Abstract
This was a 52-week, double-blind, extension study in which COPD patients previously treated with twice-daily (BID) aclidinium bromide 200 μg or 400 μg during a 12-week lead-in study (ACCORD COPD I) continued the same treatment, while patients previously receiving placebo were rerandomized (1:1) to aclidinium 200 μg or 400 μg BID. The primary objective of this study was to evaluate the long-term safety and tolerability of aclidinium treatment. Efficacy outcomes included bronchodilation, health status, and rescue medication use. A total of 467 patients completed the lead-in study and 291 patients consented to participate in the extension. At study end, the percentages of patients who reported a treatment-emergent adverse event (TEAE) were similar for both treatments (200 μg, 77.4%; 400 μg, 73.7%). Incidence of anticholinergic TEAEs was low and similar for both treatments, with dry mouth reported in only 1 patient (400 μg). Cardiac TEAEs were reported by a similarly low percentage of patients (<5% for any event in any group) with no apparent dose dependence. Improvements from baseline in lung function were greatest for patients who received continuous aclidinium treatment and those who were rerandomized from placebo to aclidinium 400 μg; these improvements were generally sustained throughout the study. Health status and overall rescue medication use was improved from baseline for both treatments. The safety profile of twice-daily aclidinium and sustained improvements in lung function and health status throughout the 52-week extension study support its use as a long-term maintenance treatment for patients with COPD. (Clinical trial registration number NCT00970268).
Introduction
An estimated 64 million people worldwide are reported to suffer from chronic obstructive pulmonary disease (COPD) (1), a major global health challenge with projected increases in prevalence, mortality, and associated healthcare costs (2). Although not fully reversible, COPD is a treatable disease, with the reduction of symptoms and improvement of health status identified as important treatment goals by the Global Initiative for Chronic Obstructive Lung Disease (GOLD) (3).
Long-acting inhaled bronchodilators are recommended by the GOLD guidelines for effective disease management (3). Aclidinium bromide is a novel inhaled long-acting muscarinic antagonist (LAMA) indicated for maintenance treatment of COPD. In patients with COPD, treatment with twice-daily (BID) aclidinium 200 μg and 400 μg provided significant improvements in lung function compared with placebo as early as the first administered dose that were sustained until the end of the 1- to 24-week studies (4–7). In addition, both doses of aclidinium were associated with significant improvement in health status and COPD symptoms, as well as a reduction in breathlessness (6, 7). The efficacy and safety profiles of twice-daily aclidinium have been previously described in clinical trials up to 6 months in duration (6, 7), but long-term data have not yet been reported for these outcomes.
Here we present the results from the 1-year extension of the ACCORD COPD I study (AClidinium in Chronic Obstructive Respiratory Disease I) (6). The primary objective of this study was to evaluate the long-term safety and tolerability of aclidinium in patients with moderate-to-severe COPD who received an additional 52 weeks of twice-daily aclidinium 200 μg or 400 μg after 12 weeks of treatment with the same aclidinium dose or with placebo. The long-term efficacy of aclidinium in pulmonary function and patient-related outcomes was also assessed in this study.
Methods
Study design
This was a Phase 3, multicenter, randomized, double-blind, parallel-group, 52-week, long-term extension study (NCT00970268) of ACCORD I (6), which was a 12-week randomized, double-blind, placebo-controlled trial conducted in patients with moderate-to-severe COPD in North America. Patients who completed the lead-in study were enrolled in the extension study if they agreed to participate in the 52-week extension. Patients who received aclidinium 200 μg or 400 μg BID during the lead-in study continued the same treatment, and patients who received placebo were rerandomized (1:1) to twice-daily aclidinium 200 μg or 400 μg in the extension study. Both aclidinium doses were administered via a novel, multidose, dry powder inhaler (Genuair®/PressairTM)*. Written informed consent was provided by the patient before initiation of any procedure in the extension study. The protocol was approved by the Institutional Review Board at each study center; the study was conducted according to International Conference on Harmonisation/Good Clinical Practice guidelines and the Declaration of Helsinki.
Study population
Key eligibility criteria for participation in the lead-in study have been described previously (6). Key exclusion criteria in this study were abnormalities in clinical laboratory values, vital signs, or electrocardiographic (ECG) results such as prolongation of QTc interval (Bazett-corrected) greater than 500 msec, clinically significant anticholinergic effects in the lead-in study, noncompliance with treatment or attending study visits during the lead-in study, and significant interruption of double-blind treatment between the end of the lead-in study and the initiation of the extension study.
The use of long-acting beta2-agonists (LABA) and other anticholinergics were prohibited during the study. Patients receiving LABA/inhaled corticosteroid (ICS) maintenance therapy prior to study entry were maintained on the ICS component alone. Albuterol and salbutamol were permitted as rescue medications; COPD background medications such as long-acting theophylline, ICS, and oral or parenteral corticosteroids (≤10 mg/day of prednisone or 20 mg every other day) were also allowed provided that treatment was stable for ≥4 weeks before study entry. Discontinuation of rescue medication, ICS, or theophylline was required ≥6 hours before a study visit.
Assessments and outcome measures
Visit 1 (enrollment) of the extension study was the final visit of the lead-in study. Safety and efficacy were evaluated during study visits at 12, 24, 36, 48, and 52 weeks after completion of the lead-in study; safety was also assessed 1 week after enrollment in the extension study. Safety assessments include those reported or observed during the extension study only, while efficacy assessments included measurements made during both the lead-in and extension studies. Safety was assessed during the extension study based on documentation of any adverse event (AE) reported by the patient or observed by the investigator (an approach consistent with AE reporting in clinical trials), clinical laboratory tests, vital signs, physical examinations, and ECGs. An AE was classified as treatment-emergent if it started on or after the date of the first dose of double-blind treatment of the extension study (until 30 days after the last treatment dose) or if it started before the first dose in the extension study and continued during the study with increased severity. Assessment of relationship to treatment of AEs was provided by the study investigator. Lung function was assessed by standardized spirometry (8); assessments were made at premorning dose (−45 and −15 minutes [−60 and −10 at enrollment]) and postmorning dose (0.5, 1.0, 2.25, and 3.0 hours) at Visit 1 of the extension study, and at 12, 24, and 52 weeks thereafter. Health status was assessed using St. George's Respiratory Questionnaire (SGRQ). Rescue medication use was assessed daily using an electronic diary, with baseline use assessed during the 1-week period prior to treatment initiation in the lead-in study. Overall use of rescue medication was assessed over a 64-week period, from the date of the first dose administered during the lead-in study to the last dose administered during the extension study.
The change from baseline (randomization visit of the lead-in study) to Week 64 (Week 52 of the extension) in morning predose (trough) FEV1 (calculated as the mean of the 2 morning predose FEV1 readings) was the primary efficacy outcome. The change from baseline to Week 64 in peak FEV1 (maximum FEV1 reading observed ≤3 hours postdose) was the secondary efficacy outcome. Additional efficacy outcomes included change from baseline by visit in trough and peak FEV1, SGRQ total and domain scores (symptoms, activity, and impact), and the percentage of patients achieving clinically important improvements in the SGRQ total score (defined as a decrease of ≥4 points) (9) at intermediate time points, as well as daily rescue medication use.
Statistical analysis
For each safety and efficacy outcome, the baseline value used for analysis was the original baseline value from the lead-in study, unless otherwise specified. All safety analyses were done using descriptive statistics and were based on the safety population (all patients who took at least one dose of treatment in the extension study). All efficacy analyses were considered exploratory and performed using the intent-to-treat (ITT) population, which consisted of all patients in the safety population who had a baseline FEV1 assessment in the lead-in study and at least one FEV1 assessment during the treatment phase of the extension study. Imputation of missing data was done by the last observation carried forward (LOCF) approach. Efficacy outcomes were analyzed using an analysis of covariance (ANCOVA) model with treatment sequence and sex as factors and baseline value and age as covariates. Rescue medication outcomes were analyzed similarly, but used only treatment sequence as a factor and baseline value as the covariate. The sample size of this extension study was dependent on the number of patients who completed the lead-in study and agreed to enroll in the extension study and was thus not based on statistical considerations.
Results
Study population
Of the 467 patients with COPD who completed the lead-in study, 291 enrolled in the extension study (). Similar proportions of patients who completed the lead-in study entered the extension study across the treatment sequences, with the lowest proportion in the placebo-aclidinium sequences (60%) versus continuous aclidinium (63%–64%). As the number of patients in the placebo group of the lead-in study who enrolled in the extension study were divided into 2 treatment groups (aclidinium 200 μg or 400 μg) upon study entry, the number of patients who were rerandomized to either aclidinium dose from placebo (n = 44, placebo-aclidinium 200 μg; n = 46, placebo-aclidinium 400 μg) was smaller compared with the number of patients who received continuous aclidinium treatment (n = 95, aclidinium 200 μg-aclidinium 200 μg; n = 106, aclidinium 400 μg-aclidinium 400 μg) at the beginning of this study.
Figure 1. Study Flow Chart. ITT = intent to treat.
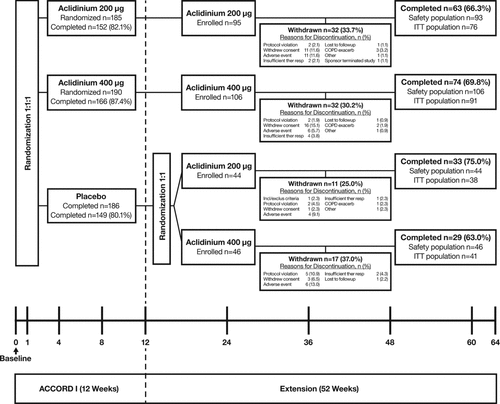
A total of 289 patients received at least 1 dose of the double-blind treatment and were included in the safety population. Of these patients, 246 had a baseline in the lead-in study and at least 1 FEV1 assessment after enrolling in the extension study and were thus included in the ITT population for efficacy analyses. Patients had a mean (SD) age of 64 (10) years and a baseline FEV1 of 1.3 (0.5) L (). Characteristics at baseline (randomization visit of the lead-in study) were generally similar for all treatment sequences () but with slightly better lung function and health status at baseline for patients switched from placebo to aclidinium 200 μg compared with patients in other treatment sequences.
Table 1. Demographic and baseline characteristics of patients in the extension studya
Safety
Treatment-emergent AEs (TEAEs) were mostly mild to moderate in severity and reported by similar percentages of patients across all treatment sequences during the 52-week extension study (). COPD exacerbation was the most frequently reported TEAE. The lowest percentage of patients reporting this TEAE was in the continuous aclidinium 400 μg treatment sequence; the highest percentage of patients reported this TEAE in the placebo-aclidinium 200 μg treatment sequence.
Table 2. Treatment-emergent adverse events (TEAEs) reported by ≥2% of total patients (number [%]; safety population)
Among all TEAEs reported in this study, such events that were considered by investigators to be related to treatment occurred in a similar percentage of patients treated with 200 μg (10.2%) or 400 μg (12.5%). TEAEs considered related to treatment that occurred with both aclidinium doses included dyspnea, headache, increased gamma-glutamyltransferase, and blood creatine phosphokinase; each of which was reported by ≤2 patients (<1.5%) for either dose.
The percentages of patients in the continuous aclidinium 200 μg or 400 μg treatment sequences who discontinued the study due to a TEAE were 15.1% and 7.5%, respectively; 11.4% and 13.0% of patients, who switched from placebo to aclidinium 200 μg or 400 μg, respectively, discontinued from the study due to a TEAE. The most commonly reported TEAE that resulted in study discontinuation was COPD exacerbation (continuous aclidinium 200 μg, 3.2%; continuous aclidinium 400 μg, 1.9%; placebo-aclidinium 200 μg, 2.3%), followed by dyspnea (continuous aclidinium 200 μg, 1.1%; placebo-aclidinium 400 μg, 2.2%) and rash (continuous aclidinium 400 μg, 0.9%; placebo-aclidinium 200 μg, 2.3%). The percentages of patients who discontinued due to a TEAE in the current study were greater than those in the 12-week lead-in study (200 μg, 6.0%; 400 μg, 4.2%); however, COPD exacerbations and dyspnea were also the most frequent TEAEs leading to treatment discontinuation in the lead-in study. These TEAEs led to discontinuation of a similar percentage of patients in the lead-in study (COPD exacerbation: 200 μg, 2.2%; 400 μg, 0.5%; dyspnea: 200 μg, 0; 400 μg, 1.1%) compared with those in the extension study.
Typically expected anticholinergic AEs occurred in small numbers of patients (<5% overall) and were reported by similar percentages of patients who received either aclidinium dose. Constipation and urinary tract infections (UTI) () were the most common anticholinergic AEs, of which only the UTI reported by 1 patient in the continuous aclidinium 400 μg treatment sequence was considered treatment related. Dry mouth was only reported by 1 patient in the extension study (placebo-400 μg); this was considered related to treatment.
The incidence of cardiac-related TEAEs was low, with any individual event being reported by <5% of patients with either aclidinium dose. The most frequently reported cardiac events were left/right bundle branch (LBB/RBB) block, atrioventricular (AV) block first degree, and congestive cardiac failure (). Two events each of LBB block and AV block first degree were reported on the first day of the extension study in patients who had received placebo in the lead-in study. These patients had been rerandomized to receive aclidinium during the extension (LBB block: 200 μg, n = 2; AV block: 200 μg, n = 1; 400 μg, n = 1) but had not yet received active treatment at the time the AE was reported. Of all the reported cardiac TEAEs, 3 patients reported such events that were considered by study investigators to be related to treatment. One patient who switched from placebo to aclidinium 200 μg reported treatment-related acute coronary syndrome. At the time of this event, the patient also had a nonserious treatment-related coronary artery disease. The same patient also reported atrial fibrillation that was considered related to treatment. Prior to study entry, this patient had a history of cardiac disorders which included coronary angioplasty and coronary artery bypass. A patient who received continuous aclidinium 400 μg reported treatment-related congestive cardiac failure and also had a history of cardiac disorders prior to entering the study. The third patient who reported a treatment-related cardiac TEAE (AV block first degree) received continuous aclidinium 400 μg treatment during the study.
Table 3. Treatment-emergent cardiac adverse events reported by ≥2 patients in the total population (number [%]; safety population)
The percentage of patients who reported on-therapy serious adverse events (SAEs) was similar between the 2 aclidinium doses (200 μg, 14.6%; 400 μg, 13.2%). COPD exacerbation was the most frequently reported SAE, with similar proportions of patients reporting this SAE for aclidinium 200 μg (3.6%) and 400 μg (4.6%). All other SAEs were reported by <3% of patients who received either aclidinium dose for any event. Three SAEs were considered by investigators to be related to study treatment, including the previously discussed treatment-related acute coronary syndrome in the patient switched from placebo to aclidinium 200 μg. A patient who received continuous aclidinium 400 μg treatment had 2 treatment-related SAEs (severe hypertension and severe hemorrhagic stroke) on the same day. The hypertension resolved 4 days after occurrence and the hemorrhagic stroke resulted in study termination 32 days after occurrence. This patient had a history of hypertension and mitral valve prolapse prior to study entry.
Two deaths were reported during the study, neither of which were considered to be related to treatment. One patient who received continuous aclidinium 200 μg treatment died due to an accidental multiple drug overdose to oxycodone and morphine. One patient who received continuous aclidinium 400 μg treatment had severe esophagitis at the time of death; no cause of death or death certificate was available.
The changes from baseline in clinical laboratory tests, vital signs, and ECG parameters were similar between the treatment sequences and not of clinical concern.
Lung function
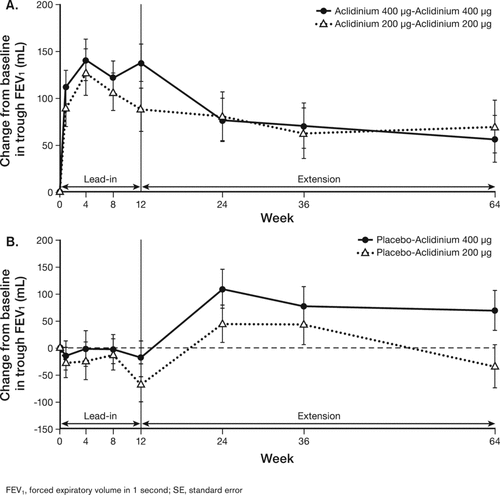
Patients who received continuous aclidinium 200 μg or 400 μg demonstrated improvements from baseline in morning predose (trough) FEV1 throughout the lead-in and extension studies, with trough FEV1 change from baseline values at study end of 69 mL and 56 mL, respectively (). Patients who were rerandomized to aclidinium 200 μg or 400 μg from placebo in the lead-in study exhibited a change from baseline in trough FEV1 of −60 mL and −18 mL, respectively, at the start of the extension study. Twelve weeks after initiation of treatment with aclidinium 200 μg or 400 μg (first time point assessed), these patients demonstrated improvements from baseline in trough FEV1 () similar to those observed at the end of the 12-week lead-in study for patients who were on continuous aclidinium treatment (). However, the improvements from baseline in trough FEV1 decreased at Week 64 for patients who switched from placebo to aclidinium 200 μg, in contrast to the generally maintained improvement observed in the other treatment arms.
Figure 2. Least squares mean (SE) change from baseline in trough FEV1 in patients on continuous aclidinium treatment (A) or switched from placebo to aclidinium (B).
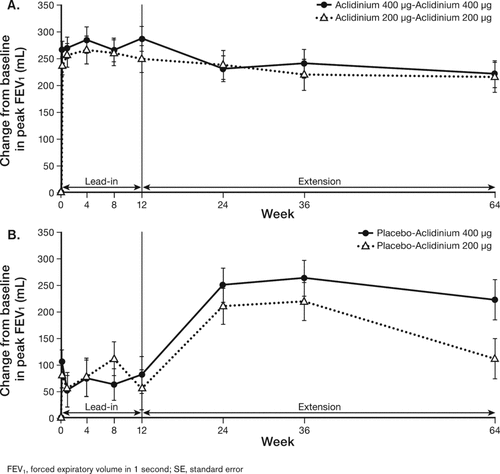
Patients on continuous aclidinium 200 μg or 400 μg throughout the lead-in and extension studies showed improvements from baseline in peak FEV1 at Week 12 (247 mL and 285 mL, respectively) that were maintained until Week 64 (213 mL and 219 mL, respectively) (). Patients who originally received placebo in the lead-in study demonstrated improvements from baseline in peak FEV1 following 12 weeks of treatment with aclidinium 200 μg or 400 μg. This improvement was maintained through study end only with the 400 μg dose () and was of a magnitude similar to what was observed in patients who were continuously treated with aclidinium 400 μg ().
Figure 3. Least squares mean (SE) change from baseline in peak FEV1 in patients on continuous aclidinium treatment (A) or switched from placebo to aclidinium (B).
Health status
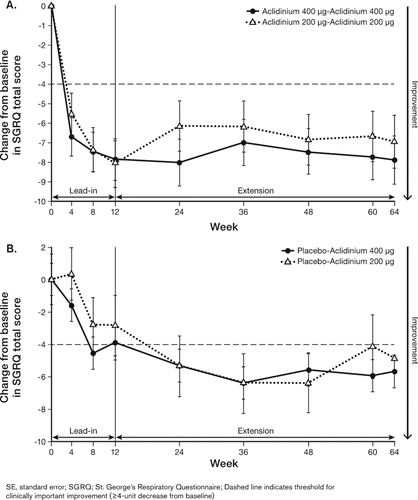
Patients who received continuous treatment with aclidinium 200 μg or 400 μg showed clinically important improvements from baseline (≥4-unit decrease) (9) in SGRQ total scores in the 12-week lead-in study and in the 52-week extension period, with numerically greater improvements observed at the end of the study with aclidinium 400 μg (−7.9 units) compared with aclidinium 200 μg (−7.0) (). Patients who received placebo during the lead-in study showed improvements from baseline in SGRQ total scores at Week 12; further numerical improvements from baseline were observed when patients were switched to aclidinium during the extension study (). After 52 weeks of aclidinium treatment, a numerically greater improvement from baseline was observed with the 400 μg dose (−5.7 units) compared with the 200 μg dose (−4.9 units) ().
Figure 4. Least squares mean (SE) change from baseline in SGRQ total score in patients on continuous aclidinium treatment (A) or switched from placebo to aclidinium (B).
Improvements from baseline in each of the 3 SGRQ domains in patients who received continuous aclidinium treatment ranged from 7.2 to 13.1 units for symptoms, 3.3 to 8.2 units for activity, and 5.8 to 7.5 for impact throughout the extension study. These were generally greater in magnitude compared with those observed in patients who switched from placebo to aclidinium (0.7 to 10.8, 2.9 to 6.9, and 2.5 to 7.4 for each of the domains, respectively).
The percentage of patients who achieved a ≥4-unit decrease from baseline in SGRQ total scores ranged from 43.2% to 65.6% for all treatment sequences throughout the extension study. At study end (Week 64), a numerically greater percentage of patients who received continuous aclidinium treatment achieved a clinically important improvement in SGRQ total scores (200 μg, 50.7%; 400 μg, 64.4%) compared with those who switched from placebo to aclidinium 200 μg (43.2%) or 400 μg (59.0%).
Rescue medication use
Over the 64-week treatment period, which included the lead-in and extension studies, mean rescue medication use for all treatment sequences (continuous aclidinium 200 μg and 400 μg, 2.6 and 2.2 puffs/day, respectively; placebo-aclidinium 200 μg and placebo-aclidinium 400 μg, 2.3 and 2.7 puffs/day, respectively) were less than at baseline ().
Discussion
Most patients with COPD require continued use of pharmacotherapies for effective management of the disease, therefore, assessing the long-term safety and efficacy of any new treatment option is essential to evaluating its therapeutic potential. Cardiovascular disease is a common co-morbidity in patients with COPD (10), highlighting the importance of evaluating cardiac safety of COPD therapies.
In this long-term extension study, both doses of twice-daily aclidinium 200 μg and 400 μg were well tolerated throughout the 52-week period following the 12-week lead-in study, with no differences in safety profiles observed between doses. Anticholinergic AEs typically expected with LAMA treatment occurred in a small number of patients in this study, with dry mouth being reported in only 1 patient. Cardiac events were infrequent and were reported by a numerically higher proportion of patients with the 200 μg dose compared with the higher dose, suggesting the absence of a dose-dependent effect. The safety profile observed with long-term aclidinium treatment in the current study appears to support the safety and tolerability of aclidinium reported in previous studies with twice-daily aclidinium ranging in duration from 12 to 24 weeks (6, 7). The tolerability of aclidinium may be due to its rapid plasma hydrolysis (11–14), which suggests that treatment with the drug may lead to fewer systemic side effects. As the lead-in study excluded patients with clinically significant cardiovascular conditions or unstable cardiac disease (6), a more comprehensive evaluation of the cardiac safety of aclidinium may be obtained from clinical studies with patients suffering from cardiovascular co-morbidities.
In terms of efficacy, patients with COPD who received continuous treatment with twice-daily aclidinium 200 μg or 400 μg showed improvements in bronchodilation that were maintained until the end of the 64-week study. These results suggest that bronchodilation efficacy reported as early as the first day of treatment in earlier studies of shorter duration (6, 7) may be sustained with long-term treatment with aclidinium.
The observed decline in lung function outcomes at study end, particularly in patients who switched from placebo to aclidinium, may have been due to a high degree of variability as a consequence of the small number of patients enrolled in the placebo-aclidinium treatment sequences. These patients (n = 44, 46) were less than half the number of the patients enrolled in the continuous aclidinium treatment sequences (n = 95, 106). In addition, the number of patients who completed the study in either placebo-aclidinium treatment sequence was also approximately half the number of patients who completed continuous aclidinium treatment, further contributing to the variability and the decline in lung function observed at Week 64. Furthermore, patients that switched from placebo to aclidinium 200 μg had slightly better lung function and health status at baseline (ie, randomization at lead-in) compared with patients in the other treatment sequences, which may have contributed to the less robust treatment efficacy observed for this group. Regardless of the initial treatment taken during the lead-in study, patients who received the aclidinium 400 μg dose demonstrated improvements in lung function in the extension study that were sustained up to study end, similar to what has been previously reported in studies with shorter treatment periods (6, 7).
Although spirometric assessments remain the primary outcome measure in COPD management, health status has been shown to better correlate with patient symptoms such as breathlessness (15, 16) and is thus an important goal in the effective management of COPD (3). In this long-term study, continuous aclidinium treatment provided the greatest magnitude of improvement in SGRQ total scores, with differences from baseline at study end ranging from 7 to 8 units. These improvements in SGRQ provide evidence for the continued efficacy of aclidinium with long-term treatment that has previously been demonstrated in aclidinium clinical studies of shorter duration (6, 7). The improvements in health status in this study, as well as the observed reduction in rescue medication use, suggest that long-term aclidinium treatment may provide substantial benefits to patients in symptom management.
One limitation of this study may have been an artificial bias in patient selection as only those who completed the lead-in study and agreed to participate in the extension study were included. This may have led to a patient population of “healthy survivors,” a phenomenon typically observed in long-term studies wherein patients with more severe disease discontinue treatment (17–19). As such, the possible contribution of the “survivor” effect on the safety and efficacy outcomes in this study would need to be further evaluated. Second, the lack of a placebo arm makes it difficult to comprehensively evaluate the full therapeutic benefits of aclidinium over one year of treatment. Nevertheless, the current study, which was conducted primarily to assess the long-term safety and tolerability of aclidinium, provides important information addressing both those questions and the persistence of therapeutic benefits.
Conclusion
In summary, these data show that long-term treatment with twice-daily aclidinium 200 μg or 400 μg was effective and well tolerated in patients with moderate-to-severe COPD, with similar safety profiles observed for both doses. Aclidinium 400 μg consistently provided improvements in bronchodilation and health status as well as reduced rescue medication use that were maintained throughout the 52-week treatment period in the extension study, supporting the effectiveness of aclidinium treatment observed in earlier studies.
Declaration of Interest Statement
This work was funded by Forest Research Institute, Inc. (FRI), a wholly owned subsidiary of Forest Laboratories, Inc., and Almirall, S.A. Anthony D’Urzo has received research, consulting and lecturing fees from GlaxoSmithKline, Sepracor, Schering Plough, Altana, Methapharma, AstraZeneca, ONO pharma, Merck Canada, Forest Laboratories, Novartis Canada/USA, Boehringer Ingelheim (Canada) Ltd, Pfizer Canada, SkyePharma, and KOS Pharmaceuticals. Edward Kerwin has served on advisory boards or speaker panels for Astra Zeneca, Ironwood, Mylan, Pearl, Pfizer, Sanofi Aventis, Sunovion, and Targacept, has received travel reimbursement from Merck, Forest, and Novartis, and has performed multicenter clinical trials for multiple pharmaceutical companies including Forest and Almirall, S.A. Stephen Rennard (SR) has received honoraria for lectures from AARC, Almirall, Am Col Osteopathic Physicians, Asan Medical Center, American Thoracic Society, California Society of Allergy, CME Incite, COPD Foundation, Creative Educational Concepts, Dey, Duke University, Forest, France Foundation, HSC Medical Education, Information TV, Lung Association, Novartis (Horsham, Nycomed, Otsuka, PeerVoice, Pfizer, Shaw Science, University of Washington, University of Alabama Birmingham, VA Sioux Falls. SR has also received honorarium for consulting with the following: ABIM, Able Associates, Adelphi Research, Align2Acton, Almirall/Prescott, APT Pharma/Britnall, Astra-Zeneca, American Thoracic Society Beilenson, Boehringer Ingelheim, Boehringer Ingelheim (ACCP), BoomCom, Britnall and Nicolini, Capital Research, Chiesi, Clarus Acuity, CommonHealth, Complete Medical Group, Consult Complete, COPDForum, DataMonitor, Decision Resources, Dunn Group, Easton Associates, Equinox, Forest, Frankel Group, Fulcrum, Gerson Lehman, Globe Life Sciences, Guidepoint, Health Advanced, Hoffman LaRoche, Informed, Insyght, KOL Connection, Leerink Swan, M. Pankove, McKinsey, MDRxFinancial, Medimmune, Merck, Novartis, Nycomed, Oriel, Osterman, Peal, Penn Technology, Pennside, Pfizer, PharmaVentures, Pharmaxis, Prescott, Price Waterhouse, Propagate, Pulmonary Reviews, Pulmatrix, Reckner Associates, Recruiting Resource, Roche, Sankyo, Schering, Schlesinger Medical, Scimed, Smith Research, Sudler and Hennessey, Summer Street Research, Talecris, Think Equity, UBC, Uptake Medical, Vantage Point Management. Esther Garcia Gil is an employee of Almirall, S.A. Thomas He and Cynthia Caracta are employees of FRI. Editorial assistance by Joy Ramos, PhD, of Prescott Medical Communications Group, Chicago, IL was funded by FRI. There are no other financial disclosures/conflicts of interest.
All the authors were involved in the creation and review of the manuscript, had full access to study data, and take full responsibility for data integrity and accuracy of all data analysis. All authors participated in the writing of this manuscript and are responsible for its content.
Acknowledgments
The authors would like to thank the investigators and the study team in each of the participating centers.
References
- World Health Organization. The global burden of disease: 2004 update [Internet]. Geneva, Switzerland: WHO Press; 2008. Available from: http://www.who.int/healthinfo/global_burden_disease/GBD_report_4update_full.pdf (accessed 21 December 11).
- Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet 2007 Sep 1;370(9589):765–773.
- Vestbo J, Hurd SS, Agusti AG, Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Pulmonary Disease, GOLD Executive Summary. Am J Respir Crit Care Med 2013 Feb;187(4):347–365.
- Fuhr R, Magnussen H, Sarem K, Efficacy of aclidinium bromide 400 microgram twice daily compared with placebo and tiotropium in patients with moderate to severe COPD. Chest 2012 Mar;141(3):745–752.
- Singh D, Magnussen H, Kirsten A, A randomised, placebo- and active-controlled dose-finding study of aclidinium bromide administered twice a day in COPD patients. Pulm Pharmacol Ther 2012 Jun;25(3):248–253.
- Kerwin EM, D’Urzo AD, Gelb AF, Lakkis H, Garcia Gil E, Caracta CF. Efficacy and safety of a 12-week treatment with twice-daily aclidinium bromide in COPD patients (ACCORD COPD I). COPD 2012 Apr;9(2):90–101.
- Jones PW, Singh D, Bateman ED, Efficacy and safety of twice-daily aclidinium bromide in COPD patients: the ATTAIN study. Eur Respir J 2012 Mar;40(4):830–836.
- Miller MR, Hankinson J, Brusasco V, Standardisation of spirometry. Eur Respir J 2005 Aug;26(2):319–338.
- Jones PW. St. George's Respiratory Questionnaire: MCID. COPD 2005 Mar;2(1):75–79.
- Huiart L, Ernst P, Suissa S. Cardiovascular morbidity and mortality in COPD. Chest 2005 Oct;128(4):2640–2646.
- Sentellas S, Ramos I, Alberti J, Aclidinium bromide, a new, long-acting, inhaled muscarinic antagonist: in vitro plasma inactivation and pharmacological activity of its main metabolites. Eur J Pharm Sci 2010 Mar;39(5):283–290.
- Jansat JM, Lamarca R, Garcia Gil E, Ferrer P. Safety and pharmacokinetics of single doses of aclidinium bromide, a novel long-acting, inhaled antimuscarinic, in healthy subjects. Int J Clin Pharmacol Ther 2009 Jul;47(7):460–468.
- Jansat JM, Lamarca R, de Miquel G, Schrodter A, Miletzki B, Gurniak M. Safety and pharmacokinetics of multiple doses of aclidinium bromide, a novel long-acting muscarinic antagonist for the treatment of chronic obstructive pulmonary disease, in healthy participants. J Clin Pharmacol 2009 Oct;49(10):1239–1246.
- Lasseter K, Dilzer S, Jansat JM, Garcia Gil E, Caracta C, Ortiz S. Safety and pharmacokinetics of multiple doses of aclidinium bromide administered twice daily in healthy volunteers. Pulm Pharmacol Ther 2012;25(2):193–199.
- Mahler DA, Tomlinson D, Olmstead EM, Tosteson AN, O’Connor GT. Changes in dyspnea, health status, and lung function in chronic airway disease. Am J Respir Crit Care Med 1995 Jan;151(1):61–65.
- Hajiro T, Nishimura K, Tsukino M, Ikeda A, Oga T, Izumi T. A comparison of the level of dyspnea vs disease severity in indicating the health-related quality of life of patients with COPD. Chest 1999 Dec;116(6):1632–1637.
- Burge PS, Calverley PM, Jones PW, Spencer S, Anderson JA, Maslen TK. Randomised, double blind, placebo controlled study of fluticasone propionate in patients with moderate to severe chronic obstructive pulmonary disease: the ISOLDE trial. BMJ 2000 May 13;320(7245):1297–1303.
- Tashkin DP, Celli B, Senn S, A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008 Oct 9;359(15):1543–1554.
- Calverley PM, Anderson JA, Celli B, Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007 Feb 22;356(8):775–789.