Abstract
AATD is a European genetic condition that has disseminated along human migration routes. The discovery, function, phenotyping methodologies and biochemical mechanisms have been led by several European countries. The variable availability of augmentation therapy has permitted a better understanding of the natural history and the ability to deliver controlled clinical trials. The establishment of a worldwide registry remains central to the future of understanding and managing AATD.
Keywords :
Introduction
Once upon a time, a long time ago (about 2,000 years), the Z mutation arose and persisted in the Swedish population. The survival benefit of the heterozygous state has never been satisfactorily explained, although the homozygous state has been suggested to increase fertility. At the time the mutation arose life expectancy was short, and smoking and atmospheric pollution were not relevant such that health (other than neonatal hepatic failure) was probably not affected, even by indoor pollution within the reproductive and active years.
With the travels of the Vikings and their social needs at landfall the Z gene was gradually disseminated throughout the Baltic region and thereafter more widely by sea and land travel () and population prevalence followed this such that there is a gradient of from 1 in 1600 in Denmark to 1 in 5000 in the United States.
Figure 1. Viking expansion from http://upload.wikimedia.org/wikipedia/commons/5/50/viking-expansion.svg.
The first clinical observations of the condition, its genetic inheritance and association with early onset and severe emphysema was therefore not surprisingly also described in Sweden in the early 1960s where its function as a proteinase inhibitor was first recognised by Carl-Bertil Laurell (Citation1). Subsequently, in collaboration with Magne Fagerhol in Norway, the use of isoelectric focusing to describe the PI typing system was developed (Citation2). Turino et al reported that the serums of individuals with severe pulmonary emphysema which are deficient in alpha-1 globulin antitrypsin are also deficient in an inhibitor of pancreatic elastase (Citation3).
Their results reinforced the concept that a form of early onset emphysema exists where tissue destruction predominates and which may be secondary to an intrinsic biochemical defect which results in primary parenchymal destruction. The extraordinarily low values of elastase inhibition in the serums of their patients suggested that elastase was the secondary factor. However, it was a further 20–30 years before the mechanism of deficiency was established in Cambridge, UK, by Lomas and co-workers (Citation4).
Following the development of augmentation therapy in the United States, it received orphan drug status in Europe and became available in some countries for individuals who fitted the moderately severe range of physiological impairment (FEV1 30–60% predicted) where a suggestion of benefit had been shown by the NIH registry (Citation5). Nevertheless, this did not receive widespread approval leaving many patients and indeed countries without augmentation therapy.
This has, in many ways, provided a major advantage in understanding the natural history of AATD and providing further supporting evidence of the efficacy of augmentation therapy. For example comparison of patient demographics between Denmark (no augmentation) and Germany (augmentation available) suggesting the latter group did better (Citation6). A German sequential study indicated that patients with a rapid decline in FEV1 showed attenuation of this process with augmentation therapy (Citation7), although this could have represented regression towards the mean. Finally, extensive databases of decline in treated and untreated patients internationally (including major input from Europe), again supports the protective benefit (Citation8).
Natural History
From 1970–1972, Sweden undertook a neonatal policy of screening for AATD. The suspected prevalence was confirmed and at the last 30 year follow up, awareness had reduced the prevalence of smoking but symptoms were becoming apparent and in those who smoked, lung function was already impaired compared to non-smokers (Citation9), although no radiological evidence of emphysema was identified on CT scanning (Citation9). Furthermore, longitudinal follow up of patients has confirmed that mortality in non-smoking, non-index cases is not significantly different from that of the general Swedish population (Citation10), although morbidity remains unknown.
In the United Kingdom, the establishment of the U.K. registry in 1997 under the auspices of ADAPT (Antitrypsin Deficiency Assessment and Programme for Treatment) has enabled extensive and longitudinal study of a large cohort of untreated (augmentation therapy) deficient subjects together with a comprehensive DNA and plasma biobank. This has enabled the study of features predictive of fast decliners (Citation11), physiological feature other than spirometry (Citation12), the features and effect of exacerbations (Citation13,14), validation of CT scan as a sensitive measure of severity (Citation15) and, importantly, the best single predictor of mortality (Citation16).
This validation, together with determination of the best parameters to measure lung density and its progression as well as automation of analysis developed by researchers in Denmark and Sweden, has enabled CT densitometry to become accepted as the primary outcome measure in clinical trials in AATD. To date, 2 such trials have been carried out of augmentation in Europe using this methodology (Citation17,18), and a third worldwide study is nearing completion.
Establishment of AIR (Alpha-1 International Registry)
Following the WHO meeting on AATD in 1996 (Citation19), key opinion leaders in Europe established AIR to increase the understanding of the condition and facilitate collaborative research projects and clinical trials.
The registry is web-based, allowing principal investigators from individual countries to upload extensive demographic data and follow up data with a defined minimal dataset. At the time of writing, the registry includes PI*Z patients from 17 countries and 4,758 patients in total (). Some of the baseline demographic data are shown in .
Figure 2. Annual accumulation of patient numbers in the AIR database.
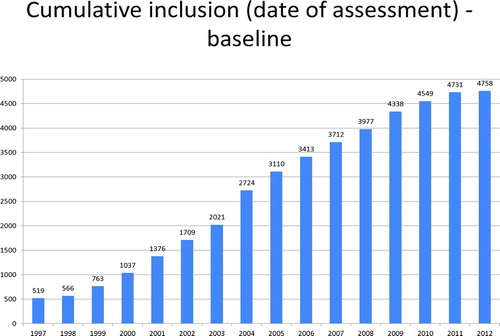
Table 1. Baseline demographics of the main groups of recognised pathophysiological genotypes in the AIR data base
Augmentation therapy is not generally available in Europe with only a few countries having this therapy reasonably available (Spain, Austria, Germany, and Italy). In others, there is limited availability with treated patients numbering less than 10 and in Belgium, treatment has been made no longer available. In the United Kingdom, as well as the Netherlands, Denmark, Sweden, Poland and the other Baltic states, augmentation is not available except on a limited named patient basis, usually for panniculitis although it often remains difficult to obtain funding from health care services even for this indication.
However, the general lack of therapy has enabled a better understanding of the natural history of AATD to be obtained. In addition, the registry has enabled 3 European framework grants to be obtained and 2 interventional studies to be completed together with contributions to further studies of intravenous and inhaled therapy. Collaborations between countries in developing technical methods for phenotyping, biomarker research and basic research studies have been facilitated by this consortium approach. Indeed, recent studies using an elastase-specific cleavage product of fibrinogen has shown promise as a biomarker of the pathogenic process and efficacy of augmentation therapy (Citation20).
Summary
AATD is a European genetic condition that has disseminated along human migration routes. The discovery, function, phenotyping methodologies and biochemical mechanisms have been led by several European countries. The variable availability of augmentation therapy has permitted a better understanding of the natural history and the ability to deliver controlled clinical trials. The establishment of a worldwide registry remains central to the future of understanding and managing AATD.
Declaration of Interest Statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Laurell C, Eriksson, S. The electrophorectic alpha-1 globulin pattern of serum in alpha 1-antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15:132–140.
- Fagerhol MK, Laurell CB. The polymorphism of ìprealbuminsî and alpha-1 antitrypsin in human sera. Clin Chim Acta 1967; 16(2):199–203.
- Turino GM, Senior RM, Garg GD, Keller S, Levi MM, Mandl I. Serum elastase inhibitor deficiency and alpha-1 antitrypsin deficiency in patients with obstructive emphysema. Science 1969; 165:709–711.
- Lomas DA, Evans DL, Finch JT, The mechanism of Z alpha-1 antitrypsin accumulation in the liver. Nature 1992; 357(6379):605–607.
- The Alpha-1 Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Respir Crit Care Med 1998; 158(1):49–59.
- Seersholm N, Does alpha-1 antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha1-antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL) alpha1-AT study group. Eur Respir J 1997; 10(10):2260–2263.
- Wencker M, Longitudinal follow-up of patients with alpha-1 protease inhibitor deficiency before and during therapy with IV alpha-1 protease inhibitor. Chest 2001; 119(3):737–744.
- Chapman KR, Augmentation therapy for alpha1 antitrypsin deficiency: a meta-analysis. COPD 2009; 6(3):177–184.
- Bernspang E, CT lung densitometry in young adults with alpha-1 antitrypsin deficiency. Respir Med 2011; 105(1):74–79.
- Tanash HA, Survival in severe alpha-1 antitrypsin deficiency (PiZZ). Respir Res 2010; 11:44
- Dawkins PA, Rate of progression of lung function impairment in alpha-1 antitrypsin deficiency. Eur Respir J 2009; 33(6):1338–1344.
- Dowson LJ, Guest PJ, Stockley RA. Longitudinal changes in physiological, radiological, and health status measurements in alpha-1 antitrypsin deficiency and factors associated with decline. Am J Respir Crit Care Med 2001; 164(10 Pt 1):1805–1809.
- Needham M, Stockley RA. Exacerbations in alpha-1 antitrypsin deficiency. Eur Respir J 2005; 25(6):992–1000.
- Vijayasaratha K, Stockley RA. Reported and unreported exacerbations of COPD: Analysis by diary cards. Chest 2008; 133:34–41.
- Parr DG, Validation of computed tomographic lung densitometry for monitoring emphysema in alpha-1 antitrypsin deficiency. Thorax 2006; 61(6):485–490.
- Dawkins PA, Predictors of mortality in alpha-1 antitrypsin deficiency. Thorax 2003; 58(12):1020–1026.
- Dirksen A, A randomized clinical trial of alpha-1 antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160 (5 Pt 1):1468–1472.
- Dirksen A, Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33(6):1345–1353.
- World Health Organization Alpha-1 antitrypsin deficiency: memorandum from a WHO meeting, available on their web site. Bull World Health Organ 1997; 75(5):397–415.
- Carter RI, The fibrinogen cleavage product alpha-Val360, a specific marker of neutrophil elastase activity in vivo. Thorax 2011; 66(8):686–691.