the patient attends regular appointments at the practice. Planned-care follow-up visits involve examinations to assess lung function parameters by spirometry. In addition, the primary care physician and/or practice staff are working with the patient to ensure adherence to recommended medication. The patient has now been receiving augmentation therapy with a purified AAT preparation for 1.5 years and receives his infusions at home. The family have become actively involved with AAT deficiency patient support groups.
Case study conclusions
Poor attention to a family history can lead to missed diagnosis in other family members.
Symptoms resulting from AAT deficiency can easily be confused with those of non-hereditary COPD or asthma.
Awareness of published guidelines for the management of AAT deficiency can facilitate time to diagnosis.
Primary care physicians can easily initiate and conduct testing for the condition; this does not require specialist intervention.
Further testing in other family members should be recommended.
Comprehensive follow-up and monitoring programs initiated by the primary care physician can improve patient quality of life.
By being alert to risk factors, markers of disease in the family background, and published guidelines, the primary care physician can improve diagnosis rates.
RAISING AWARENESS OF AAT DEFICIENCY
Diagnosis rates cannot be improved if physicians and patients have little or no understanding of AAT deficiency. Greater disease awareness needs to be promoted in primary care through the development and widespread implementation of specific medical education programs and activities endorsed by professional societies. Furthermore, the wider dissemination of ATS/ERS guidelines () (Citation2) within primary care practices is required. Patient groups such as the Alpha-1 Foundation (www.alphaone.org/), the Alpha-1 Association (www.alpha1.org/), and AlphaNet (www.alphanet.org/) are also in a strong position, through easily comprehensible information sources, to promote a greater understanding of the condition among individuals who have been diagnosed and to ensure that their families seek testing. There is evidence that the promotion of disease awareness coupled with the offer of a diagnostic test can result in the identification of a significant number of individuals with AAT deficiency (Citation12).
CONCLUSIONS
AAT deficiency is a common and underrecognised disease. The earlier a diagnosis is made, the sooner an individual can begin treatment and make the necessary lifestyle changes to improve prognosis and quality of life. This review has identified a number of impediments to better detection rates and has suggested practical processes that may improve diagnosis rates and clinical follow-up. From the guidelines to the front lines, primary care physicians have an important role to play in raising awareness of AAT deficiency and improving long-term care in these patients. They can act as educator and counselor as well as physician to provide optimal care for individuals and their families.
Declaration of interest
Dr. Fromer is a consultant for Talecris Biotherapeutics, Inc., and has received honoraria for advisory boards and speakers bureau for CSL Behrings, Inc. The author alone is responsible for the content and writing of the paper.
ACKNOWLEDGMENT
This manuscript was supported by a grant from Talecris Biotherapeutics, Inc.
Technical editorial assistance was provided under the direction of the author by Susan Robinson, PhD, of PAREXEL and was supported by Talecris Biotherapeutics, Inc.
REFERENCES
- Janoff A. Inhibition of human granulocyte elastase by serum alpha-1-antitrypsin. Am Rev Respir Dis 1972; 105:121–122.
- American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Demeo DL, Silverman EK. Alpha1-antitrypsin deficiency. 2: genetic aspects of alpha(1)-antitrypsin deficiency: phenotypes and genetic modifiers of emphysema risk. Thorax 2004; 59:259–264.
- Seersholm N, Kok-Jensen A, Dirksen A. Decline in FEV1 among patients with severe hereditary alpha 1-antitrypsin deficiency type PiZ. Am J Respir Crit Care Med 1995; 152:1922–1925.
- Campos MA, Wanner A, Zhang G, Sandhaus RA. Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest 2005; 128:1179–1186.
- Stoller JK, Aboussouan LS. Alpha1-antitrypsin deficiency. Lancet 2005; 365:2225–2236.
- Stoller JK, Fromer L, Brantly M, Stocks J, Strange C. Primary care diagnosis of alpha-1 antitrypsin deficiency: issues and opportunities. Cleve Clin J Med 2007; 74:869–874.
- Rosenthal TC. The medical home: growing evidence to support a new approach to primary care. J Am Board Fam Med 2008; 21:427–440.
- de Serres FJ, Blanco I, Fernandez-Bustillo E. Estimating the risk for alpha-1 antitrypsin deficiency among COPD patients: evidence supporting targeted screening. COPD 2006; 3:133–139.
- Fleming LE, Oquendo S, Bean JA, Tamer R, Finn S, Wanner A. Pilot detection study of alpha(1) antitrypsin deficiency in a targeted population. Am J Med Genet 2001; 103:69–74.
- Luisetti M, Massi G, Massobrio M, Guarraci P, Menchicchi FM, Beccaria M, Balbi B. A national program for detection of alpha 1-antitrypsin deficiency in Italy. Gruppo I.D.A. Respir Med 1999; 93:169–172.
- Bals R, Koczulla R, Kotke V, Andress J, Blackert K, Vogelmeier C. Identification of individuals with alpha-1-antitrypsin deficiency by a targeted screening program. Respir Med 2007; 101:1708–1714.
- Martinez FJ, Raczek AE, Seifer FD, Conoscenti CS, Curtice TG, D'Eletto T, Cote C, Hawkins C, Phillips AL. Development and initial validation of a self-scored COPD Population Screener Questionnaire (COPD-PS). COPD 2008; 5:85–95.
- Rahaghi F, Ortega I, Rahaghi N, Oliveira E, Ramirez J, Smolley L, Stoller JK. Physician alert suggesting alpha-1 antitrypsin deficiency testing in pulmonary function test (PFT) results. COPD 2009; 6:26–30.
- Needham M, Stockley RA. Alpha 1-antitrypsin deficiency. 3: Clinical manifestations and natural history. Thorax 2004; 59:441–445.
- Wewers MD, Casolaro MA, Sellers SE, Swayze SC, McPhaul KM, Wittes JT, Crystal RG. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N Engl J Med 1987; 316:1055–1062.
- Seersholm N, Wencker M, Banik N, Viskum K, Dirksen A, Kok-Jensen A, Konietzko N. Does alpha1-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha1-antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL) alpha1-AT study group. Eur Respir J 1997; 10:2260–2263.
- The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha 1-antitrypsin. Am J Respir Crit Care Med 1998; 158:49–59.
- Wencker M, Banik N, Buhl R, Seidel R, Konietzko N. Long-term treatment of alpha1-antitrypsin deficiency-related pulmonary emphysema with human alpha1-antitrypsin. Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL)-alpha1-AT-study group. Eur Respir J 1998; 11:428–433.
- Dirksen A, Dijkman JH, Madsen F, Stoel B, Hutchinson DC, Ulrik CS, Skovgaard LT, Kok-Jensen A, Rudolphus A, Seersholm N, Vrooman HA, Reiber JH, Hansen NC, Heckscher T, Viskum K, Stolk J. A randomized clinical trial of alpha 1-antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160:1468–1472.
- Dirksen A, Piitulainen E, Parr DG, Deng C, Wencker M, Shaker SB, Stockley RA. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1345–1353.
- Stockley RA, Deng C, Piitulainen E, Parr DG, Shaker SB, Wencker M, Stolk J, Stoel BC, Dirksen A. Therapeutic efficacy of alpha-1 antitrypsin (AAT) augmentation therapy on the loss of lung tissue: an integrated analysis [abstract]. Am J Respir Crit Care Med Apr 2008; 177.