Abstract
Background: Pulmonary hypertension (PH) in COPD carries a poor prognosis. Statin therapy has been associated with numerous beneficial clinical effects in COPD, including a possible improvement in PH. We examined the association between statin use and pulmonary hemodynamics in a well-characterized cohort of patients undergoing evaluation for lung transplantation. Methods: We conducted a cross-sectional analysis of 112 subjects evaluated for lung transplant with a diagnosis of COPD. Clinical characteristics, pulmonary function, cardiac catheterization findings and medical comorbidities were compared between statins users and non-users. Results: Thirty-four (30%) subjects were receiving statin therapy. Statin users were older and had an increased prevalence of systemic hypertension and coronary artery disease (CAD). Mean pulmonary arterial pressure (mPAP) in the statin group was lower [26 ± 7 vs 29 ± 7 mmHg, p = 0.02], as was pulmonary artery wedge pressure (PAWP) [12 ± 5 vs. 15 ± 6 mmHg, p = 0.02]. Pulmonary vascular resistance did not differ between the groups. In multiple regression analysis, statin use was associated with a 4.2 mmHg (95% CI: 2 to 6.4, p = <0.001) lower PAWP and a 2.6 mmHg (95% CI: 0.3 to 4.9, p = 0.03) reduction in mPAP independent of PAWP. Conclusions: In patients with severe COPD, statin use is associated with significantly lower PAWP and mPAP. These finding should be evaluated prospectively.
INTRODUCTION
The presence of pulmonary hypertension (PH) in COPD is well recognized as an independent prognostic indicator (Citation1,2). In addition to increased mortality, PH in COPD is associated with increased hospitalization rates (Citation3) and decreased exercise capacity (Citation4). Mechanisms for the development of pulmonary hypertension in COPD include both lung parenchymal and vascular pathologies (Citation5). Conventionally, PH in COPD has been thought to be primarily related to hypoxic vasoconstriction. However, other and perhaps more important mechanisms may include structural remodeling and endothelial dysfunction of the pulmonary vasculature (Citation6,7), elevated left heart filling pressures associated with cardiovascular disease, and mechanical factors related to increased alveolar and intrathoracic pressures (Citation8,9).
Experience with therapies proven beneficial in pulmonary arterial hypertension (PAH) have been disappointing in patients with COPD, with studies revealing either lack of clinical benefit or even harmful effects (Citation10–13). Observational studies suggest that statin therapy may be associated with benefits which are not explained through traditional lipid-lowering mechanisms (Citation14). These pleiotropic effects are usually attributed to a reduction in isoprenoid synthesis and consequent reduction in Rho and other small G-protein signaling pathways. Statins have been shown to inhibit progression of emphysema and pulmonary hypertension associated with tobacco exposure and chronic hypoxia in murine models (Citation15–17). Beneficial effects described in COPD have included a slower decline in pulmonary function (Citation18), reduced frequency of exacerbations and intubations (Citation19), and improved exercise capacity coupled with reduced pulmonary pressures estimated by echocardiography (Citation20). In this study, we examined the relationship between statin use and cardio-pulmonary hemodynamics through a cross-sectional analysis of COPD patients underging lung transplant evaluation.
METHODS
The methods of this study were reviewed and approved by our institution's investigational review board. Two hundred and eighty-nine consecutive COPD patients evaluated at our center for lung transplantation from 1995 to 2009 were screened for inclusion in the analysis. Subjects were excluded for the following reasons: 7 had no diagnosis of COPD, and 23 had an additional lung disease such as interstitial lung disease or sarcoidosis. Of the remaining 259 subjects, hemodynamics were available in 112. The following data were recorded: demographics, anthropometrics, pulmonary function, medical comorbidities, medications, fasting lipid profile values, and results of right and left heart catheterization performed as part of the transplant evaluation. Lipid profiles were determined using the Friedewald calculation. Diagnoses such as diabetes mellitus, congestive heart failure, hypertension, and hyperlipidemia were defined by mention in the medical record or by medication use. Coronary artery disease (CAD) by angiography, was defined as either ≥50% stenosis of the left main coronary vessel, ≥70% stenosis in another vessel, or revascularization intervention.
Statistical analysis
Continuous data are expressed as mean (SD), and categorical data are presented as counts and percentages. Univariate analyses included Student's t-test, Chi2-test, and Fisher's exact tests as appropriate. Pearson correlation coefficient was used to assess the relationship between continuous variables. Two separate multiple linear regression models were built to assess predictors of mPAP and PAWP. Covariates were included in the regression model if they varied by statin use at the 0.1 significance level in univariate analyses or were thought to be of clinical importance. Reverse stepwise regression modeling was performed with a p-value inclusion threshold of 0.2 to generate the final multiple regression models. Underlying modeling assumptions were checked graphically. Analysis was performed using Stata Statistical Software, Release 10.0 (Stata Corporation; College Station, TX).
RESULTS
Clinical characteristics of the study subjects are described in . Subjects were primarily Caucasian with severe COPD reflected by a mean FEV1% predicted of 21 ± 8. Ninety percent used supplemental oxygen. Ninety-two percent of subjects were found to have mean pulmonary arterial pressure (mPAP) ≥20 mmHg, and 66% of these also had pulmonary arterial wedge pressure (PAWP) ≤ 15 mmHg. Fifty seven percent were found to have mPAP ≥25 mmHg, and 53% of these also had PAWP ≤ 15 mmHg. Six percent of our subjects were found to have mPAP ≥ 40 mmHg, and all but one of these had PAWP >15 mmHg. Coronary angiographic data were available in 96 subjects. Nineteen percent of subjects had significant CAD at catheterization. The presence of CAD was not associated with differences in mPAP or PAWP.
Table 1 Clinical characteristics of study subjects
Thirty percent of subjects were receiving statin therapy. Of those with CAD, 67% were using a statin. The specific agents were primarily atorvastatin (53%) and simvastatin (35%). Systemic hypertension, hyperlipidemia, and angiographically proven CAD were present in a significantly higher proportion of statin users and this group was older. Despite these comorbidities, mPAP in the statin group was lower (26 ± 7 vs. 29 ± 7 mmHg; p = 0.02). This difference in mPAP was accompanied by a similarly lower value of PAWP in those patients receiving statin therapy (12 ± 5 vs. 15 ± 6 mmHg, p = 0.02) and not by differences in cardiac output or pulmonary vascular resistance (). Statin users also had a slightly higher FVC% predicted, a somewhat lower oxygen requirement, and were more likely to be taking a calcium channel blocker. After adjustment for age, race, gender, BMI, hypertension, use of calcium channel blockers, diuretic use, pulmonary function (FEV1 and DLCO), PaCO2, oxygen requirement, and presence of CAD, statin use was associated with a 5.2 mmHg lower mPAP (95% CI, 1.9 to 8.5) and a 4.3 mmHg lower PAWP (95% CI, 2.0 to 6.6) ().
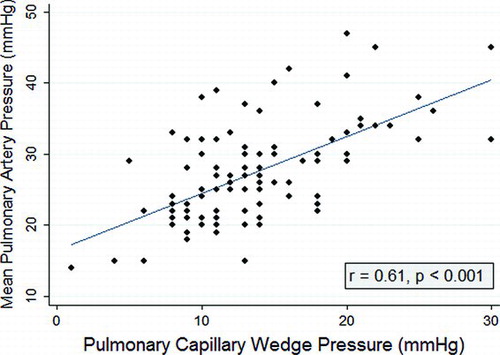
There was a strong linear correlation between mPAP and PAWP (). PAWP, age, BMI, and non-use of statin were the only significant determinants of mPAP in the univariate analysis (). No significant association was observed between mPAP and any pulmonary function variable. A marginal trend was observed with oxygen requirement. Statin use, lower BMI, and Caucasian race were associated with lower PAWP in the univariate analysis (). Variables evaluated in the stepwise model for mPAP included statin use, PAWP, BMI, age, race, calcium channel blocker use, and oxygen use in liters per minute. Reverse stepwise regression modeling demonstrated persistence of statin effect after controlling for PAWP (). BMI, age, and PAWP were also significant independent determinants of mPAP. Variables evaluated in the stepwise model for PAWP included statin use, age race, BMI, CAD, HTN, calcium channel blocker use, diuretic use, and oxygen use in liters per minute. Only non-statin use and BMI were significant independent determinants of PAWP.
Figure 1 Scatterplot with Pearson's Correlation Between Pulmonary Arterial Wedge Pressure and Mean Pulmonary Arterial Pressures.
DISCUSSION
Our study is the first to show an association between statin use and reduced pulmonary arterial and wedge pressures obtained invasively in subjects with COPD. Furthermore, we demonstrated these reductions to be independent of relevant clinical factors, and that the reduction in mPAP persisted after controlling for PAWP.
PH is defined as a mean pulmonary arterial pressure (mPAP) ≥ 25 mmHg, but PH complicating pulmonary disease is defined by many experts in the field as a mPAP ≥ 20 mmHg (Citation21). Pulmonary pressures in our cohort were similar to those previously reported in comparable patient populations. We found a 92% prevalence of mPAP ≥ 20 mmHg, which is quite similar to that reported by Scharf et al., who found a prevalence of 91% at this threshold in patients evaluated for lung volume reduction surgery (Citation22). We also found mPAP ≥ 25 mmHg in 57% of subjects, which is similar to the prevalence of 50% at this higher threshold described in a cohort of 215 patients evaluated for lung volume reduction surgery or lung transplantation (Citation23). Subjects with PAWP > 15 mmHg (32 out of 247) were excluded from that study. Six percent of our subjects were found to have mPAP ≥ 40 mmHg, which is much higher than the 2.7% prevalence described in a large cohort of patients with COPD (Citation24). This difference may be explained by the greater severity of illness in our subjects whose mean percent predicted FEV1 was 21% versus 33% observed in that study.
Table 2 Regression models of correlates to pulmonary artery wedge pressure
Table 3 Regression models of correlates to mean pulmonary artery pressure
Table 4 Regression models of correlates to mean pulmonary artery pressure and pulmonary artery wedge pressure
Numerous studies have consistently found a strong relationship between mPAP and PAWP in COPD (Citation22,23,25). Among 120 patients in the National Emphysema Treatment Trial (NETT), PAWP exceeded 12 mmHg in over 60% and 15 mmHg in 38% (Citation22), which is comparable to the 60% and 29% at these respective thresholds in our cohort. Multiple regression analysis found that PAWP, along with FEV1 and DLCO predicted mPAP (Citation22). Similarly, PAWP was strongly and independently related to mPAP, along with PaO2 and the alveolar-arterial oxygen gradient, in a large group of COPD subjects referred for lung volume reduction surgery or lung transplantation (Citation23). We could not detect a relationship between pulmonary function variables and mPAP, a finding noted in other series (Citation25). We found only a weak association between oxygen requirement and mPAP. However, this may have been due to clustering of these values and underrepresentation of subjects without hypoxemia, as 90% of patients in the current study used supplemental oxygen. Arterial blood gas measurements while breathing room air were not available for most subjects.
The basis for the high prevalence of elevated PAWP in our series, as well as others (Citation22,23) is likely multifactorial. It is well recognized that cardiovascular disease (CVD) is highly prevalent in COPD and accounts for a substantial proportion of morbidity and mortality (Citation26). We found a weak relationship between PAWP and systemic HTN, but not CAD. Subclinical systemic vascular disease in the form of atherosclerosis (Citation27) with consequent endothelial dysfunction and arterial stuffiness (Citation28) may lead to LV diastolic dysfunction. The strong influence of BMI on PAWP noted here is consistent with the recognized increased risk of non-systolic heart failure with obesity (Citation29). Mean pulmonary arterial pressures and PAWP typically rise with exercise in advanced COPD. This rise has been shown to occur with hyperventilation alone (Citation30), indicating a contribution from air trapping and consequent hyperinflation. We found no association between pulmonary function measures of hyperinflation and resting mPAP or PAWP. In the series of Scharf et al., mPAP was not related to lung volumes or emphysema scores by computed tomography (Citation22).
Statins have numerous beneficial effects in CVD that could account for the lower PAWP observed in this study. Their use is associated with small reductions in blood pressure (Citation31–33) as well as reduced left ventricular hypertrophy and fibrosis both in experimental animal models and in humans (Citation34–36). Statins increase arterial distensibility (Citation32, 37) and induce regression of aortic atherosclerosis (Citation38), likely through beneficial effects on endothelial function (Citation39–43). At a cellular level, normal myocardial relaxation requires calcium scavenging through a process dependent upon sarcoplasmic reticulum Ca2+ ATPase (SERCA) activity, the impairment of which has been associated with diastolic dysfunction (Citation44). Endothelin may also promote diastolic dysfunction by a mechanism involving induction of intracellular alkalosis which in turn affects myofilament Ca2+ sensitivity. Statins could improve diastolic function through their beneficial effects on SERCA (Citation45) as well as reduced endothelin activity in the heart (Citation46).
Despite the high prevalence of elevated PAWP in PH associated with COPD, the primary pathophysiologic mechanism is felt to be structural remodeling of the small pulmonary arteries, which occurs early in the disease (Citation47). Recent data from the NETT trial found a strong correlation between mPAP and the cross-sectional area of the small pulmonary vessels by computed tomography (Citation48). Statins possess numerous properties that would be expected to retard pulmonary vascular remodeling, including inhibition of vascular smooth muscle cell (VSMC) proliferation and migration, induction of VSMC apoptosis, and have been shown to be effective in several animal models of PH (Citation15,16,49,50). Despite no effect on PVR, our observation of reduced mPAP independently of PAWP in statin users suggests that these drugs may reduce pulmonary vascular remodeling in COPD.
The association between age and mPAP, but not PAWP, was unexpected. Advanced age is well known to correlate with left ventricular diastolic dysfunction (Citation51,52), and so a correlation with PAWP would have been predicted. Longitudinal studies have shown a mPAP rise of 0.4 to 0.6 mmHg per year in patients with COPD (Citation53,54), which is in line with our β coefficient estimate. Thus, increased age with consequent longer disease duration, may be associated with more pulmonary vascular remodeling. Cross sectional studies, however, are significantly affected by survival bias and rarely demonstrate this correlation. A large series presented by Bishop et al., including 595 subjects with COPD, found no correlation between mPAP and age (Citation55).
The primary limitation of this study is the retrospective nature of the data collection. Statin use was nonrandom, and although regression models were able to control for many clinical differences, it is still possible that unknown confounders exist. Significantly more patients receiving statin therapy were taking calcium channel blockers. It is unlikely, however, that this could have affected mPAP or PAWP in our population as long-term studies have shown no significant change in these values with their use in COPD (Citation12) and there was no association between calcium channel blocker use and mPAP or PAWP in our population. The clinical implications of these findings are not clear, particularly since no effects were observed on resting cardiac index or PVR. A recent placebo-controlled trial demonstrated a 52% increase in treadmill exercise time and significant improvement in Borg dyspnea score associated with a reduction in systolic PAP assessed by Doppler echocardiography after 6 months of pravastatin therapy (Citation20).
CONCLUSIONS
In patients with severe COPD, statin use is associated with significantly lower mPAP and PAWP despite older age and a higher prevalence of medical comorbidities such as HTN and coronary disease. Improvements in pulmonary hemodynamics may represent an addition to the growing list of potential benefits of statin use in COPD (Citation56). Prospective clinical trials are required to assess the long-term clinical impact of statin therapy in this population.
Disclosures: None of the authors have any conflicts of interest to disclose. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Oswald-Mammosser M, Weitzenblum E, Quoix E, Moser G, Chaouat A, Charpentier C, Prognostic factors in COPD patients receiving long-term oxygen therapy. Importance of pulmonary artery pressure. Chest 1995; 107(5):1193–1198.
- Weitzenblum E, Hirth C, Ducolone A, Mirhom R, Rasaholinjanahary J, Ehrhart M. Prognostic value of pulmonary artery pressure in chronic obstructive pulmonary disease. Thorax 1981; 36(10):752–758.
- Kessler R, Faller M, Fourgaut G, Mennecier B, Weitzenblum E. Predictive factors of hospitalization for acute exacerbation in a series of 64 patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999; 159(1):158–164.
- Sims MW, Margolis DJ, Localio AR, Panettieri RA, Kawut SM, Christie JD. Impact of pulmonary artery pressure on exercise function in severe COPD. Chest 2009; 136(2):412–419.
- Macnee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Part One. Am J Respir Crit Care Med 1994; 150(3):833–852.
- Barbera JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J 2003; 21(5):892–905.
- Magee F, Wright JL, Wiggs BR, Pare PD, Hogg JC. Pulmonary vascular structure and function in chronic obstructive pulmonary disease. Thorax 1988; 43(3):183–189.
- Finkelstein J, Cha E, Scharf SM. Chronic obstructive pulmonary disease as an independent risk factor for cardiovascular morbidity. Int J Chron Obstruct Pulmon Dis 2009; 4:337–349.
- Huiart L, Ernst P, Suissa S. Cardiovascular morbidity and mortality in COPD. Chest 2005; 128(4):2640–2646.
- Melot C, Hallemans R, Naeije R, Mols P, Lejeune P. Deleterious effect of nifedipine on pulmonary gas exchange in chronic obstructive pulmonary disease. Am Rev Respir Dis 1984; 130(4):612–616.
- Rietema H, Holverda S, Bogaard HJ, Marcus JT, Smit HJ, Westerhof N, Sildenafil treatment in COPD does not affect stroke volume or exercise capacity. Eur Respir J 2008; 31(4):759–764.
- Saadjian AY, Philip-Joet FF, Vestri R, Arnaud AG. Long-term treatment of chronic obstructive lung disease by Nifedipine: an 18-month haemodynamic study. Eur Respir J 1988; 1(8):716–720.
- Stolz D, Rasch H, Linka A, Di Valentino M, Meyer A, Brutsche M, A randomised, controlled trial of bosentan in severe COPD. Eur Respir J 2008; 32(3):619–628.
- Almuti K, Rimawi R, Spevack D, Ostfeld RJ. Effects of statins beyond lipid lowering: potential for clinical benefits. Int J Cardiol 2006; 109(1):7–15.
- Girgis RE, Li D, Zhan X, Garcia JG, Tuder RM, Hassoun PM, Attenuation of chronic hypoxic pulmonary hypertension by simvastatin. Am J Physiol Heart Circ Physiol 2003; 285(3):H938–H945.
- Girgis RE, Mozammel S, Champion HC, Li D, Peng X, Shimoda L, Regression of chronic hypoxic pulmonary hypertension by simvastatin. Am J Physiol Lung Cell Mol Physiol 2007; 292(5):L1105–L1110.
- Lee JH, Lee DS, Kim EK, Choe KH, Oh YM, Shim TS, Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am J Respir Crit Care Med 2005; 172(8):987–993.
- Alexeeff SE, Litonjua AA, Sparrow D, Vokonas PS, Schwartz J. Statin use reduces decline in lung function: VA Normative Aging Study. Am J Respir Crit Care Med 2007; 176(8):742–747.
- Blamoun AI, Batty GN, DeBari VA, Rashid AO, Sheikh M, Khan MA. Statins may reduce episodes of exacerbation and the requirement for intubation in patients with COPD: evidence from a retrospective cohort study. Int J Clin Pract 2008; 62(9):1373–1378.
- Lee TM, Chen CC, Shen HN, Chang NC. Effects of pravastatin on functional capacity in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Clin Sci (Lond) 2009; 116(6):497–505.
- Weitzenblum E, Chaouat A, Canuet M, Kessler R. Pulmonary hypertension in chronic obstructive pulmonary disease and interstitial lung diseases. Semin Respir Crit Care Med 2009; 30(4):458–470.
- Scharf SM, Iqbal M, Keller C, Criner G, Lee S, Fessler HE. Hemodynamic characterization of patients with severe emphysema. Am J Respir Crit Care Med 2002; 166(3):314–322.
- Thabut G, Dauriat G, Stern JB, Logeart D, Levy A, Marrash-Chahla R, Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest 2005; 127(5):1531–1536.
- Chaouat A, Bugnet AS, Kadaoui N, Schott R, Enache I, Ducolone A, Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005; 172(2):189–194.
- Chaouat A, Savale L, Chouaid C, Tu L, Sztrymf B, Canuet M, Role for interleukin-6 in COPD-related pulmonary hypertension. Chest 2009; 136(3):678–687.
- Sin DD, Man SF. Impact of cancers and cardiovascular diseases in chronic obstructive pulmonary disease. Curr Opin Pulm Med 2008; 14(2):115–121.
- Iwamoto H, Yokoyama A, Kitahara Y, Ishikawa N, Haruta Y, Yamane K, Airflow limitation in smokers is associated with subclinical atherosclerosis. Am J Respir Crit Care Med 2009; 179(1):35–40.
- Maclay JD, McAllister DA, Mills NL, Paterson FP, Ludlam CA, Drost EM, Vascular dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2009; 180(6):513–520.
- Desai A, Fang JC. Heart failure with preserved ejection fraction: hypertension, diabetes, obesity/sleep apnea, and hypertrophic and infiltrative cardiomyopathy. Heart Fail Clin 2008; 4(1):87–97.
- Harris P, Segel N, Green I, Housley E. The influence of the airways resistance and alveolar pressure on the pulmonary vascular resistance in chronic bronhcitis. Cardiovasc Res 1968; 2(1):84–92.
- Borghi C. Interactions between hypercholesterolemia and hypertension: implications for therapy. Curr Opin Nephrol Hypertens 2002; 11(5):489–496.
- Ferrier KE, Muhlmann MH, Baguet JP, Cameron JD, Jennings GL, Dart AM, Intensive cholesterol reduction lowers blood pressure and large artery stiffness in isolated systolic hypertension. J Am Coll Cardiol 2002; 39(6):1020–1025.
- Glorioso N, Troffa C, Filigheddu F, Dettori F, Soro A, Parpaglia PP, Effect of the HMG-CoA reductase inhibitors on blood pressure in patients with essential hypertension and primary hypercholesterolemia. Hypertension 1999; 34(6):1281–1286.
- Indolfi C, Di Lorenzo E, Perrino C, Stingone AM, Curcio A, Torella D, Hydroxymethylglutaryl coenzyme A reductase inhibitor simvastatin prevents cardiac hypertrophy induced by pressure overload and inhibits p21ras activation. Circulation 2002; 106(16):2118–2124.
- Nishikawa H, Miura S, Zhang B, Shimomura H, Arai H, Tsuchiya Y, Statins induce the regression of left ventricular mass in patients with angina. Circ J 2004; 68(2):121–125.
- Patel R, Nagueh SF, Tsybouleva N, Abdellatif M, Lutucuta S, Kopelen HA, Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation 2001; 104(3):317–324.
- Smilde TJ, Van Den Berkmortel FW, Wollersheim H, van Langen H, Kastelein JJ, Stalenhoef AF. The effect of cholesterol lowering on carotid and femoral artery wall stiffness and thickness in patients with familial hypercholesterolaemia. Eur J Clin Invest 2000; 30(6):473–480.
- Corti R, Fuster V, Fayad ZA, Worthley SG, Helft G, Smith D, Lipid lowering by simvastatin induces regression of human atherosclerotic lesions: two years’ follow-up by high-resolution noninvasive magnetic resonance imaging. Circulation 2002; 106(23):2884–2887.
- Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation 2004; 109(23 Suppl 1):III39–III43.
- Landmesser U, Bahlmann F, Mueller M, Spiekermann S, Kirchhoff N, Schulz S, Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation 2005; 111(18):2356–2363.
- Laufs U, Fata VL, Liao JK. Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. J Biol Chem 1997; 272(50):31725–31729.
- O'Driscoll G, Green D, Taylor RR. Simvastatin, an HMG-coenzyme A reductase inhibitor, improves endothelial function within 1 month. Circulation 1997; 95(5):1126–1131.
- Wilkinson IB, Qasem A, McEniery CM, Webb DJ, Avolio AP, Cockcroft JR. Nitric oxide regulates local arterial distensibility in vivo. Circulation 2002; 105(2):213–217.
- Alpert NR, LeWinter M, Mulieri LA, Hasenfuss G. Chemomechanical Energy Transduction in the Failing Heart. Heart Failure Reviews 1999; 4(3):281–295.
- Zheng X, Hu SJ. Effects of simvastatin on cardiac performance and expression of sarcoplasmic reticular calcium regulatory proteins in rat heart. Acta Pharmacol Sin 2005; 26(6):696–704.
- Saka M, Obata K, Ichihara S, Cheng XW, Kimata H, Nishizawa T, Pitavastatin improves cardiac function and survival in association with suppression of the myocardial endothelin system in a rat model of hypertensive heart failure. J Cardiovasc Pharmacol 2006; 47(6):770–779.
- Peinado VI, Pizarro S, Barbera JA. Pulmonary vascular involvement in COPD. Chest 2008; 134(4):808–814.
- Matsuoka S, Washko GR, Yamashiro T, Estepar RS, Diaz A, Silverman EK, Pulmonary hypertension and computed tomography measurement of small pulmonary vessels in severe emphysema. Am J Respir Crit Care Med 2010; 181(3):218–225.
- Nishimura T, Vaszar LT, Faul JL, Zhao G, Berry GJ, Shi L, Simvastatin rescues rats from fatal pulmonary hypertension by inducing apoptosis of neointimal smooth muscle cells. Circulation 2003; 108(13):1640–1645.
- Taraseviciene-Stewart L, Scerbavicius R, Choe KH, Cool C, Wood K, Tuder RM, Simvastatin causes endothelial cell apoptosis and attenuates severe pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2006; 291(4):L668–L676.
- Fischer M, Baessler A, Hense HW, Hengstenberg C, Muscholl M, Holmer S, Prevalence of left ventricular diastolic dysfunction in the community. Results from a Doppler echocardiographic-based survey of a population sample. Eur Heart J 2003; 24(4):320–328.
- Samdarshi TE, Taylor HA, Edwards DQ, Liebson PR, Sarpong DF, Shreenivas SS, Distribution and determinants of Doppler-derived diastolic flow indices in African Americans: the Jackson Heart Study (JHS). Am Heart J 2009; 158(2):209–216.
- Kessler R, Faller M, Weitzenblum E, Chaouat A, Aykut A, Ducolone A, “Natural history” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med 2001; 164(2):219–224.
- Weitzenblum E, Sautegeau A, Ehrhart M, Mammosser M, Hirth C, Roegel E. Long-term course of pulmonary arterial pressure in chronic obstructive pulmonary disease. Am Rev Respir Dis 1984; 130(6):993–998.
- Bishop JM, Cross KW. Use of other physiological variables to predict pulmonary arterial pressure in patients with chronic respiratory disease. Multicentre study. Eur Heart J 1981; 2(6):509–517.
- Janda S, Park K, FitzGerald JM, Etminan M, Swiston J. Statins in COPD: A systematic review. Chest 2009; 136(3):734–743.