
Abstract
The study of rare diseases is compromised by its rarity. The establishment of national and international registries can overcome many of the problems and be used for many monogenetic conditions with relatively consistent outcomes and thus lead to a consistency of clinical management by centres of excellence.
However, in Alpha-1 antitrypsin deficiency (AATD), the outcome is highly variable in terms of the organ(s) most affected and the diversity of disease penetration and progression. This creates the added difficulty of understanding the disease sufficiently to monitor and advise the patients to the standard required and importantly design and deliver clinical trials that address the many facets of the disease.
The development of research registries and centres of excellence provides the necessary expertise and data to further the understanding and management of diseases like AATD though with significant cost implications. The ADAPT programme was established in 1996 with extensive core funding to enable patients to be seen from all regions of the United Kingdom as an addition to the National Health Service without appointment time constraints and with the purpose of collecting extensive state of the art demographics.
The model has proven to be highly productive providing new insights especially into the lung disease, generating and delivering on clinical trials and importantly establishing active patient groups and participation.
Introduction
Rare diseases remain rare to individual doctors and their clinical understanding of the nature, impact, progression and management can therefore remain limited. The establishment of registries and especially centres of excellence makes patients and data less rare to individuals working in those centres, thus facilitating the development of collective understanding of disease variations, impact on the patient and their lifestyle and importantly the interpretation of treatment effects and the design and delivery of clinical trials.
Many registries have been established over the years in countries throughout the world and largely lead by individuals with a special interest. Although such registries have enabled some understanding of the nature and prognosis of the clinical features of AATD, none has been established a priori to develop “state of the art” assessment of all the aspects of lung disease and its impact, to determine the best way to monitor patients, how this could predict prognosis and importantly patient quality of life. Such basic data would be crucial in the design and delivery of interventional studies. In addition with the emerging interest in biomarkers and gene analyses there was also a need to establish and extensive biobank. However the success of such biobanks is intimately dependant upon the extent and accuracy of the clinical/physiological and radiological characterisation.
In 1996 following several meetings with the then Bayer Biologicals, University of Birmingham and Queen Elizabeth Hospital Birmingham developed an innovative and unique relationship to establish a UK registry for alpha-1-antitrypsin (AAT) deficiency with Professor E. Campbell (Salt Lake City, Utah) as co-lead investigators. Funding was initially established with a rolling contract to be reviewed before the last year of funding and renewed as appropriate providing longer term stability than is usual in grant supported research.
The development and validation of dried blood spot sampling for AAT, level, phenotype and genotyping was to be based in the USA and clinical work in the UK.
The programme was designed to answer several key questions:
1. What is the clinical phenotype of AATD?
2. How does this affect patients health and lifestyle?
3. How does the condition progress and how should it be monitored?
4. What feature/s, including exacerbations affect progression?
5. What is the role of CT densitometry scanning in understanding and monitoring disease?
6. How can we design and deliver a robust clinical trial of therapeutic efficacy?
7. Can we predict long-term outcome at baseline?
With these questions at the background we wished also to deliver a diagnostic and educational programme for patients and their families.
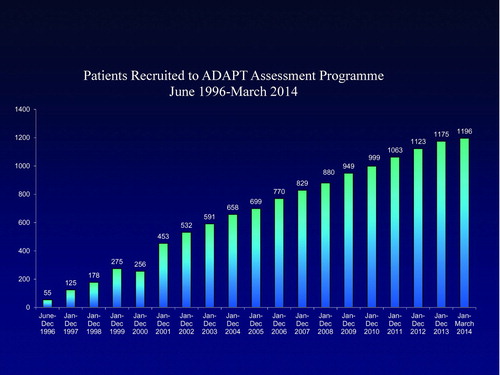
Recruitment started in 1997 following an announcement at the British Thoracic Society and has continued until recently. Figure shows the overall recruitment of Pi Z and SZ individuals as well as other rare severe deficiency variants. At the present time we have 930 ZZ and 135 SZ highly characterised patients on the database. This database has been central to the whole programme and was designed by both this author and Professor E. Campbell, based on the current state of the art concepts of COPD and its’ relevant features.
Figure 1. Recruitment to the ADAPT programme.
Patients have been seen generally once a year, at which time full demographics are collected including health status questionnaires, full post bronchodilator lung function testing, CT scanning as indicated clinically or for research purposes, routine bloods for haematology and liver function, research bloods for potential biomarkers, whole blood for DNA, sputum for quantitative culture and diary cards for monitoring exacerbations. More latterly vascular stiffness and periodontal disease have also been monitored.
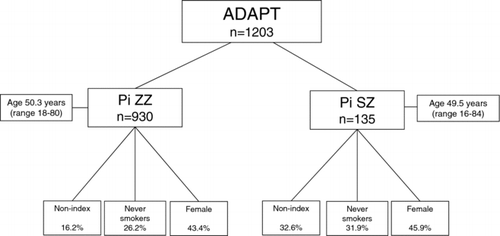
Other investigations such as CT follow-up of abnormalities, liver ultrasound and biopsies were carried out when indicated, in collaboration with the patient's local medical services. Figure shows the basic patient demographics of the data base for the PiZZ and SZ subjects. This model was also used to develop the Alpha-1-International Registry (AIR) involving over 20 countries although the latter registry was established with a minimal (though mandatory) rather than full data set.
Figure 2. The basic demographics for the 1,203 patients who have undergone at least one baseline assessment since the start of the registry in 1996 is shown just for the PiZZ and SZ genotypes.
Success of the Registry
Of note has been the long-term commitment of the patients who have attended on an annual basis from all parts of the UK indicating that the registry also addressed important issues from a patients perspective and at least the likely paucity of information available elsewhere. This in itself facilitated the development of an active patient group closely allied to the ADAPT registry. The importance and success of the registry can be monitored in the documented achievements towards delivering answers to the initial questions posed.
Clinical Phenotype
It was believed that AATD was predominantly associated with basal pan lobular emphysema presenting at a younger age than the non deficient COPD. Testing was widened to all AATD patients with a diagnosis of COPD indicating a wide variety of clinical phenotypes including those with predominantly apical emphysema (Citation1), a high prevalence of bronchiectasis (Citation2), variable exacerbations that impacted on health status (Citation3) and physiological decline (Citation4) and airways colonisation and inflammation (Citation5) that was similar to usual COPD although the inflammation was greater in AATD (Citation6).
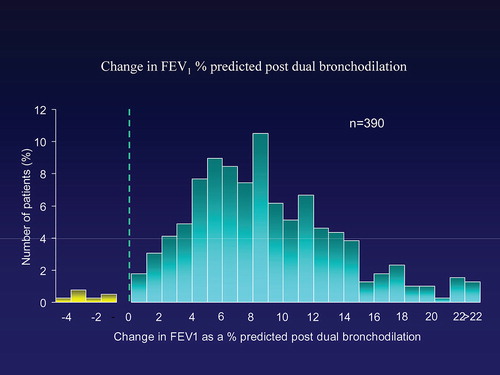
Spirometry indicated a fair degree of reversible airflow obstruction (Citation7) to combined bronchodilator therapy (Figure ) that also related to progression (Citation8) but although the obstruction related to defects in gas transfer, which reflects the emphysema pathology better than spirometry, there was discordance with some patients only having abnormal spirometry and others with gas transfer defects (Citation9). This reflects the distribution of the emphysema and not its presence (Citation1,Citation10). This discordance of physiology was most noted in siblings where there was no correlation between pairs for spirometry but where there was for gas transfer and upper zone emphysema (Citation11).
Figure 3. Bronchodilator reversibility to combined beta2agonist and anticholinergic agent is shown for the first 390 patients (PiZZ) as the proportion showing each% increase in FEV1.
CT densitometry
From the outset a major objective was to evaluate CT densitometry for the assessment and monitoring of disease as this is the most direct in vivo measure of emphysema. These data confirmed that CT densitometry correlated well with health status, lung function (especially gas transfer) and exercise capacity (Citation12). In addition CT densitometry was shown to influence symptomatology and recovery from exacerbations and to be the best independent predictor of mortality (Citation13). This data placed CT densitometry above routine spirometry in the monitoring of disease progression (specifically the emphysema component) leading to it becoming an accepted outcome measure in trials of augmentation therapy (Citation14,15). In addition the distribution of the emphysema on CT scan explained (at least in part) some of the physiological discordance between spirometry and gas transfer (Citation1).
Monitoring of progression/natural history
The extensive registry permits cross-sectional and longitudinal assessment of the disease characteristics and their progression. The inclusion of both index and non-index patients, siblings, smokers and never smokers together with extensive demographics provides a unique opportunity to study the phenotypic variations and factors that influence progression.
Retrograde analysis of data from the never smokers enables a snapshot of the natural history to be obtained. The data indicates that conventional spirometry only starts to deviate from normality in this patient group in the mid 40s (Citation16) whereas tests of gas transfer start to deviate from normal in the late 20s together with CT densitometry (Citation16). This suggests that early in the disease, monitoring of gas transfer is more likely to identify subjects who are deteriorating earlier than conventional spirometry. This data is supported by the real-time monitoring of patients in the Swedish registry identified at birth. Now in their 40s, spirometry remains normal whereas the average gas transfer is now approximately 85% predicted with some now outside the normal range (Citation17).
Longitudinal studies in the ADAPT registry have demonstrated an effect of exacerbations (Citation8) and bronchodilator reversibility on progression of COPD as defined by spirometry. However progression is far from certain even in those who present with established physiological impairment. On average CT densitometry, spirometry and gas transfer progress, although this may not occur for individuals. At the severe end of the disease spectrum FEV1 seems to stabilise (Citation4) perhaps reflecting a survivor effect. However in these subjects the decline in gas transfer is greatest perhaps reflecting progression of the emphysema from the lower to upper zones. This clearly has implications for augmentation therapy which is usually dictated by initial spirometry (Citation18). The ability to monitor patients closely (particularly in a largely untreated (no augmentation therapy available in the UK population) raising the issue that such therapy should be individualised and depend on factors other than FEV1 (Citation19).
Patient management
Registry data provides clinical experience in monitoring and prognosis that assists realistic advice on outlook for patients. Within the ADAPT registry of approximately 1200 deficient patients over up to 18 years it has been possible to reassure patients about the likelihood of liver failure. Sensitive tests of the liver function such as Gamma GT can be abnormal in about 20% of patients who do not drink or drink within government guidelines but in up to 40% of patients who exceed these consumption guidelines (Citation20). In this publication the data indicated that Gamma GT was inversely related to the FEV1, was increased in subjects with chronic bronchitis and was higher in patients who died from non-liver causes. These data suggested that the lung itself was a significant source of plasma Gamma GT (Citation20).
However, abnormalities of other routine liver function tests are unusual and even when deteriorating may not reflect liver failure. In our registry we encountered 5 such patients and in 2 the cause was gall stones and only 3 required liver transplantation. More recently a single patient with an unsuspected primary hepatocellular carcinoma was identified as part of a study of the use of routine liver scans in patient assessment. The Alpha fetoprotein level was also raised suggesting that, even though uncommon in our patient cohort, this biomarker should perhaps be also measured regularly to identify such patients.
This national data with clinical experience and observation provides robust information for reassuring individual patients who have been recruited from a respiratory cohort and their relatives. The experience of patients and relatives recruited because of presentation with liver abnormalities remains largely unknown and establishing such a registry with extensive lung assessment should be a priority.
Registry data helps provide supportive evidence of the likely benefit and cost effectiveness of augmentation therapy. The ADAPT registry provides detailed information about patients that can be used in several ways:
1. Patient selection for investigative studies. Patients with chronic bronchitis have been ideal for studies of the increased inflammation in the airways (Citation6) and the beneficial anti-inflammatory effect of augmentation (Citation21).
2. Deep knowledge of the patient provides the optimal way to identify patients that fulfil the stringent entry criteria for clinical trials and ADAPT has provided substantial numbers for augmentation trials (Citation15), alveolar repair studies (Citation22) and inhaled AAT studies (Citation23).
3. Detailed knowledge of the patients and natural history of progression can be used in conjunction with other robust registry data to identify the potential benefit of augmentation (Citation24) and health economics if required. In addition increased prevalence of other unknown co-morbidities can be determined with reasonable confidence (Citation25).
4. The interest of registries and the collaboration between the clinicians, researchers and patients generates strong patient support groups such as the Alpha-1 Alliance UK. These groups ensure continued understanding of patient impact and aims, as well as strong support for other affected individuals. The registry has led to regular patient/clinician symposia and clearer understanding between the aims of both groups leading to more relevant patient orientated research programs.
5. The deep phenotyping of registry patients has proven essential in the understanding of genetic modifiers and the ADAPT database has not only identified some (Citation26.Citation27) but also provides a replication resource for similar studies in other cohorts that is an essential component of such studies.
6. The deep phenotyping and longitudinal follow up has enabled the development and study of pathophysiological biomarkers for both cross-sectional associations and longitudinal ones. Many of the clinical associations such as reversibility of airflow obstruction, exacerbations and Gamma GT have been mentioned above. More recently an in vivo footprint of the generally accepted key proteinase (neutrophil elastase) has been studied. This assay is dependant of the activity of neutrophil elastase on fibrinogen prior to its inhibition and hence a reflection of its activity in the tissues. The Aa val 360 cleavage peptide shows good cross-sectional relationships to lung radiological and physiological measures of Emphysema as well as response to augmentation therapy (Citation28). However baseline values only relate to emphysema progression early in the disease suggesting that as emphysema progresses the inflammatory events become more complex (Citation29). The implications are that early treatment with augmentation may be more effective than late intervention and this may also explain the lack of effect of augmentation therapy on the emphysema progression in the mid and upper zones despite and effect in the lower zones seen in the EXACTLE study (Citation30). The results are consistent with a more complex process than a simple elastase/anti-elastase imbalance in regions other than the lower zones of the lung.
7. ADAPT has not only proven directly important for the design and implementation of clinical trials but is also an important International resource. The extensive data base enables patients to be selected to closely match patients with such data available in other registries. This alone can provide comparisons of outcomes between health care services in different countries and importantly at present the natural history of patients who do not currently have access to augmentation therapy. This provides observational data to assess health economics and progression and survival data that can supplement formal clinical trials in evaluation of the benefits of intervention therapies.
8. Finally ADAPT has provided and insight into the complexities of what has long been believed to be a simple monogenetic disease and its’ monitoring. Indeed many of the lessons learnt and in particular the development of CT densitometry has now become central to ongoing studies in usual COPD and its’ outcomes.
Summary
This unique industry/academic registry has led to a much clearer understanding of AATD patients, the disease, it progression and management. Regular 6 monthly meetings between experts in the pharmaceutical group and the academic researchers has led to a truly translational research programme with clearer understanding of many aspects of health care delivery including economics and trial design based on relevance to patients and health care providers as well as licensing authorities. This initial and unique programme has been continued by Talecris and until recently by Grifols without whom this registry and its output would not have existed.
Establishment of the registry has enabled further research funding to be obtained from MRC, NIHR, West Midlands Chest Fund, BBC CLRN, the Alpha-1 Foundation, EU Grants from FP5 and FP7 and CSL Behring thus providing added value to the core funding provided initially by Bayer Biologicals. ADAPT remains the most comprehensive and unique registry with which the natural history of the disease can still be studied.
ADAPT has trained and employed over 30 research fellows and post doctoral scientists. Our gratitude to the Bayer Biologicals team that had the vision and trust to establish the programme including K. Allenby, David Spencer (Bayer Biologicals), John Humphries, mark Forshag and Amy McGrath (Talecris) and more latterly Les Garlinghouse and Angela Davies (Grifols) cannot be underestimated nor can that of the UK AATD patients.
Declaration of Interest Statement
RAS has acted as adviser to CSL Behringer, Grifols, Kamada and Baxter, and participated in clinical trials of treatment in AAT deficiency funded by Talecris and Kamada. Received unrestricted grant funding, lecture fees and travel from Grifols and CSL Behring.
The author is responsible for the content and writing of the paper.
References
- Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in α-1-antitrypsin deficiency influences lung function impairment. Amer J Respir Crit Care Med 2004; 170(11):1172–1178.
- Needham M, Stockley RA. Exacerbations in α-1-antitrypsin deficiency. Eur Respir J 2005; 25:992–1000.
- Dawkins PA, Dawkins CL, Wood AM, et al. Rate of progression of lung function impairment in alpha-1-antitrypsin deficiency. Eur Respir J 2009; 33:1338–1344.
- Stockley RA, Hill AT, Hill SL, Campbell EJ. Bronchial inflammation. Its relationship to colonising microbial load and α1-antitrypsin deficiency. Chest 2000; 117(suppl):291S–293S.
- Hill AT, Bayley DL, Campbell EJ, et al. Airways inflammation in chronic bronchitis: the effects of smoking and α1-antitrypsin deficiency. Eur Respir J 2000; 15:886–890.
- Dowson LJ, Guest PJ, Hill SL, et al. High-resolution computed tomography scanning in α1-antitrypsin deficiency: relationship to lung function and health status. Eur Respir J 2001; 17:1097–1104.
- Dowson LJ, Guest PJ, Stockley RA. Longitudinal changes in physiological, radiological, and health status measurements in a1-antitrypsin deficiency and factors associated with decline. Am J Respir Crit Care Med 2001; 164:1805–1809.
- Ward H, Turner AM, Stockley RA. Spirometric and gas transfer discordance in alpha-1-antitrypsin deficiency; patient characteristics and progression. Chest 2014; 145(6):1316–1324.
- Holme J, Stockley RA. Radiologic and clinical features of COPD patients with discordant pulmonary physiology: Lessons from α-1-antitrypsin deficiency. Chest 2007; 132:909–915.
- Wood AM, Needham M, Simmonds MJ, et al. Phenotypic differences in alpha 1 antitrypsin deficient sibling pairs may relate to genetic variation. J COPD 2008; 5:353–359.
- Parr DG, Guest PG, Reynolds JH, et al. Prevalence and impact of bronchiectasis in α1-antitrypsin deficiency. Amer J Respir Crit Care Med 2007; 176:1215–1221.
- Dowson LJ, Guest PJ, Hill SL, et al. High-resolution computed tomography scanning in α1-antitrypsin deficiency: relationship to lung function and health status. Eur Respir J 2001; 17:1097–1104.
- Vijayasaratha K, Stockley RA. Relationship between frequency, length, and treatment outcome of exacerbations to baseline lung function and lung density in alpha-1 antitrypsin-deficient COPD. Inter J COPD 2012; 7:789–796.
- Dirksen A, Dijkman JH, Madsen F, et al. A randomised clinicl trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160(5 Pt 1):1468–1472.
- Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha-1-antitrypsin deficiency. Eur Respir J 2009; 33:1345–1353.
- Holme J, Stockley JA, Stockley RA. Age related development of respiratory abnormalities in non-index α−1 antitrypsin deficient studies Respir Med 2013; 107:387–393.
- Piitulainen E, Montero LC, Nystedt-Düzakin M, Stoel BC, Sveger T, Wollmer P, Tanash HA, Diaz S. Lung function and CT densitometry in subjects with alpha-1-antitrypsin deficiency and healthy controls at 35 years of age. COPD 2014; Oct 3 Epub ahead of print.
- American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. American Thoracic Society; European Respiratory Society. Am J Respir Crit Care Med 2003; 168(7):818–900.
- Stockley RA, Miravitlles M, Vogelmeier C. Augmentation therapy for alpha-1 antitrypsin deficiency: towards a personalised approach. Orphanet J Rare Dis 2013; 8:149.
- Holme J, Dawkins PA, Stockley EK, et al. Studies of gamma-glutamyl 1 transferase in alpha-1 antitrypsin deficiency. COPD 2010; 7(20):126–132.
- Stockley RA, Bayley DL, Unsal I, Dowson L. The effect of augmentation therapy on bronchial inflammation in α1-antitrypsin deficiency. Am J Respir Crit Care Med 2002; 165:1494–1498.
- Stolk J, Cooper BG, Stoel B, et al. Retinoid treatment of emphysema in patients on the Alpha-1 International Regsitry. The REPAIR study: study design, methodology and quality control of study assessments. Ther Adv Respir Dis 2010; 4(6):319–332.
- International study evaluating the safety and efficacy of inhaled human alpha-1 antitrypsin (AAT) in alpha-1 antitrypsin deficient patients with emphysema. 2010 Clinical Trials Gov NCT01217671.
- Chapman KR, Stockley RA, Dawkins C, et al. Augmentation therapy for alpha-1 antitrypsin deficiency: A meta-analysis. J COPD 2009; 6(3):177–184.
- Stone H, Pye A, Stockley RA. Disease associations in alpha-1-antitrypsin deficiency. Respir Med 2014; 108:338–343.
- Wood AM, Simmonds MJ, Bayley DL, et al. The TNF alpha gene relates to clinical phenotype in alpha-1-antitrypsin deficiency. Respir Res 2008; 9(52):1–8.
- McAloon CJ, Wood AM, Gough SC, Stockley RA. Matrix metalloprotease polymorphisms are associated with gas transfer in alpha 1 antitrypsin deficiency. Ther Adv Respir Dis 2009; 3:23–30.
- Carter RI, Mumford RA, Treonze KM, et al. The fibrinogen cleavage product Aa-Val360, a specific marker of neutrophil elastase activity in vivo. Thorax 2011; 66:686–691.
- Carter R, Ungurs M, Pillai A, et al. The relationship of the fibrinogen cleavage biomarker Aα-Val 360 with disease severity and activity in lpha-1-antitrypsin deficiency. Chest 2015; in press.
- Parr DG, Dirksen A, Piitulainen E, et al. Exploring the optimum approach to the use of CT densitometry in a randomised placebo-controlled study augmentation therapy in alpha-1-antitrypsin deficiency. Respir Res 2009; 10:75.