Abstract
Chronic obstructive pulmonary disease (COPD) is a multicomponent condition that is characterized by partially reversible airflow obstruction. Serum surfactant protein D (SP-D) is synthesized by type II pneumocytes and Clara cells and participates in surfactant homeostasis and pulmonary host defense. Serum levels of SP-D are raised in individuals with COPD but there is no correlation between the serum level of SP-D and the severity of airflow obstruction. Serum SP-D is present in different forms that may have more utility as a biomarker for COPD. We report here the development of new monoclonal antibodies to full length and cleaved SP-D. We have assessed these and existing antibodies in 98 individuals with COPD recruited to the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) cohort. Our data show that neither monoclonal antibodies to full length nor cleaved SP-D provide additional information over that obtained with a polyclonal antibody. Moreover, levels of serum nitrosylated-SP-D did not correlate with serum level of SP-D or any clinical phenotype of COPD. The measurement of modified SP-D is of limited value in characterising individuals with COPD.
INTRODUCTION
Surfactant Protein D (SP-D) is a large hydrophilic protein that is a member of the collectin family (Citation1). It consists of a short N-terminal domain, a collagen domain, a hydrophobic neck and a calcium-dependant carbohydrate recognition domain (CRD). The association of 3 SP-D molecules, through the formation of a triple helical collagen region and stabilizing interchain disulfide bonds, forms basic subunits, which then associate into dodecamers linked by their N-terminal regions (cruciform structure). SP-D is secreted into the lungs by both type II alveolar cells and by Clara cells (Citation2).
It participates in the pulmonary host defense as well as surfactant homeostasis. In particular, SP-D can facilitate microbacterial agglutination (Citation3, 4), play a role in the clearance of pathogens (Citation5) and apoptotic cells (Citation6), modulate the cellular immune response (Citation7), have a direct effect on microbacterial growth (Citation8), modulate the allergic response (Citation9), play a role in the regulation of inflammation (Citation10, 11) and bind and help to clear DNA from the lungs (Citation12). A recombinant fragment of SP-D (composed of the neck and the CRD) can prevent emphysema and chronic inflammation in SP-D knock-out mice (Citation13).
As SP-D is predominantly synthesized within the respiratory tract, it has been evaluated as a potential biomarker in small numbers of individuals with community acquired pneumonia (Citation14), drug induced lung disease (Citation15, 16), interstitial fibrosis (Citation17), and allergic bronchopulmonary aspergillosis in cystic fibrosis (Citation18). The concentration of SP-D is raised in the serum and reduced in bronchoalveolar lavage from individuals with chronic obstructive pulmonary disease (COPD) (Citation19, 20).
SP-D is elevated in smoking and in COPD but there is no significant correlation between serum levels of SP-D and the severity of COPD (as assessed by forced expiratory volume in 1 second (FEV1)) (Citation20). However, there is a marked decrease in serum SP-D following treatment of individuals with COPD with both inhaled and oral corticosteroids (Citation20, Citation21). It seems likely that SP-D is acting as a marker of lung permeability with the protein leaking into the circulation as a result of exposure to cigarette smoke and the enhanced inflammation in individuals with COPD. However, it is unclear whether the assays are detecting full length or fragments of SP-D (Citation22). Indeed, small fragments may be more sensitive to changes in permeability and so be more useful as biomarkers of COPD. Moreover, SP-D may be a target of nitrosylation during the immune or inflammatory response, making nitrosylated SP-D a potentially interesting biomaker for lung disease.
We have developed novel monoclonal antibodies and have evaluated existing antibodies to assess whether fragments of SP-D and the nitrosylation of SP-D are useful biomarkers of COPD.
MATERIALS AND METHODS
Antibodies
The polyclonal rabbit anti-mouse IgG and anti-rabbit antibodies linked to HRP were purchased from Sigma-Aldrich (Dorset, UK). The polyclonal rabbit anti-SP-D antibody was produced against a recombinant fragment of human SP-D (Citation23). The anti-S-nitroso-cystein (SNO) antibody was purchased from Autogen Bioclear (Nottingham, UK). The epitopes recognized by the monoclonal anti-SP-D antibodies 246–2, 246–4 and 245–1 are summarized in (Citation24).
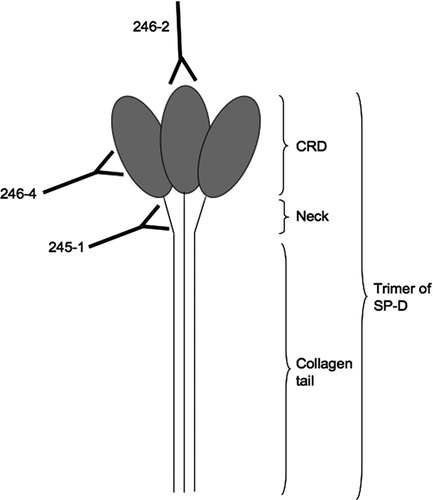
Figure 1 Binding of monoclonal antibodies to epitopes on SP-D. 245–1 binds to the neck area of SP-D, which may become less accessible when SP-D is polymerised. 246–4 binds to the lateral side of the CRD and blocks the binding of gp340 to SP-D. 246–2 binds to the lectin site and reduces the binding of viruses to SP-D.
Purification of SP-D
SP-D was synthesized from CHO cells containing a cDNA encoding full length human SP-D (Citation3). The protein was captured from conditioned medium using anion ion exchange chromatography to concentrate the sample and remove glucose. Following extensive washing to remove impurities, the SP-D was eluted from the anion exchange column, diluted and calcium was added to a final concentration of 5 mM. The SP-D was then affinity-purified on maltose-agarose as described previously (Citation3).
Cleavage assay
Two micrograms of SP-D were incubated with increasing concentrations of neutrophil elastase linked to beads of agarose (Elastin Product Company (Owensville, USA)) (4 mg/ml) for 30 min at 37°C in 10 mM Tris pH 7.4, 150 mM NaCl. The samples were then centrifuged at 13,000g to remove the beads. Aliquots of cleaved SP-D were separated by 12% w/v acrylamide NuPage gels with MES buffer (Invitrogen (Paisley, UK)). The proteins were visualized by silver staining or by staining with Simply Blue SafeStain (Invitrogen).
Production of the anti-SP-D monoclonal antibodies
Monoclonal antibodies specific to fragments of SP-D were prepared by immunizing 2 mice (Harlan Sera-Lab (Belton, UK)) with a mix of full length SP-D and SP-D cleaved with neutrophil elastase. Mice were immunized subcutaneously on day 0 with 20 μg of antigen with Freunds Complete Adjuvant following the collection of a pre-immunization bleed. The immunization was boosted on day 7, 14 and 28 with 20 μg of subcutaneous antigen with Freunds Incomplete Adjuvant and test bleed 1 was collected on day 21. The mice received an intravenous pre-fusion boost with 20 μg of antigen on day 33. On day 35, the mice were then bled and the spleens were removed.
The production of hybridoma cell lines was carried out as described previously (Citation25). In brief, the spleens were removed from both mice, mixed and fused with P3-X63-Ag8.653 myeloma cells. After fusion, cells were cultured in Hypoxanthine Aminopterin Thymidine (HAT) medium in 96-well plates for 12 days. The hybridoma clones were screened by antigen-mediated ELISA using a mix of full length and neutrophil elastase cleaved SP-D as the antigen. Selected clones were sub-cloned by limited dilution and expanded as cell lines.
Antigen-mediated ELISA
All steps were carried out at room temperature with 50 μl solution/well unless otherwise stated. Plates were coated overnight at 4°C with SP-D, cleaved SP-D or neutrophil elastase at 2 μg/ml in coating buffer (15 mM Na2CO3, 35 mM NaHCO3). The wells were then washed with washing buffer (PBS, 0.05% v/v Tween20) and blocked for 2 h with 300 μl/well of blocking buffer (PBS, 3% w/v BSA, 0.05% v/v Tween20). Plates were washed and either undiluted culture media supernatant or supernatant diluted in blocking buffer was added to each well for 2 h. The plates were washed a further 3 times and bound monoclonal antibodies were detected with rabbit anti-mouse HRP antibody (1:20,000 in blocking buffer) for 1 h. The reaction was developed for 10 min with TMB (tetramethylbenzidine) substrate solution (Invitrogen), stopped with 1 M H2SO4 and then HRP activity was measured in a plate reader at 450 nm.
SP-D Sandwich ELISA
All steps were carried out at room temperature using 50 μl/ well unless otherwise stated. Plates were coated overnight at 4°C with rabbit polyclonal anti-SP-D antibody at 1.6 μg/ml in coating buffer (15 mM Na2CO3, 35 mM NaHCO3). The wells were then washed with washing buffer (PBS, 0.05% v/v Tween20) and blocked for 2 h with 300 μl/well of blocking buffer (PBS, 3% w/v BSA, 0.05% v/v Tween20). Plates were washed and standards and samples were diluted in washing buffer and incubated in the wells for 2 h.
After washing, the wells were incubated with either 246–4, 246–2, 245–1 or 1F4 monoclonal antibodies (1 μg/ml) diluted in blocking buffer for 2 h. Bound monoclonal antibodies were detected with rabbit anti-mouse HRP antibody (1:20,000 in blocking buffer) for 1 h. The reaction was developed for 10 min with TMB substrate solution (Sigma-Aldrich), stopped with 1 M H2SO4 and then HRP activity was measured in a plate reader (Molecular Devices, Thermo-max microplate reader) at 450 nm. All ELISAs were performed in triplicate and bagplots were drawn with R version 2.10.0. The inter-assay coefficient of variation (CV) was measured for each antibody and each sample using the equation: CV = (standard deviation / mean) × 100. The average of the CV for each sample was measured and used as the intra-assay CV of that assay.
Western blot analysis of serum SP-D
SP-D was partly purified from serum by adding 500 μl 10 mM Tris, 150 mM NaCl, 20 mM CaCl2 and 30 μl maltose-agarose beads to 20 μl of serum. After incubation for 30 min at room temperature, the mix was centrifuged at 13,000 g and beads were collected and separated by 12% w/v NuPage gels with MES buffer (Invitrogen) followed by transfer onto Immobilon-P membrane (Millipore (Watford, UK)) in 25 mM Tris, 192 mM glycine, 20% v/v methanol at 200 mA for 2 h.
Membranes were blocked overnight at 4°C in PBS with 0.05% w/v Tween 20 (PBS-T) with 5% w/v skimmed dried milk. They were then incubated with either the polyclonal rabbit anti-SP-D antibody or the monoclonal mouse anti-SP-D antibodies (1 μg/ml) in 5% w/v milk for 2h at room temperature. The membranes were washed 3 times for 5 min and twice for 10 min in PBS-T before being incubated with Horseradish peroxidase-linked anti-rabbit or anti-mouse IgG (according to the primary antibody used) (Sigma-Aldrich) for 2h at room temperature. The antibodies were diluted 1/20,000 in PBS-T with 5% w/v milk. Membranes were washed as before, incubated for 5 min with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific (Cramlington, UK)) and developed on film.
S-Nitrosylation (SNO) sandwich ELISA
All steps were carried out at room temperature and using 50 μl/well unless otherwise stated. Plates were coated overnight at 4°C with 246–5 anti-SP-D antibody (diluted 1/1000) in coating buffer (15 mM Na2CO3, 35 mM NaHCO3). The wells were then washed with washing buffer (PBS, 0.05% v/v Tween20) and blocked for 2 h with 300 μl/well of blocking buffer (PBS, 3% w/v BSA, 0.05% v/v Tween20). The plates were washed and samples were diluted in washing buffer and incubated for 2 h. After washing, the wells were incubated with rabbit anti-S-nitrosylation (SNO) antibody (diluted 1/1000) diluted in blocking buffer for 2 h. Bound monoclonal antibodies were detected with rabbit anti-rabbit HRP antibody (1:20,000 in blocking buffer) for 1 h. The reaction was developed for 10 min with TMB substrate solution, stopped with 1 M H2SO4 and then HRP activity was measured in a plate reader at 450 nm. The monoclonal antibody 246–5 was chosen for this assay as it gave the highest signal of all the antibodies tested.
The evaluation of COPD longitudinally to identify predictive surrogate endpoints (ECLIPSE) cohort (Clinicaltrials.gov identifier NCT00292552, appendix 1)
ECLIPSE is a multicentre 3-year longitudinal prospective study to identify novel endpoints in COPD (Citation20, Citation26). Individuals aged 40–75 years were recruited to the study if they had a smoking history of ≥10 pack-years, a post-bronchodilator ratio between forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) ≤0.7 and GOLD stage II (FEV1 50–80% predicted), III (FEV1 30–50% predicted) or IV (FEV1 <30% predicted) COPD (Citation27). Smoking (≥10 pack-years) and non-smoking (<1 pack-year) control subjects were enrolled if they were aged 40–75 years and had normal lung function (post-bronchodilator FEV1 > 85% predicted and FEV1/FVC > 0.7).
All subjects underwent standardized spirometry following the inhalation of 180 mcg (2 puffs) of salbutamol and all were offered a low dose computed tomography (CT) scan of the chest. The CT scans were evaluated at the central imaging unit at the University of British Columbia in Vancouver. The extent of the emphysema was assessed in two ways. First, it was independently scored by two radiologists who were blind to the individual's lung function. Emphysema was reported as none (grade 0), trivial (grade I), mild (grade II), moderate (grade III), severe (grade IV) and very severe (grade V) if it affected <5%, 5–25%, 25–50%, 50–75%, and >75% of the lungs, respectively.
A consensus reading was obtained when there was a difference of more than 1 emphysema category between the 2 observers. Otherwise the average of the 2 readings was used in the analysis. Secondly, emphysema was assessed by the percentage of the lung with attenuation below -950 HU using the Pulmonary Workstation 2.0 software (VIDA Diagnostics, Inc., Iowa City, IA). Ninety-eight stable subjects were selected as having SP-D levels in the lowest (48 individuals) and highest (50 individuals) quartile when measured at baseline (t = 0, day 1, visit 1). Subjects were considered as stable if they were not taking oral steroids and/or antibiotics within 4 weeks of the visit being evaluated.
RESULTS
Production of antigen for immunization
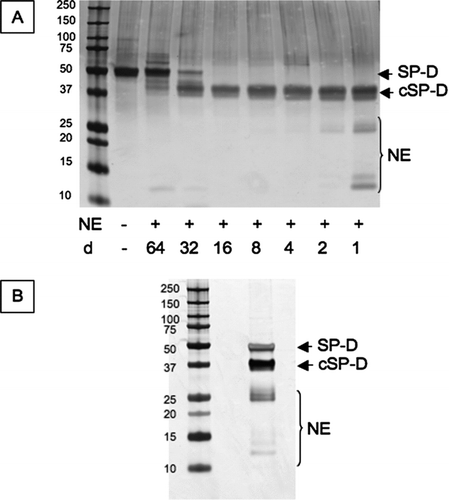
Two micrograms of purified SP-D were cleaved with increasing concentration of neutrophil elastase linked to beads of agarose for 30 min at 37°C (). A final ratio of neutrophil elastase:SP-D of 4:1 was selected and the reaction scaled up to produce sufficient cleaved protein to immunize 2 mice (20 μg per injection per mouse). There was a faint 12kDa band of cleaved SP-D at lower neutrophil elastase:SP-D ratios (, lane 3 and 4). This was also present in the final mix of antigen. Following incubation the mixture was centrifuged at 13,000 g for 5 minutes to remove agarose beads (). Neutrophil elastase can dissociate from the beads to give a fragment of 25.6 kDa. It can also autolyse into 2 fragments of 14 and 15 kDa. These fragments were not removed from the mix during the centrifugation step and so the mice were immunized with a mixture of full length SP-D, cleaved SP-D and residual neutrophil elastase.
Figure 2 Cleavage of SP-D by neutrophil elastase. (A) 2 μg SP-D were cleaved at 37°C for 30 minutes with different dilutions (d) of neutrophil elastase (NE). This was then scaled up to prepare the antigen to immunise the mice (B). The proteins were separated on 12% w/v acrylamide SDS-PAGE and then visualised by silver staining. cSP-D: cleaved SP-D.
Immunization of mice
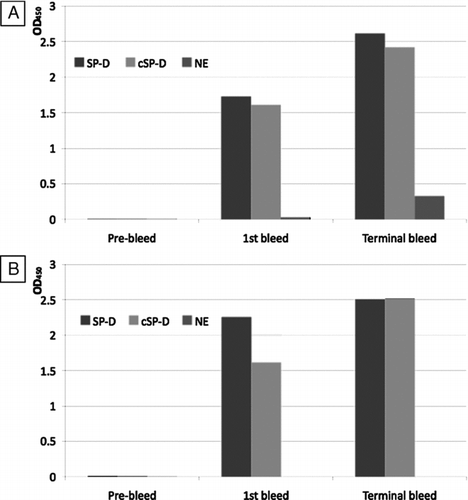
The mice were immunized as detailed in the methods section. The presence of antibodies was assessed in the pre-immunization bleed (day 0), the test bleed 1 (day 21) and the terminal bleed (day 35) by antigen-mediated ELISA against full length SP-D, cleaved SP-D and neutrophil elastase (). Neither of the mice had antibodies to either full length SP-D, cleaved SP-D or neutrophil elastase prior to immunization. Antibodies were present to full length and cleaved SP-D but not neutrophil elastase at day 21. At the terminal bleed, both mice had developed antibodies to full length and cleaved SP-D whilst one mouse had developed low levels of antibodies to neutrophil elastase.
Figure 3 Screening of immunisation bleeds. The pre-immunisation bleed (day 0), first bleed (day 21) and terminal bleed (day 35) of each mouse ((A) –mouse 1; (B) –mouse 2) were used in antigen-mediated ELISA against SP-D, neutrophil elastase cleaved SP-D (cSP-D) and neutrophil elastase (NE). Values showed are maximum OD450 obtained.
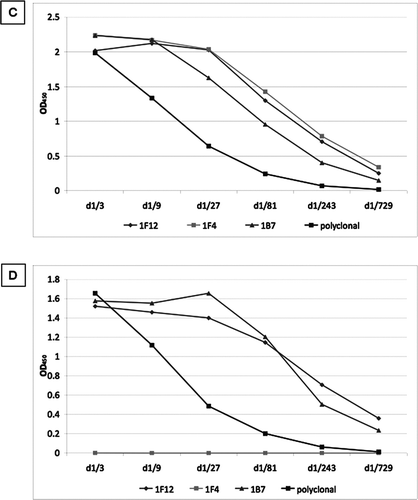
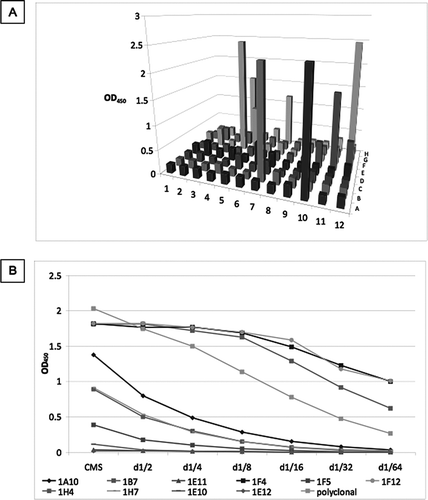
The fused cells were plated in 96-well plates and grown for 12 days. The culture media supernatants (CMS) were assessed by antigen-mediated ELISA (), and positive wells were expanded and assessed by limited dilution (). Clones 1E11, 1F5, 1E10 and 1E12 were lost. After further expansion clones 1A10, 1H4, 1H7 were also lost. The remaining clones were screened against full length SP-D () and cleaved SP-D (). None of the clones detected neutrophil elastase (data not shown). All the clones recognized both cleaved and full length SP-D apart from 1F4, which recognized only the full length protein. This clone was selected for further evaluation.
Figure 4 Screening of culture media supernatant (CMS) by antigen-mediated ELISA. (A) Fused cells were plated on a 96-well plate. CMS from all wells was screened against a mix of 1μg/ml of full length SPD and 1 μg/ml neutrophil elastase cleaved SP-D. Clones were named according to their position on the plate. (B) Positive wells were expanded in 12-well plates and rescreened against a mix of 1μg/ml of full length SPD and 1 μg/ml cleaved SP-D. Clones 1E11, 1F5, 1E10 and 1E12 were lost. After further expansion clones 1A10, 1H4, 1H7 were also lost. The remaining clones were screened against (C) full length SP-D and (D) cleaved SP-D. A polyclonal anti-SP-D antibody was used as a positive control (diluted 1/1000). d1/x: dilution of CMS. None of the antibodies detected neutrophil elastase.
Characterization of the monoclonal antibody 1F4 and other commercial anti-SP-D antibodies
Full length and cleaved SP-D were separated on SDS-PAGE, in reduced and non-reduced conditions, followed by staining with SafeStain () or Western blots analysis ( B-F). Western blots were undertaken using the polyclonal antibody control (), monoclonal antibody 1F4 () as well as other commercially available anti-SP-D monoclonal antibodies (246–4 (), 246–2 () and 245–1 ()). Although presenting slight differences in their recognition pattern, all monoclonal antibodies apart from 245–1 recognize full length SP-D in both the reduced and non-reduced state but not neutrophil elastase-cleaved SP-D. Monoclonal antibody 245–1 recognizes the different forms of full length and cleaved SP-D in both reduced and non-reduced conditions in a pattern that is similar to that of the polyclonal antibody.
Figure 5 Characterisation of novel and commercially available antibodies. Full length SP-D and neutrophil elastase cleaved SP-D (cSP-D) were separated by a 10% w/v SDS-PAGE with or without 200mM DTT and (A) stained with Simply Blue SafeStain, or blotted with (B) polyclonal anti-SP-D antibody, (C) 1F4, (D) 246–4, (E) 246–2 or (F) 245–1 monoclonal antibodies.
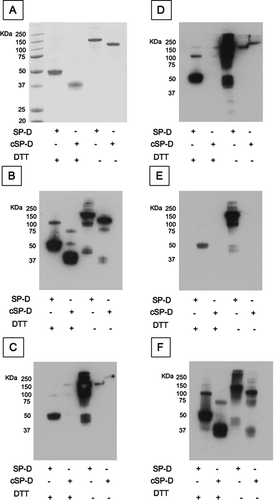
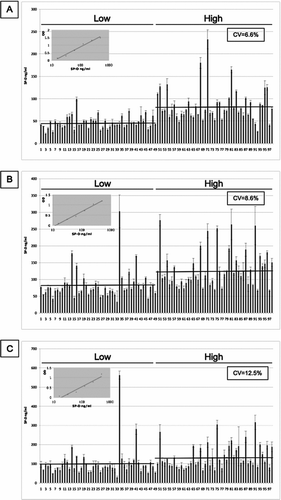
Figure 6 SP-D levels in 98 samples from the ECLIPSE cohort. SP-D levels were measured in serum samples by sandwich ELISA with 4 different antibodies ((A) 246–4, (B) 1F4, (C) 246–2 and (D) 245–1). CV: inter-assay coefficient of variation. Inserted graph: Representative standard curve. Low/high: Samples were chosen for their low/high SP-D levels according to previous measurements. Bars represent the average values. The mean values for the high and low samples were 83.431 (±37.1) and 46.899 (±14.16) ng/ml for monoclonal antibody 246–4 (A), 128.602 (±56.21) and 82.172 (±43.37) ng/ml for monoclonal antibody 1F4 (B), 133.858 (±62.2) and 102.348 (±78.13) ng/ml for monoclonal antibody 246–2 (C) and 0.021 (±0.0152) and 0.018 (±0.0109) units for monoclonal antibody 245–1 (D). Monoclonal antibody 245–1 did not recognise purified full length SP-D in ELISA and thus, no standard curve was constructed. Eight serum samples were added to each ELISA plate to control for intra-plate variation.
Measure of SP-D levels in serum samples
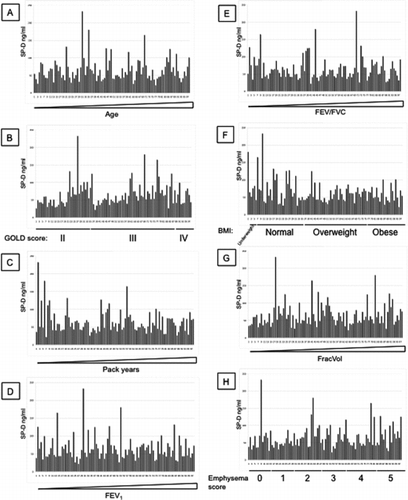
It is important to determine whether any of the monoclonal antibodies give a different measure of serum SP-D from that reported previously (Citation20). The 4 monoclonal antibodies in were used to measure serum SP-D in 98 samples from individuals with COPD from the ECLIPSE cohort (). The samples were drawn at the 6-month visit. All patients were stable, and were selected as having SP-D levels in the lowest (48 individuals) and highest (50 individuals) quartile when measured at baseline (t = 0, day 1, visit 1) (ratio of low:high 0.257) (Citation20). For all antibodies, the average SP-D levels for the low samples are lower than the average SP-D levels in the high samples. The range of values was similar to that obtained when the levels were measured at baseline in this group of individuals (data not shown). However, the differences between the mean values for the high and low samples was greater for antibodies 246–4 (83.431 (±37.1) and 46.899 (±14.16) ng/ml, ratio of low:high : 0.562) and 1F4 (128.602 (±56.21) and 82.172 (±43.37) ng/ml, ratio of low:high : 0.639) than for 246–2 (133.858 (±62.2) and 102.348 (±78.13) ng/ml, ratio of low:high : 0.765). There was little difference between the high and low values for antibody 245–1 (0.021 (±0.0152) and 0.018 (±0.0109) units, ratio of low:high : 0.857) suggesting that this is measuring something different. Moreover, some samples show markedly different values with different antibodies.
These results were then assessed with increasing age, GOLD stage, the number of pack-years of cigarette smoked, FEV1, FEV1/FVC ratio, BMI, FracVol or CT score of emphysema for each monoclonal antibody (Additional File 1–4). There was no correlation between any of these characteristics and SP-D levels as confirmed by Spearman correlation (). Moreover, there was no association between the level of SP-D measured with these antibodies and exacerbations of COPD.
Table 1 Spearman correlation for the monoclonal antibodies 1F4, 245–1, 246–2 and 246–4 and the anti-SNO antibody with the components of the COPD phenotype
The results from different antibodies were then compared and outliers were determined using bagplots (). The bagplot is a 2D generalization of the boxplot. A bivariate median is calculated and a “bag” containing 50% of the observations with greatest bivariate depth is determined. The bag is expanded 3 times thus creating the “loop”. Values found outside this loop are regarded as too far from the central bulk of the data and so are flagged as outliers. Samples 21, 34, 41, 51, 54, 65, 68, 71, 75, 81, 91 and 95 were flagged as outliers in the bagplots. Sample 34 was an outlier in all comparisons, whereas samples 21, 54 and 95 were outliers in only one comparison (245–1 vs 246–2, 245–1 vs 246–4 and 245–1 vs 246–4, respectively). All other outliers were identified in several comparisons.
Characteristics of outliers by western blot analysis
The difference in the mean values of SP-D () and the identification of outliers may indicate that different antibodies are detecting different conformers of SP-D. These were evaluated by SDS-PAGE and western blot analysis in comparison with a polyclonal antibody (). All antibodies detect a 50 kDa band corresponding to the full length SP-D monomer. The polyclonal antibody also recognizes a 160 kDa band that is likely to represent a trimer of SP-D as well as a 130 kDa band that may represent a cleaved trimer. All the monoclonal antibodies detect a 25 kDa band, with 246–2 showing the greatest affinity. A higher band of 30 kDa was also present in this Western blot and seems to be stronger in sample with the highest quartile value of SP-D.
However, it was also present in some outliers and some samples with the lowest quartile value of SP-D. All the monoclonal antibodies also detect the 130 kDa band, but with much less intensity than the polyclonal antibody, as well as a smaller 90 kDa band. All samples, outliers and non-outliers, present the same pattern of bands apart from outlier 41 that shows an additional band at 100 kDa with each antibody. The blots are consistent with findings from lung lavage from individuals with cystic fibrosis, in which there were higher molecular mass bands as well as cleaved SP-D (Citation23).
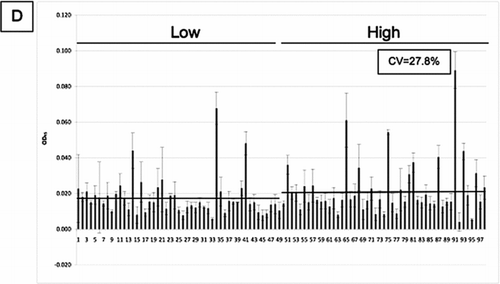
Measure of nitrosylated-SP-D levels
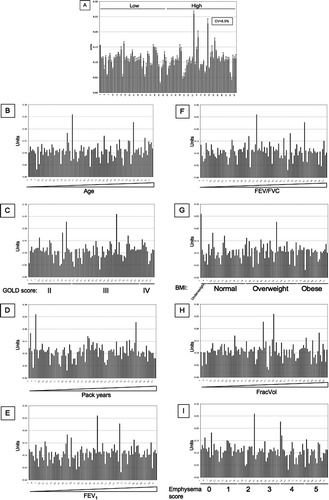
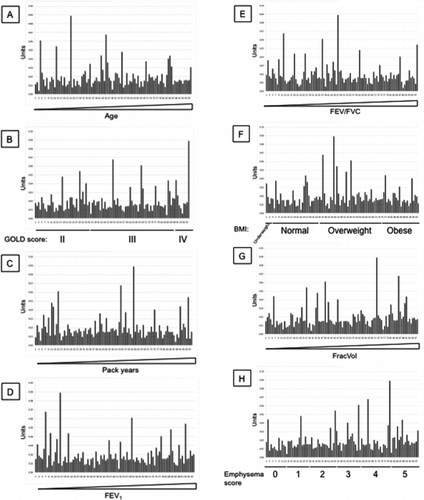
Finally, we assessed if nitrosylation of SP-D is a useful biomarker for COPD, and whether this would give a signal that is different from the new and existing monoclonal antibodies. SNO-SP-D was measured in an ELISA that used a monoclonal anti-SP-D antibody (246–5) as the capture antibody, and a polyclonal anti-SNO antibody as the detection antibody. SNO-SP-D levels were measured in the same ECLIPSE samples as before (). All measurements were standardized by the measurement of SNO-SP-D levels in 8 healthy controls. There was no association between the level of nitrosylated SP-D in individuals with high and low levels of SP-D. Moreover, there was no association of SNO-SP-D levels with increasing age (), GOLD stage (), the number of pack-years of cigarettes smoked (), FEV1 (Figure 9E), FEV1/FVC ratio (Figure 9F), BMI (Figure 9G), FracVol (Figure 9H). The findings were confirmed by Spearman correlation (). There was a weak but significant correlation between SNO-SP-D levels and the radiologist score of emphysema (Figure 9I and Table 1).
DISCUSSION
COPD is a complex disease that is characterized by only partially reversible airflow obstruction (Citation28). However, dyspnoea, quality of life and frequency of exacerbations do not correlate well with the standard measures of lung function, such as FEV1, that are used to classify the disease. SP-D is a pulmonary protein that is involved in the innate immune response (Citation7) and in inflammation (Citation10, 11). Serum levels are raised in current and former smokers but are even higher in individuals with COPD (Citation20).
Serum SP-D does not correlate with the severity of disease as assessed by FEV1 and CT scores of emphysema. However, raised levels have a weak association with exacerbations of COPD (Citation20). The aim of the current study is to establish whether existing or novel monoclonal antibodies can improve the utility of serum SP-D as a biomarker of COPD. Support for this hypothesis comes from the demonstration that SP-D can be cleaved by elastase in the lung in response to inflammation (Citation22, 23). This then leaks in the circulation. Antibodies to small fragments may be more sensitive in reporting components of the COPD phenotype. We have therefore produced monoclonal antibodies against full length and cleaved SP-D and assessed their utility in individuals with COPD.
Antibodies were generated that recognized full length as well as cleaved SP-D. However, 1F4 was of a particular interest as it had no affinity for cleaved SP-D and was specific for full length SP-D. Monoclonal antibody 1F4 was compared to several commercially available antibodies (246–2, 246–4, 245–1) as well as a polyclonal antibody in Western blot analysis in order to characterize any potential differences between these antibodies. 1F4 had a similar recognition pattern to all existing monoclonal antibodies. They all recognized the full-length form of SP-D. The only exception was 245–1, which recognized the full length and cleaved fragment, which was similar to the specificity of the polyclonal antibody.
We then used these monoclonal antibodies to measure SP-D in 98 serum samples from individuals with COPD recruited to the ECLIPSE cohort. Blood samples are taken regularly in patients in this study. Samples obtained from the first visit were used to measure serum SP-D with a commercially available ELISA (Citation20). We have used samples taken from patients 6 months after the start of the study. Samples were selected from individuals with serum levels of SP-D that were in the highest and lowest quartile. This is reasonable as serum levels of SP-D are relatively stable over time in individuals with stable disease (Citation20).
Figure 7 Statistical comparison of the values obtained with 5 different antibodies by bagplot. *: Bivariate median, dark grey: bag containing 50% of the observations with greatest bivariate depth, light grey: loop being the bag expanded 3 times, dot: Outlier being values outside the loop. (A) 1F4 vs 245–1, (B) 1F4 vs 246–4, (C) 1F4 vs 246–2, (D) 246–2 vs 246–4, (E) 245–1 vs 246–4, (F) 245–1 vs 246–2.
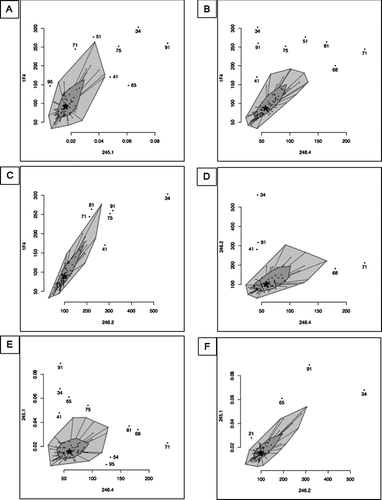
Figure 8 Western blot analysis of the outliers and controls (Low: lowest quartile, High: highest quartile of SP-D levels). Serum SP-D was purified by binding to maltose-agarose beads and analysed by western blot with (A) a polyclonal anti-SP-D antibody, (B) 1F4, (C) 246–4, (D) 246–2 or (E) 245–1 antibody.
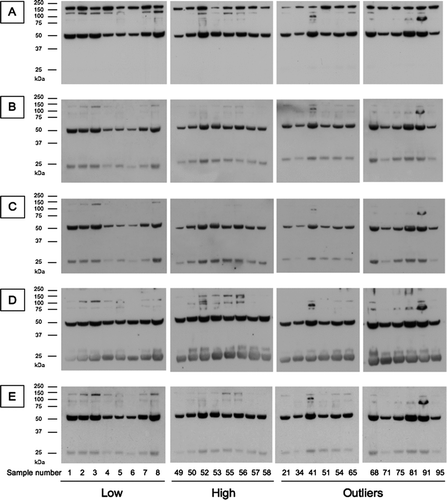
Figure 9 SNO-SP-D levels in 98 samples from the ECLIPSE cohort. (A) SNO-SP-D levels were measured in serum samples by sandwich ELISA. CV: Inter-assay coefficient of variation. Low/high: Samples chosen for their low/high SP-D levels according to previous measurements. Values were then arranged by increasing age (B), GOLD stages (C), increasing pack years (D), increasing FEV1 (E), increasing FEV1/FVC (F), BMI (G), increasing FracVol (H) and emphysema scores (I).
Additional File 1 SP-D levels measured with antibody 246–4 were arranged by increasing age (A), GOLD stages (B), increasing pack years (C), increasing FEV1 (D), increasing FEV1/FVC (E), BMI (F), increasing FracVol (G) and emphysema scores (H).
Additional File 2 SP-D levels measured with antibody 1F4 were arranged by increasing age (A), GOLD stages (B), increasing pack years (C), increasing FEV1 (D), increasing FEV1/FVC (E), BMI (F), increasing FracVol (G) and emphysema scores (H).
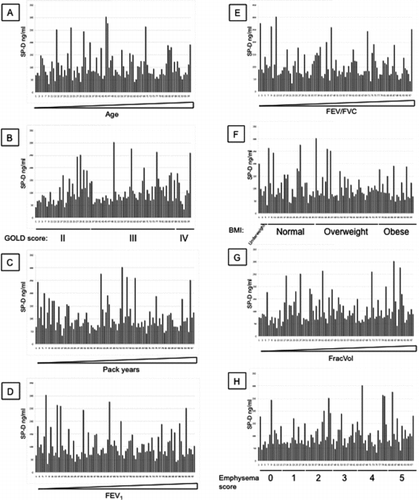
Additional File 3 SP-D levels measured with antibody 246–2 were arranged by increasing age (A), GOLD stages (B), increasing pack years (C), increasing FEV1 (D), increasing FEV1/FVC (E), BMI (F), increasing FracVol (G) and emphysema scores (H).
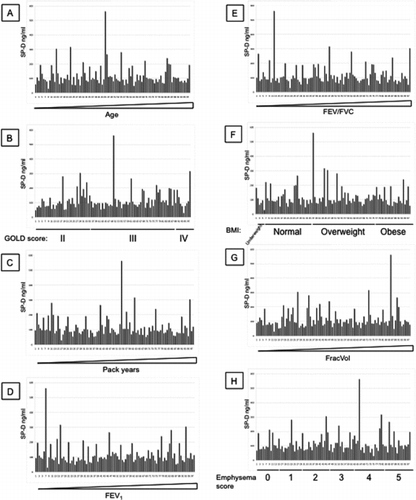
Additional File 4 SP-D levels measured with antibody 245–1 were arranged by increasing age (A), GOLD stages (B), increasing pack years (C), increasing FEV1 (D), increasing FEV1/FVC (E), BMI (F), increasing FracVol (G) and emphysema scores (H).
The concentrations of SP-D measured with our novel monoclonal antibody were compared with the values obtained with the commercial monoclonal antibodies. Although average values of the low and high quartile samples retain the same profile of low and high levels of SP-D, there were wide differences with some of the antibodies. Monoclonal antibodies 246–4 and 1F4 reported a larger difference between the high and low SP-D levels than monoclonal antibodies 246–2 and 245–1. There was no correlation between the value of SP-D reported by any of the antibodies and either age, GOLD stage of COPD, the number of pack-years of cigarette smoked, FEV1, FEV1/FVC ratio, BMI, FracVol, CT score of emphysema or exacerbations of COPD. It is possible that SP-D or the fragments of SP-D fluctuate with disease activity rather than the severity of COPD. However, it is difficult to measure disease activity in COPD.
The difference between the antibodies was then assessed with a bi-dimensional bagplots. This allowed us to highlight several outliers. Those outliers were further characterized by western blot analysis using a polyclonal anti-SP-D antibody as well as all the monoclonal antibodies used to measure the levels of SP-D (). There was no major difference between the Western blot analysis of SP-D of the outliers and the non-outliers, apart from a 100 kDa band in outlier 41. However, SP-D was purified from serum samples prior to the Western blot, and so it is possible that some of the smaller fragments of SP-D were lost. It is unclear what is being recognized by the antibodies in the outlier samples. Our previous western blot analysis of full length and cleaved SP-D () has shown that all the monoclonal antibodies recognized the full length SP-D, whilst monoclonal antibody 245–1 recognized both full length and cleaved SP-D. This difference in the western blot profiles must result from differences in partly purified serum SP-D and the purified recombinant protein.
The levels of SNO-SPD present were also assessed to establish if they are a useful biomarker of COPD. Production of nitric oxide (NO) results in the nitrosylation of proteins such as SP-D. NO production is regulated during the immune response (Citation29) and serves as a marker of inflammation in individuals with asthma (Citation30, 31, Citation32). Thus, the nitrosylation of SP-D may provide useful information in individuals with COPD.
Moreover, nitrosylation modifies the polymerization of SP-D and so may enhance its pro-inflammatory action (Citation33). SNO-SP-D levels were measured in the 98 ECLIPSE samples used previously. There was no significant association between SNO-SP-D and plasma levels of SP-D, age, GOLD stage of COPD, pack-years of cigarette smoked, FEV1, FEV1/FVC ratio, BMI, and exacerbation of COPD. There was only weak correlation with the radiologist score of emphysema. In conclusion, we have shown that the generation of new monoclonal antibodies does not improve the use of serum SP-D as a biomarker of stable COPD.
DECLARATION OF INTEREST
DAL has received grant support, consultancy fees and honoraria from GSK. BEM, RMT-S and LDE are employees of GSK. AD, EM, JP, JM, GLS, UH, HWC, BCT have no conflict of interest. The authors alone are responsible for the content and writing of the paper.
ACKNOWLEDGMENTS
We would like to thank Anne Presanis from MRC Biostatistics Unit, Cambridge for her help with the Bagplots. This work was funded by GlaxoSmithKline. JP was supported by the Spanish government (grants PR2009–0165 and MCINN SAF2010–19087).
Appendix 1.
List of principal investigators and centers participating in ECLIPSE (NCT00292552).
Bulgaria: Yavor Ivanov, Pleven; Kosta Kostov, Sofia.
Canada: Jean Bourbeau, Montreal, Que Mark Fitzgerald, Vancouver, BC; Paul Hernandez, Halifax, NS; Kieran Killian, Hamilton, On; Robert Levy, Vancouver, BC; Francois Maltais, Montreal, Que; Denis O'Donnell, Kingston, On.
Czech Republic: Jan Krepelka, Praha.
Denmark: JØrgen Vestbo, Hvidovre.
Netherlands: Emiel Wouters, Horn-Maastricht.
New Zealand: Dean Quinn, Wellington.
Norway: Per Bakke, Bergen.
Slovenia: Mitja Kosnik, Golnik.
Spain: Alvar Agusti, Jaume Sauleda, Palma de Mallorca.
Ukraine: Yuri Feschenko, Kiev; Vladamir Gavrisyuk, Kiev; Lyudmila Yashina, Kiev; Nadezhda Monogarova, Donetsk.
United Kingdom: Peter Calverley, Liverpool; David Lomas, Cambridge; William MacNee, Edinburgh; David Singh, Manchester; Jadwiga Wedzicha, London.
United States of America: Antonio Anzueto, San Antonio, TX; Sidney Braman, Providence, RI; Richard Casaburi, Torrance CA; Bart Celli, Boston, MA; Glenn Giessel, Richmond, VA; Mark Gotfried, Phoenix, AZ; Gary Greenwald, Rancho Mirage, CA; Nicola Hanania, Houston, TX; Don Mahler, Lebanon, NH; Barry Make, Denver, CO; Stephen Rennard, Omaha, NE; Carolyn Rochester, New Haven, CT; Paul Scanlon, Rochester, MN; Dan Schuller, Omaha, NE; Frank Sciurba, Pittsburgh, PA; Amir Sharafkhaneh, Houston, TX; Thomas Siler, St. Charles, MO, Edwin Silverman, Boston, MA; Adam Wanner, Miami, FL; Robert Wise, Baltimore, MD; Richard ZuWallack, Hartford, CT.
Steering Committee: Harvey Coxson (Canada), Lisa Edwards (GlaxoSmithKline, USA), Katharine Knobil (Co-chair, GlaxoSmithKline, UK), David Lomas (UK), William MacNee (UK), Edwin Silverman (USA), Ruth Tal-Singer (GlaxoSmithKline, USA), Jørgen Vestbo (Co-chair, Denmark), Julie Yates (GlaxoSmithKline, USA).
Scientific Committee: Alvar Agusti (Spain), Peter Calverley (UK), Bartolome Celli (USA), Courtney Crim (GlaxoSmithKline, USA), Gerry Hagan (GlaxoSmithKline, UK), William MacNee (Chair, UK), Stephen Rennard (USA), Ruth Tal-Singer (GlaxoSmithKline, USA), Emiel Wouters (The Netherlands), Julie Yates (GlaxoSmithKline, USA).
REFERENCES
- Rust K, Grosso L, Zhang V, Chang D, Persson A, Longmore W, Cai GZ, Crouch E. Human surfactant protein D: SP-D contains a C-type lectin carbohydrate recognition domain. Arch Biochem Biophys 1991; 290(1):116–126.
- Crouch E, Rust K, Marienchek W, Parghi D, Chang D, Persson A. Developmental expression of pulmonary surfactant protein D (SP-D). Am J Respir Cell Mol Biol 1991; 5(1):13–18.
- Hartshorn K, Chang D, Rust K, White M, Heuser J. Crouch E. Interactions of recombinant human pulmonary surfactant protein D and SP-D multimers with influenza A. Am J Physiol 1996; 271(5 Pt 1): L753–762.
- Ferguson JS, Voelker DR, McCormack FX, Schlesinger LS. Surfactant protein D binds to Mycobacterium tuberculosis bacilli and lipoarabinomannan via carbohydrate-lectin interactions resulting in reduced phagocytosis of the bacteria by macrophages. J Immunol 1999; 163(1):312–321.
- Hartshorn KL, Crouch E, White MR, Colamussi ML, Kakkanatt A, Tauber B, Shepherd V, Sastry KN. Pulmonary surfactant proteins A and D enhance neutrophil uptake of bacteria. Am J Physiol 1998; 274(6 Pt 1):L958–969.
- Vandivier RW, Ogden CA, Fadok VA, Hoffmann PR, Brown KK, Botto M, Walport MJ, Fisher JH, Henson PM, Greene KE. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: Calreticulin and CD91 as a common collectin receptor complex. J Immunol 2002; 169(7):3978–3986.
- Borron PJ, Crouch EC, Lewis JF, Wright JR, Possmayer F, Fraher LJ. Recombinant rat surfactant-associated protein D inhibits human T lymphocyte proliferation and IL-2 production. J Immunol 1998; 161(9):4599–4603.
- Wu H, Kuzmenko A, Wan S, Schaffer L, Weiss A, Fisher JH, Kim KS, McCormack FX. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J Clin Invest 2003; 111(10):1589–1602.
- Strong P, Reid KB, Clark H. Intranasal delivery of a truncated recombinant human SP-D is effective at down-regulating allergic hypersensitivity in mice sensitized to allergens of Aspergillus fumigatus. Clin Exp Immunol 2002; 130(1):19–24.
- Crouch E, Wright JR. Surfactant proteins A and D and pulmonary host defense. Annu Rev Physiol 2001; 63:521–554.
- Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, Greene KE, Henson PM. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 2003; 115(1):13–23.
- Palaniyar N, Nadesalingam J, Clark H, Shih MJ, Dodds AW, Reid KB. Nucleic acid is a novel ligand for innate, immune pattern recognition collectins surfactant proteins A and D and mannose-binding lectin. J Biol Chem 2004; 279(31):32728–32736.
- Knudsen L, Ochs M, Mackay R, Townsend P, Deb R, Muhlfeld C, Richter J, Gilbert F, Hawgood S, Reid K, Clark H. Truncated recombinant human SP-D attenuates emphysema and type II cell changes in SP-D deficient mice. Respir Res 2007; 8:70.
- Leth-Larsen R, Nordenbaek C, Tornoe I, Moeller V, Schlosser A, Koch C, Teisner B, Junker P, Holmskov U. Surfactant protein D (SP-D) serum levels in patients with community-acquired pneumonia small star, filled. Clin Immunol 2003; 108(1):29–37.
- Miyata M, Sakuma F, Fukaya E, Kobayashi H, Rai T, Saito H, Kasukawa R, Suzuki S. Detection and monitoring of methotrexate-associated lung injury using serum markers KL-6 and SP-D in rheumatoid arthritis. Intern Med 2002; 41(6):467–473.
- Umetani K, Abe M, Kawabata K, Iida T, Kohno I, Sawanobori T, Kugiyama K. SP-D as a marker of amiodarone-induced pulmonary toxicity. Intern Med 2002; 41(9):709–712.
- Ohnishi H, Yokoyama A, Kondo K, Hamada H, Abe M, Nishimura K, Hiwada K, Kohno N. Comparative study of KL-6, surfactant protein-A, surfactant protein-D, and monocyte chemoattractant protein-1 as serum markers for interstitial lung diseases. Am J Respir Crit Care Med 2002; 165(3):378–381.
- Krane M, Griese M. Surfactant protein D in serum from patients with allergic bronchopulmonary aspergillosis. Eur Respir J 2003; 22(4):592–595.
- Sims MW, Tal-Singer RM, Kierstein S, Musani AI, Beers MF, Panettieri RA, Haczku A. Chronic obstructive pulmonary disease and inhaled steroids alter surfactant protein D (SP-D) levels: a cross-sectional study. Respir Res 2008; 9:13.
- Lomas DA, Silverman EK, Edwards LD, Locantore NW, Miller BE, Horstman DH, Tal-Singer R. Serum surfactant protein D is steroid sensitive and associated with exacerbations of COPD. Eur Respir J 2009; 34(1):95–102.
- Sin DD, Man SF, Marciniuk DD, Ford G, FitzGerald M, Wong E, York E, Mainra RR, Ramesh W, Melenka LS, Wilde E, Cowie RL, Williams D, Gan WQ, Rousseau R. The effects of fluticasone with or without salmeterol on systemic biomarkers of inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2008; 177(11):1207–1214.
- Cooley J, McDonald B, Accurso FJ, Crouch EC, Remold-O'Donnell E. Patterns of neutrophil serine protease-dependent cleavage of surfactant protein D in inflammatory lung disease. J Leukoc Biol 2008; 83(4):946–955.
- Duvoix A, Mackay RM, Henderson N, McGreal E, Postle A, Reid K, Clark H. Physiological concentration of calcium inhibits elastase-induced cleavage of a functional recombinant fragment of surfactant protein D. Immunobiology 2011; Epub ahead of print. doi: 10.1164/reem.201008-12200C.
- Tecle T, White MR, Sorensen G, Gantz D, Kacak N, Holmskov U, Smith K, Crouch EC, Hartshorn KL. Critical role for cross-linking of trimeric lectin domains of surfactant protein D in antiviral activity against influenza A virus. Biochem J 2008; 412(2):323–329.
- Perez J, Garrido O, Cifuentes M, Alonso FJ, Estivill-Torrus G, Eller G, Nualart F, Lopez-Avalos MD, Fernandez-Llebrez P, Rodriguez EM. Bovine Reissner's fiber (RF) and the central canal of the spinal cord: An immunocytochemical study using a set of monoclonal antibodies against the RF-glycoproteins. Cell Tissue Res 1996; 286(1):33–42.
- Vestbo J, Anderson W, Coxson HO, Crim C, Dawber F, Edwards L, Hagan G, Knobil K, Lomas DA, MacNee W, Silverman EK, Tal-Singer R. Evaluation of COPD longitudinally to identify predictive surrogate end-points (ECLIPSE). Eur Respir J 2008; 31(4):869–873.
- American Thoracic Society. Standardization of spirometry. Am J Respir Crit Care Med 1994; 152:1107–1136.
- Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, Fukuchi Y, Jenkins C, Rodriguez-Roisin R, van Weel C, Zielinski J. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2007; 176(6):532–555.
- Atochina-Vasserman EN, Gow AJ, Abramova H, Guo CJ, Tomer Y, Preston AM, Beck JM, Beers MF. Immune reconstitution during Pneumocystis lung infection: disruption of surfactant component expression and function by S-nitrosylation. J Immunol 2009; 182(4):2277–2287.
- Keen C, Gustafsson P, Lindblad A, Wennergren G, Olin AC. Low levels of exhaled nitric oxide are associated with impaired lung function in cystic fibrosis. Pediatr Pulmonol 45(3):241–248.
- Smith AD, Cowan JO, Brassett KP, Herbison GP, Taylor DR. Use of exhaled nitric oxide measurements to guide treatment in chronic asthma. N Engl J Med 2005; 352(21):2163–2173.
- Syk J, Unden AL, Alving K. Relationship between exhaled nitric oxide and IgE sensitisation in patients with asthma: influence of steroid treatment. Clin Respir J 2009; 3(3):143–151.
- Guo CJ, Atochina-Vasserman EN, Abramova E, Foley JP, Zaman A, Crouch E, Beers MF, Savani RC, Gow AJ. S-nitrosylation of surfactant protein-D controls inflammatory function. PLoS Biol 2008; 6(11):e266.