
Abstract
Background: Since 1999, as part of the Alpha1 International Registry (AIR), the Canadian Alpha-1 Antitrypsin Deficiency (AATD) Registry has maintained demographic and medical information volunteered by AATD individuals.
Methods: We undertook a retrospective chart review to describe the characteristics of registry participants. Inclusion criteria were ZZ phenotype or other severe deficiency and written consent. We reviewed baseline medical records and annual follow-ups, conducted by mail.
Results: The number of registrants ranged from 8.7 per million in British Columbia and Ontario to 1.3 per million in Quebec. Similarly, the rate of augmentation therapy use ranged from 7.7 per million in British Columbia to 0.1 per million in Quebec. 290 patients (146 males), most PiZZ, were enrolled by 2013. Patients with lung disease reported symptoms onset at (mean ± SD) 40 ± 11 years but were diagnosed as AATD at 47 ± 10 years. Typical patients were ex-smokers with respiratory symptoms, severely reduced FEV1, an accelerated rate of FEV1 decline, and minimal bronchodilator response. A subgroup diagnosed by liver disease or familial screening was younger and had better preserved lung function but a similar rate of FEV1 decline. There were 63 deaths, of which 29 were lung-related and 6 were liver-related. Average age at death was 60.5 ± 11.2 years.
Discussion: Most patients experience a diagnostic delay of seven years after symptom onset, a period during which lung health may deteriorate further. There is marked regional variation in the rate of diagnosis and specific therapy usage for AAT in Canada.
Introduction
Alpha-1 antitrypsin (AAT) is a serine antiprotease produced by the liver and delivered via the systemic circulation. A major physiologic function appears to be protection of lung tissue from inflammatory injury caused by neutrophil elastases and other proteases. Single base pair mutations of the AAT gene are common and when an individual has two alleles thus affected, conformational changes during production of the AAT glycoprotein are altered. This hinders excretion by the liver, with consequent accumulation of AAT glycoprotein in the hepatocyte and markedly reduced circulating levels. Affected individuals are at increased risk of pulmonary emphysema and/or liver cirrhosis (Citation1). AATD is a co-dominant genetic trait, whose prevalence is much greater than the number of individuals diagnosed. It is estimated that 1 in 6,000 of the Caucasian population in North America is affected (Citation2) of whom only 5% have been diagnosed (Citation3), making AATD the second most common type of genetic lung disease after cystic fibrosis (Citation4) with the deficient Z allele frequency around 2–3% (Citation5).
Diagnosis is often delayed in patients with pulmonary symptoms because symptoms are typical of common obstructive lung diseases such as asthma in younger individuals or chronic obstructive pulmonary disease (COPD) in older individuals and screening for AATD is not considered (Citation3). Many patients describe shortness of breath on exertion as early as age 30, when pulmonary function tests still appear normal (Citation6). On average, AATD individuals lose lung function at an accelerated rate of 70–120 mL/year as compared to the normal rate of decline of 30 mL/year (Citation7). The ATS/ERS statement on standards for the diagnosis and management of individuals with AATD recommends screening for the disease by measuring a serum level in patients with COPD or those with asthma if lung function is persistently abnormal, a recommendation recently echoed by the Canadian Thoracic Society (Citation8,9). Prolonged neonatal jaundice and unexplained cirrhosis in adults also warrant testing. Once diagnosed, screening of family members, especially siblings, is recommended.
The Canadian Alpha-1 Antitrypsin Deficiency Registry was formed in 1999 as part of an international effort to facilitate research in this comparatively uncommon disorder by creating a database of AATD individuals (Citation10). This effort fulfilled the World Health Organization recommendation (made in 1996) for creation of such a database to assist researchers to connect with willing A1ATD subjects, thus making adequately powered studies into this condition feasible (Citation11). This retrospective chart review describes the diagnosis, treatment and disease progression of the Canadian registry participants.
Methods
The only inclusion criterion was a confirmed phenotype of PiZZ, PiSZ or other severe deficiency (serum level <11 μmol). Once patients gave consent and permission to release their medical information, their family doctor or respiratory specialist provided us with a baseline medical history report. Follow up surveys were mailed directly to participants annually. The contents of these reports have been described previously (Citation10). When we learned of any deaths amongst registered subjects (Canada lacks a national registry that would provide such data to the investigators), the date and cause of death were requested from the referring physician. Data were collected between April, 1999 and December 2013, with the exception of the St. George Respiratory Questionnaire, added in January 2006 (Citation12). All registry participants provided written informed consent to share their data with the registry and its operation was approved by the Research Ethics Board of the University Health Network, Toronto.
Rates of forced expired volume in one second (FEV1) decline were calculated using least squares linear regression for subjects who had at least two measurements of FEV1, at least one year apart. Quality of life was estimated by score on the most recently completed version of the SGRQ; we looked for correlation between this score and age at first symptoms, age at diagnosis, smoking history, baseline FEV1% predicted, FEV1 rate of decline and specific therapy by infusion of alpha-1 antitrypsin at the time of the questionnaire. To explore further regional trends in diagnosis and management, additional data were sought from the manufacturer of augmentation therapy (formerly Talecris Canada) to better understand the use of augmentation therapy by AATD individuals in Canada, whether or not they had enrolled in the Registry (personal communication, Graeme Marney, Talecris Biotherapeutics, Canada).
Results
Within 151 months, 290 subjects (146 males) were registered. Figure shows the enrollment pattern of these subjects over time and their distribution by Canadian province. Also shown in this figure is the total number receiving augmentation therapy in each province, as reported by the manufacturer of augmentation therapy distributed in Canada (Prolastin, Grifols, formerly Talecris Canada [Citation13]). There was marked regional variation in the number of patients registered and in the number of patients receiving augmentation therapy. Excluding the smallest provinces and territories (PEI, Yukon, Northwest Territories and Nunavit), the number of registrants ranged from a high of 8.7 per million in British Columbia and Ontario to a low of 1.3 per million in Quebec. Similarly, the rate of augmentation therapy use ranged from a high of 7.7 per million in British Columbia to a low of 0.1 per million in Quebec.
Figure 1. Distribution of registered members, and usage of augmentation therapy (italics), given in absolute number and number per million (brackets).
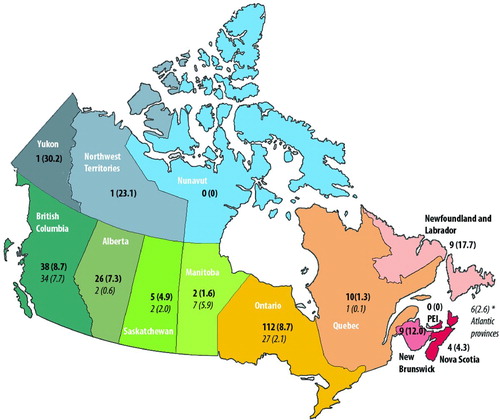
The mean response rate for annual follow-up forms was 60 ± 30%. Tobacco consumption was greater amongst men (18 ± 19 pack-years vs 12 ± 13 pack-years; p < 0.007). Despite this difference, neither baseline FEV1 nor rate of FEV1 decline were significantly different between men and women. Upon controlling for smoking history, gender accounted for 17% of the variation in FEV1 decline, with men tending to decline more rapidly than women, but this trend was not statistically significant (p < 0.071).
The distribution of phenotypes and corresponding AAT levels can be seen in Table . The majority of patients were of the phenotype PiZZ. 42 had been treated with intravenous augmentation therapy for at least one year, with a mean treatment duration of 8 ± 5 years. Of the 72 patients with a baseline FEV1 between 35 and 65% predicted, only 17 had ever received this treatment.
Table 1. General characteristics of registry participants (n = 29)
Of the 211 patients with pulmonary function test data available, 35% had a bronchodilator response of ≥ 12% (expressed as percent improvement from baseline). However, having a response over 12% did not predict a significantly faster rate of FEV1decline than for those with less reversibility (-65 vs –58 mL/year, p < 0.6). As well, patients with a baseline FEV1 under 40% predicted did not have a higher degree of reversibility than those with better preserved lung function (9 vs 11% change, p < 0.4). At baseline, 62% of patients were not working regularly, of whom 58% cited their disease, and 29% cited their age as the reason for unemployment.
Lung disease
Lung disease led to the AATD diagnosis in 248 subjects. The characteristics of these patients are shown in Table . Amongst current and ex-smokers, the average smoking history was 23 ± 13 pack-years. Forty-two patients had been treated with augmentation therapy and had a rate of FEV1 decline of -44.4 ± 58.9 mL/year while receiving therapy. Forty-one subjects were using long-term oxygen therapy at baseline. There were 33 self-reported lung transplants, 3 liver transplants and 15 lung volume reduction surgeries.
Table 2. Characteristics of patients diagnosed due to lung disease
Liver disease
The presence of liver disease led to the diagnosis of AATD in 23 patients. Age at diagnosis was bimodal with diagnoses occurring commonly in the first 10 years of life and again in middle age (data not shown). Four patients had the phenotype SZ and were diagnosed at ages 10 and 11. Liver enzymes were available at baseline for 17 patients. Physicians indicated that ALT, ASAT, GGT and ALP were elevated in 67, 60, 42 and 67% of this population. Of 5 patients with pulmonary function data available, the baseline FEV1 was 89 ± 40% predicted, and the rate of FEV1 decline was -135 ± 231 mL/year. No patients with liver disease were receiving augmentation therapy. There were 3 self-reported liver transplants and no lung transplant.
Family screening / Other diseases
The 43 non-index cases were diagnosed by family screening or due to other diseases at an average age of 41 ± 16 years. Twenty-two were ex-smokers with a history of 18 ± 22 pack-years. Baseline FEV1was 90 ± 30% predicted, with untreated and augmented declines in this subgroup of -58.3 ± 65.0 mL/year and -39.2 ± 49.7 mL/year, respectively. This subgroup had an evident gender difference in smoking history, with men smoking an average of 19 pack-years and women only smoking 4 (p < 0.047), although this was not associated with significant differences in age at diagnosis (p < 0.925), baseline FEV1 (p < 0.192) or rate of FEV1 decline (p < 0.115). Nine patients were receiving augmentation therapy and 1 had also undergone lung volume reduction surgery.
Quality of life, survival and mortality
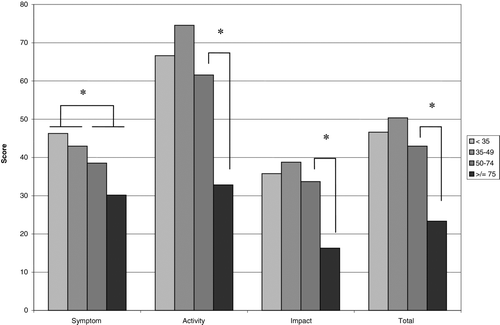
Two hundred and ninety patients completed the SGRQ at least once. Their total scores on the most recent questionnaire correlated significantly with baseline FEV1% predicted (r = -0.53, p < 0.001), smoking history in pack-years (r = 0.34, p < 0.001) and age at diagnosis (r = 0.245, p < 0.05). The correlations held for each subsection of the SGRQ except for symptom score which was not associated with the age at diagnosis. The activity score had the highest correlation coefficient with all three of these variables, even higher than the total SGRQ score (r = -0.54, r = 0.37, r = 0.26). When the results of the SGRQ were stratified according to FEV1, scores on each subsection changed most markedly between the group with 100–75% predicted FEV1 and that with 74–50% predicted FEV1 (Figure ). The exception was in the symptom sub-score, in which a significant change in score was only seen when comparing patients falling on either side of 50% FEV1 predicted. SGRQ score was not correlated with age at first symptoms or rate of lung function decline. Patients on augmentation therapy (n = 24) at the time of questionnaire had a score of 50 ± 14, while those not receiving augmentation scored 34 ± 22 points (p < 0.00005). Augmentation status was significantly correlated with SGRQ even when controlling for baseline FEV1% predicted (p = 0.014).
Figure 2. SGRQ scores at most recent follow-up for 106 patients, sorted by FEV1% predicted.
Discussion
There is pronounced regional disparity in the number of patient referrals to the registry, with Ontario having the highest number, followed by British Columba; the number of patients receiving augmentation therapy was highest in British Columbia. In contrast, Quebec had the lowest rate of patient referrals and specific therapy use. Typical Canadian patients with AATD have the phenotype PiZZ, began to suffer respiratory symptoms at 38 years of age and did not have a specific diagnosis for approximately seven years. Smokers are diagnosed younger than non-smokers, but have much lower FEV1% predicted at baseline. On average, men had longer smoking histories than women, and a trend towards faster FEV1 decline, suggesting they have a higher risk of lung damage prior to diagnosis, and faster deterioration even after smoking cessation.
Regional differences in registry referral and the use augmentation therapy rates is unlikely to reflect variation in disease prevalence but rather the locations of centers with an interest in this condition; variation in provincial funding policies concerning augmentation therapy and genetic testing; variation in the availability of specialist respiratory care; differences in funding for lung function testing; and, language barriers. Paradoxically, a slightly higher prevalence of AATD might be expected in Quebec given that the disorder is of highest prevalence in those of northern European background but is comparatively rare in those of African or Asian descent. Thus, patients with lung disease secondary to alpha-1 antitrypsin deficiency face different probabilities of receiving augmentation therapy when indicated depending on their province of domicile. The most significant factor appears to be public funding for augmentation therapy, currently provided by only four of ten provinces. Private insurance funding, however, is available to a subset of the population across Canada.
The characteristics of our members are in similar to those described in other national registries including the diagnostic delay after the onset of respiratory symptoms (3,Citation14–16). The diagnostic delay can be attributed to both low awareness and lingering uncertainty about the role of specific therapy. The first Canadian Thoracic Society statement concerning specific augmentation therapy recommended that further research was required before augmentation therapy be considered for clinical use (Citation17). In contrast, the concurrent American Thoracic Society statement recommended the clinical use of augmentation therapy for all deficient individuals with significant pulmonary disease (Citation18). An updated Canadian Thoracic Society statement cites evidence which supports consideration of augmentation therapy in individuals with rapid rates of lung function decline (Citation9) and a meta-analysis of available evidence also supports use in patients between 30–65% predicted FEV1 (Citation19), uncertainty about this intervention persists. Our registry has provided data that suggests preservation of lung function (FEV1) amongst augmented versus non-augmented patients seen in Canada (Citation20).
Specific AATD therapy aside, other benefits to earlier diagnosis are lifestyle changes such as smoking cessation and limiting occupational exposures, which can lower the number of hospital visits by these patients (Citation5). Diagnosis also helps indirectly by prompting closer observation and screening of family members for the disease. The differences we described between smokers and non-smokers and between men and women coincide with previous reports (Citation3,Citation21).
Patients who smoke without knowing their pre-disposition to emphysema will do irreversible damage to their lungs, which in turn will have a negative impact on their future quality of life and employability. Men may be at a greater risk due to longer average smoking history. Overall, the most impaired domain of the SGRQ was Activity, which is in line with the findings of a recent AlphaNet report (Citation22). Due to the low awareness of and/or referrals to the registry among liver specialists in Canada, we did not have enough registry members diagnosed due to liver disease to draw any meaningful conclusions. Our small patient numbers in this group also reflects the fact that only 10% of patients with AATD develop will have liver disease at infancy (Citation23,24). All three liver transplants in this group were from patients diagnosed with adult onset cirrhosis.
Some limitations to our study should be noted. Participation in our registry is purely voluntary and takes place only if physicians and/or their AATD patients contact the registry. Many diagnosed patients have not shared their data with the registry and our subset of diagnosed individuals may not be representative of the population as a whole. However, we suspect that our data are not markedly skewed at least with respect to regional variation. We note that the use of a specific therapy (data obtained independently of registry enrolment) follows a pattern similar to registry enrolment.
However, registered individuals have been diagnosed and may thus represent the more symptomatic and severely affected AATD individuals. There remain many undiagnosed patients, both symptomatic and asymptomatic in Canada who are not members of the Canadian registry. Currently, awareness raising programs have begun to be organized by the Canadian patient support group. The long-term implications of these educational activities remains to be seen, but past precedent has shown that similar events organized in the United States have had some success in improving recognition of this disease (Citation16).
The role of the registry in Canada has also been to foster research and research collaboration. Previous reviews of our registry data have contributed to the AIR registry overview (Citation25) and to a meta-analysis of augmentation therapy's impact on lung function (Citation19). It has also permitted enrolment of patients in completed or ongoing multi-centre trials of therapy (Citation26,27) {NCT01983241; clinicaltrials.gov}. An educational review article has informed Canadian physicians of the diseases common presenting features, screening strategies and available resources for confirmatory genotyping or phenotyping (Citation28). With the assistance of the registry, an active Canadian patient organization has been founded (Alpha-1 Canada) and maintains an active educational website (www.alpha1canada.ca), ongoing educational webcasts and a program of advocacy.
In conclusion, the state of AATD individuals in Canada most clearly reveals a need for more vigilant symptom recognition and increased screening for this disease. These steps have the potential to improve health outcomes, preserve quality of life, and allow for important life planning for affected individuals.
Funding
The authors wish to acknowledge the support of the UHN Foundation and the UHN Alpha1 Antitrypsin Research Fund. Professor Chapman is supported by the University Health Network’s GSK-CIHR Research Chair in Respiratory Health Care Delivery.
Declaration of Interest Statement
In the past 3 years, KRC has received compensation for consulting with AstraZeneca, Baxter, Boehringer-Ingelheim, CSL Behring, GlaxoSmithKline, Grifols, Kamada, Novartis, Nycomed, Roche, and Telacris; has undertaken research funded by Amgen, AstraZeneca, Baxter, Boehringer-Ingelheim, CSL Behring, Forest Labs, GlaxoSmithKline, Grifols, Novartis, Roche and Takeda; and has participated in continuing medical education activities sponsored in whole or in part by AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Grifols, Merck Frosst, Novartis, Pfizer and Takeda. He is participating in research funded by the Canadian Institutes of Health Research operating grant entitled: Canadian Cohort Obstructive Lung Disease (CanCOLD). Professor Chapman holds the GSK-CIHR Research Chair in Respiratory Health Care Delivery at the University Health Network, Toronto, Canada. The remaining authors declare no conflicts of interest.
The authors alone are responsible for the content and writing of the paper.
References
- Fregonese L, Stolk J. Hereditary alpha-1-antitrypsin deficiency and its clinical consequences. Orphanet J Rare Dis 2008; 3:16.
- Abboud RT, Ford GT, Chapman KR. Alpha1-antitrypsin deficiency: a position statement of the Canadian Thoracic Society. Can Respir J 2001; 8(2):81–88.
- Campos MA, Wanner A, Zhang G, Sandhaus RA. Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest 2005; 128(3):1179–1186.
- Richmond RJ, Zellner KM. Alpha1-antitrypsin deficiency: incidence and implications. Dimens Crit Care Nurs 2005; 24(6):255–260.
- Stoller JK, Fromer L, Brantly M, et al. Primary care diagnosis of alpha-1 antitrypsin deficiency: issues and opportunities. Cleve Clin J Med 2007; 74(12):869–874.
- Bernspang E, Sveger T, Piitulainen E. Respiratory symptoms and lung function in 30-year-old individuals with alpha-1-antitrypsin deficiency. Respir Med 2007; 101(9):1971–1976.
- Sherman CB, Xu X, Speizer FE, et al. Longitudinal lung function decline in subjects with respiratory symptoms. Am Rev Respir Dis 1992; 146(4):855–859.
- American Thoracic Society/European Respiratory Society Statement: Standards for the Diagnosis and Management of Individuals with Alpha-1 Antitrypsin Deficiency. Amer J Respir Crit Care Med 2003; 168(7):818–900.
- Marciniuk DD, Hernandez P, Balter M, et al. Alpha1 antitrypsin deficiency targeted testing and augmentation therapy: A Canadian Thoracic Society clincal practice guideline. Can Respir J 2012; 19(2):109–116.
- Stockley RA, Luisetti M, Miravitlles M, Piitulainen E, Fernandez P. Ongoing research in Europe: Alpha One International Registry (AIR) objectives and development. Euro Respir J 2007; 29(3):582–586.
- Alpha 1-antitrypsin deficiency: memorandum from a WHO meeting. Bull.World Health Organ 1997; 75(5):397–415.
- Jones PW, Quirk FH, Baveystock CM, Littlejohns P. A self-complete measure of health status for chronic airflow limitation: The St. George's Respiratory Questionnaire. Am. Rev.Respir. Dis. 1992; 145:1321–1327.
- Bradi AC, Chapman KR. Update from the Alpha-1 Canadian Registry. Am J Respir Crit Care Med. 2009; 179: A3498.
- Miravitlles M, Vidal R, Barros-Tizon JC, et al. Usefulness of a national registry of alpha-1-antitrypsin deficiency. The Spanish experience. Respir Med. 1998; 92(10):1181–1187.
- McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of alpha 1-antitrypsin deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Chest. 1997; 111(2):394–403.
- Stoller JK, Sandhaus RA, Turino G, et al. Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest 2005; 128(4):1989–1994.
- Ad Hoc Committee on Alpha-1-Antitrypsin Replacement Therapy of the Standards Committee CTS. Current status of alpha-1-antitrypsin replacement therapy: recommendations for the management of patients with severe hereditary deficiency. Can Med Asso J 1992; 146:841–844.
- American Thoracic Society. Guidelines for the approach to the patient with severe hereditary alpha-1-antitrypsin deficiency. Am.Rev.Respir Dis 1989; 140(5):1494–1497.
- Chapman KR, Stockley RA, Dawkins C, Wilkes MM, Navickis RJ. Augmentation therapy for alpha1 antitrypsin deficiency: a meta-analysis. COPD 2009; 6(3):177–184.
- Chapman KR, Bradi AC, Paterson D, Navickis RJ, Wilkes MM. Slower lung function decline during augmentation therapy in patients with alpha1-antitrypsin deficiency (A1ATD): results from the Canadian AIR Registry. Am J Respir Crit Care Med 2005;170: 3824.
- Vreim CE, Wu M, Crystal RG, et al. Survival and FEV 1 decline in individuals with severe deficiency of ‡ 1 -antitrypsin. Amer J Respir Crit Care Med 1998; 158(1):49–59.
- Campos MA, Alazemi S, Zhang G, et al. Clinical characteristics of subjects with symptoms of alpha1-antitrypsin deficiency older than 60 years. Chest. 2009; 135(3):600–608.
- Wall M, Moe E, Eisenberg J, et al. Long-term follow-up of a cohort of children with alpha-1-antitrypsin deficiency. J Pediat 1990; 116(2):248–251.
- Sveger T, Eriksson S. The liver in adolescents with alpha 1-antitrypsin deficiency. Hepatology 1995; 22(2):514–517.
- Stockley RA, Luisetti M, Miravitlles M, Piitulainen E, Fernandez P, Alpha One International Registry g. Ongoing research in Europe: Alpha One International Registry (AIR) objectives and development. Eur Respir J. 2007;29(3): 582–586.
- Stolk J, Stockley RA, Stoel BC, et al. Randomized controlled trial for emphysema with a selective agonist of the gamma type retinoic acid receptor. Eur Respir J 2012.
- Chapman KR, Burdon JGW, Piitulainen E, et al. IV Alpha1 Antitrypsin (A1AT) preserves lung density in homozygous alpha1 antitrypsin deficiency (A1ATD); A randomized, placebo-controlled trial. C20. Late breaking abstracts in clinical trials. American Thoracic Society, International Conference Abstracts 2013; A6069–A6069.
- Brode SK, Ling SC, Chapman KR. Alpha-1 antitrypsin deficiency: a commonly overlooked cause of lung disease. Canadian Medical Association Journal 2012; 184(12):1365–1371.