Abstract
The receptor protein tyrosine phosphatase T PTPρ is the most frequently mutated tyrosine phosphatase in human cancer. PTPρ mediates homophilic cell-cell aggregation. In its extracellular region, PTPρ has cell adhesion molecule–like motifs, including a MAM domain, an immunoglobulin domain, and four fibronectin type III (FNIII) repeats. Tumor-derived mutations have been identified in all of these extracellular domains. Previously, the authors determined that tumor-derived mutations in the MAM and immunoglobulin domains of PTPρ reduce homophilic cell-cell aggregation. In this paper, the authors describe experiments in which the contribution of the FNIII repeats to PTPρ-mediated cell-cell adhesion was evaluated. The results demonstrate that deletion of the FNIII repeats of PTPρ result in defective cell-cell aggregation. Furthermore, all of the tumor-derived mutations in the FNIII repeats of PTPρ also disrupt cell-cell aggregation. These results further support the hypothesis that mutational inactivation of PTPρ may lead to cancer progression by disrupting cell-cell adhesion.
INTRODUCTION
Cancer progression and metastasis is in part influenced by changes in cell adhesion and tyrosine phosphorylation mediated by cell adhesion molecules (CAMs) (CitationPaschos et al. 2009; CitationWheelock et al. 2008), as well as tyrosine kinases and tyrosine phosphatases (CitationFreiss and Vignon 2004; CitationOstman et al. 2006). Receptor protein tyrosine phosphatases (RPTPs) comprise a family of proteins that have extracellular CAM domains coupled to cytoplasmic tyrosine phosphatase domains, and thus can impact cancer progression by regulating both cell adhesion and tyrosine phosphorylation (CitationBrady-Kalnay 2001; CitationOstman et al. 2006).
The type IIB PTPμ-like subfamily of RPTPs includes PTPμ, PTPρ, PTPκ, and PCP-2 (PTPλ) (CitationAricescu et al. 2008; CitationBrady-Kalnay 2001). The extracellular segment of the PTPμ-like RPTPs contain motifs found in CAMs, including a MAM domain, an immunoglobulin (Ig) domain, and four fibronectin type III (FNIII) repeats (CitationAricescu et al. 2008; CitationBrady-Kalnay 2001; CitationOstman et al. 2006). PTPμ subfamily members bind homophilically (i.e., the “ligand” for PTPμ is an identical PTPμ molecule on an adjacent cell) (CitationBrady-Kalnay et al. 1993; CitationCheng et al. 1997; CitationGebbink et al. 1993; CitationSap et al. 1994; CitationYu et al. 2008; CitationZondag et al. 1995). The Ig domain of PTPμ is responsible for promoting homophilic interactions (CitationBrady-Kalnay and Tonks 1994), and proper cell surface localization (CitationDel Vecchio and Tonks 2005). However, both the MAM and Ig domains as well as the first two FNIII repeats are required for efficient cell-cell adhesion of PTPμ (CitationAricescu et al. 2006, Citation2007, Citation2008; CitationBrady-Kalnay and Tonks 1994; CitationCismasiu et al. 2004; CitationZondag et al. 1995). PTPρ and PTPκ also mediate homophilic cell-cell adhesion (CitationSap et al. 1994; CitationYu et al. 2008). However, PCP2 has not yet been demonstrated to mediate cell-cell adhesion or aggregation, although in vitro binding studies indicate it can bind homophilically (CitationCheng et al. 1997).
All type IIB RPTPs are potential tumor suppressor genes (CitationBurgoyne et al. 2009a; CitationFlavell et al. 2008; CitationMcArdle et al. 2001; CitationYan et al. 2006). Notably, PTPρ is the most frequently mutated phosphatase gene in human cancers, including colon, lung, skin, and stomach cancers (CitationForbes et al. 2008; CitationWang et al. 2004). Defective adhesion mediated by PTPρ may be at least one mechanism of cancer progression. In fact, cancer derived mutations in the MAM and Ig domains of PTPρ are defective in mediating cell-cell aggregation (CitationYu et al. 2008).
In addition to the tumor-derived mutations in the MAM and Ig domains of PTPρ previously described (CitationWang et al. 2004; CitationYu et al. 2008), the majority of extracellular domain mutations are located in the FNIII repeats of PTPρ (CitationWang et al. 2004). Given the importance of FNIII repeats in PTPμ-mediated adhesion and aggregation (CitationAricescu et al. 2007), we tested the contribution of the FNIII repeats of PTPρ to PTPρ-mediated adhesion. We determined that deletion constructs where each of the extracellular domains of PTPρ is removed, including the FNIII repeats, are defective in mediating cell-cell aggregation of Sf9 cells. Furthermore, when we engineered the cancer-derived mutations in the FNIII repeats of these PTPρ mutant proteins, they also result in defective cell-cell aggregation. These results demonstrate the importance of the FNIII repeats in contributing to proper cell-cell adhesion and suggest that adhesion mediated by PTPρ is likely important for the tumor suppressor activity of PTPρ.
MATERIALS AND METHODS
Baculovirus Generation
Baculoviruses were generated by transfecting cells with the BaculoGold Linearized Baculovirus DNA (BD Biosciences, San Diego, CA) according to the manufacturer’s protocol. The baculovirus for wild-type PTPρ was previously described (CitationYu et al. 2008).
Sf9 Cell Culture
Insect Sf9 cells from (BD Biosciences) were maintained at 27°C in Graces Insect Medium (BD Biosciences). The medium was supplemented with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT) and gentamicin (MP Biomedicals, Solon, OH). Viruses were added to Sf9 cell medium to induce the expression of proteins of interest. Cells were harvested 30 h postinfection for further analysis.
Generation of PTPRT Deletion Constructs
The PTPρ-ΔMAM, PTPρ-ΔIg, PTPρ-ΔFNIII-1, PTPρ-ΔFNIII-2, PTPρ-ΔFNIII-3, and PTPρ-ΔFNIII-4 constructs were generated by fusion polymerase chain reaction (PCR) using the full-length PTPRT pVL1393-V5-His vector as the template. The primers used for the constructs were as follows:
PTPρ-ΔMAM
5′-GAAGATCTATGGCGAGCCTCGCCGCG-3′
5′-CCGGAATTCGGATCCTGGGGCGCTCTGAGCCCGGG-3′
PTPρ-ΔIg
5′-GCGTGGGAGGCTCTCTGCATGGATGAGCAAGGAC-3′
5′-CAATGGGTCTTGCTCATCCATGCAGAGAGCCTCCCACGCCCATTG-3′
PTPρ-ΔFNIII-1
5′-CTGCACACTTGGTCCTGGTTTTCACGATCAGCTCCGCGTA-3′
5′-TACGCGGAGCGTCGTGAAAACCAGGACCAAGTGTGCAG-3′
PTPρ-ΔFNIII-2
5′-CCGCCAGGGCCTCCCCTCACCGAGGAAGACGTTCCAGGAG-3′
5′-CTCCTGGAACGTCTTCCTCGGTGAGGGGAGGCCCTGGCGG-3′
PTPρ-ΔFNIII-3
5′-GAGGAGCTGGTGGTGCAGACTAAAATTTCAGCTCCATCCA-3′
5′-TGGATGGAGCTGAAATTTTAGTCTGCACCACCAGCTCCTC-3′
PTPρ-ΔFNIII-4
5′-GTCACCACTCGGATTGCCACCGGTGCCTCCACCCAGAATT-3′
5′-AATTCTGGGTGGAGGCACCGGTGGCAATCCGAGTGGTGAC-3′
Immunoblotting
Sf9 cells were lysed in a standard lysis buffer (20 mM Tris·HCl, pH 7.5, 1% Triton X-100, 150 mM sodium chloride, 2 mM benzamidine, 2.5 μg/ml aprotinin, 2.5 μg/ml leupeptin, and 0.5 μg/ml pepstatin). Samples were solubilized in 2× sodium dodecyl sulfate (SDS) loading buffer and separated by SDS-polyacrylamide gel electrophoresis (PAGE). Samples were transferred to a nitrocellulose membrane (Whatman, Piscataway, NJ) and immunoblotted with SK18 (CitationBrady-Kalnay et al. 1993), which cross-reacts with PTPρ.
Cell Surface Expression of PTPρ
Cell surface expression of PTPρ protein in baculovirus-infected Sf9 cells was determined using a modified biotin-avidin reaction with the Cell Surface Protein Isolation Kit (catalog no. 89991; Pierce, Rockford, IL), as previously described (CitationYu et al. 2008). Briefly, Sf9 cell medium was removed and the flasks were incubated with rocking for 30 min at 4°C in 5ml of 0.25 mg/ml Sulfo-NHS-SS-Biotin in 1× phosphate-buffered saline (PBS). The cells were mechanically removed from the flask, centrifuged, washed, and lysed according to the manufacturer’s protocols. To isolate cell surface expressed PTPρ, the cell lysate was added to a NeutrAvidin Gel column and incubated for 60 min at room temperature (RT) with rocking prior to washing and eluting proteins with 200 µl of sample buffer containing a final concentration of 55 mM dithiothreitol (DTT) for 60 min at RT. The flow through was run on 6% SDS-PAGE gels and transferred to nitrocellulose. Total cell surface protein levels were normalized by immunoblotting cell lysates with an anti-actin antibody (JLA20). The normalized cell surface protein was run on 6% SDS-PAGE gels, transferred to nitrocellulose, and immunoblotted with antibodies to PTPµ that also recognized PTPρ (SK18), followed by secondary antibody conjugated to horseradish peroxidase (HRP). The HRP signal was detected using a Fluor-S MAX MultiImager (Bio-Rad Laboratories, Hercules, CA).
Aggregation Assays
Sf9 cells were harvested by trituration. The cell suspensions were added to glass scintillation vials and incubated at 27°C at 90 rpm in a gyratory shaker for 30 min. For a given condition, the cell suspension was transferred to a 100-mm Petri dish either prior to (0 time point) or 30 min after aggregation. Images were captured at 10× magnification with a Nikon TE-200 inverted fluorescent microscope (Tokyo, Japan). To quantify cell aggregates, pictures of four randomly selected fields were taken per dish. The area of each object (single cells and aggregates) was determined by the Metamorph software (Molecular Devices) using an autothreshold setting for light objects and appropriate size filters that allowed the counting of cells and aggregates but not debris. The four replicates were combined to yield average readings per condition. The percent aggregation was then calculated as the average aggregate area at 30 min minus the average aggregate area at 0 min divided by the average aggregate area at 30 min [(N30 – N0)/N30]. A minimum of three independent experiments was performed per condition. Statistical significance was determined in Microsoft Excel using Student’s t test. Error bars indicate standard error.
Site-Directed Mutagenesis
The I395V, Y412F, R453C, Q479E, S492F, N510K, T605M, V648G, A707T, and L708P tumor-derived mutations (CitationWang et al. 2004) were generated by fusion PCR using the full-length PTPRT pVL1393-V5-His vector as the template. The mutagenic primers used were as follows:
I395V
5′-TGGCCCACAGAACGTGGAAGTCGTAGACATCAGAGCC-3′
5′-CTTCCACGTTCTGTGGGCCA-3′
Y412F
5′-CAGTGGGAGCCCTTCGGCTTCGCGGTGACCCGCTGCCAT-3′
5′-AAGCCGAAGGGCTCCCACTG-3′
R453C
5′-CTACACCCTGCGAGGCCTGTGCCCCTTCATGACCATCCG-3′
5′-ACAGGCCTCGCAGGGTGTAG-3′
Q479E
5′-GAGCGAGGAGCTGGTGGTGGAGACTGAGGAAGACGTTCC-3′
5′-CCACCACCAGCTCCTCGCTC-3′
S492F
5′-TGTTCCTCTAGAATTCATCCAAGGGGGGCCCT TT-3′
5′-ATGAATTCTAGAGGAACA-3′
N510K
5′-TCCAGTGGAAACCTCCCAAGGAGACCAATGGGGTCATCA-3′
5′-CTTGGGAGGTTTCCACTGGA-3′
T605M
5′-CCATTGAATGAGACAGACATGACC ATCACAGTGATGCTG-3′
5′-ATGTCTGTCTCATTCAATGG-3′
V648G
5′-ATTATTGAGTGCTTTTC GGGGCCCGTGAGCTATCGGAAT-3′
5′-CCCGAAAAGCACTCAATAAT-3′
A707T
5′-CTACAGCATCTACTTCCAGACACTCAGCAAAGCCAATGG-3′
5′-TCTGGAAGTAGATGCTGTAG-3′
L708P
5′-AGCATCTACTTCCAGGCACCCAGCAAAGCCAATGGAGAG-3′
5′-GGTGCCTGGAAGTAGATGCT-3′
Mapping of PTPρ Mutations onto the PTPμ Crystal Structure
The sequence of PTPμ and PTPρ were aligned using CLUSTALW. Mutations probed in this study were mapped using this sequence alignment onto the PTPμ structure (PDB ID: 2V5Y). The figure was prepared utilizing PYMOL (Schrödinger). (http://pymol.org).
RESULTS
Deletions in the MAM, Ig, and FNIII Repeat Domains of PTPρ Result in Defective Cell-Cell Aggregation
To test whether a protein mediates cell-cell aggregation or adhesion, one has to express the protein of interest in the nonadhesive cells. Otherwise, one cannot measure the aggregation or adhesion of a single molecule in the sea of other adhesion molecules naturally expressed in adhesive cells. This is the main reason that the nonadhesive Sf9 or Drosophila S2 cells are widely used models for such studies. To our knowledge, nearly all cell lines of epithelial origin are adhesive cells and thus are not suitable for these studies. Introduction of putative adhesion molecules into nonadhesive Drosophila S2 insect cells has been used to demonstrate adhesive functions for fasciclin II, connectin, Dtrk, ARK, Neuroglian, and capricious molecules directly (CitationBellosta et al. 1995; CitationHortsch et al. 1995; CitationNose et al. 1992; CitationPulido et al. 1992; CitationShinza-Kameda et al. 2006; CitationSnow et al. 1989). In a similar approach, we and others demonstrated that the full-length form of PTPμ induced aggregation, via a homophilic binding mechanism, when expressed in nonadhesive Sf9 insect cells, which are derived from the Fall army worm Spodoptera frugiperda (CitationBrady-Kalnay et al. 1993; CitationGebbink et al. 1993; CitationZondag et al. 1995). We have also used this system for analyzing PTPRT/PTPρ (CitationYu et al. 2008). Examples of other cell adhesion molecules that have been studied in Sf9 cells include L1 (CitationGouveia et al. 2008), galectin-3 (CitationInohara and Raz 1995), and C-CAM1 (CitationPhan et al. 2001). Insect S2 and Sf9 cells are extremely useful systems to analyze adhesive mechanisms of cell adhesion molecules.
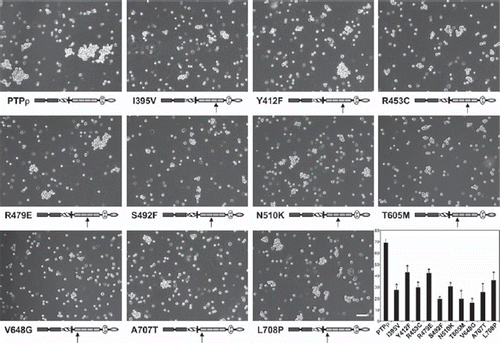
The MAM domain, Ig domain, and first two-FNIII repeats are essential for PTPμ cell-cell aggregation (CitationAricescu et al. 2006; CitationBrady-Kalnay and Tonks 1994; CitationZondag et al. 1995). To test whether these domains are the minimal essential regions required for PTPρ cell-cell aggregation, baculoviral PTPρ deletion constructs of the MAM domain, Ig domain, and each of the four individual FNIII repeats were made and expressed in nonadhesive Sf9 insect cells (). All the PTPρ deletion constructs were expressed and appropriately trafficked to the cell surface (). To test whether these domains contribute to PTPρ-dependent adhesion, Sf9 cells were evaluated for their ability to mediate cell-cell aggregation when expressing the deletion constructs. Aggregation assays with Sf9 cells expressing the deletion constructs were conducted alongside aggregation assays using Sf9 cells expressing wild-type PTPρ, which mediates cell-cell aggregation () and serves as a positive control (CitationYu et al. 2008). Deletion of any of the extracellular domains of PTPρ resulted in defective cell-cell aggregation compared to aggregation of wild-type PTPρ expressing Sf9 cells (). Deletion of the fourth FNIII repeat was the least severe, where ∼23% aggregation was observed. Deletion of the MAM domain, Ig domain, and FNIII repeat 1, 2, or 3 resulted in 5–16% aggregation. When the level of aggregation was quantified, we determined that all of the deletion constructs resulted in a large and statistically significant reduction in cell-cell aggregation (; p < .005). Together with our previous study, these results demonstrate that all of the extracellular domains of PTPρ are necessary for PTPρ-mediated cell-cell aggregation.
Figure 1. PTPρ deletion constructs are all expressed at the cell surface. Constructs containing deletions of one extracellular domain of PTPρ each were generated and expressed in Sf9 cells. Total cellular protein and cell surface protein were isolated and immunoblotted with the SK18 antibody.
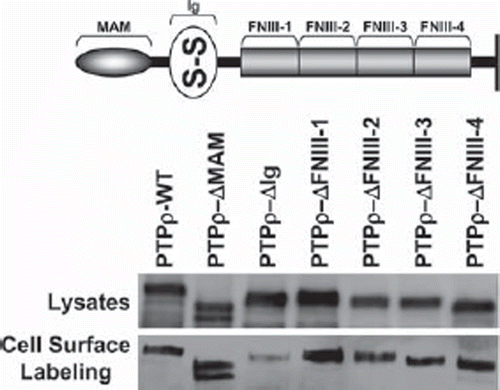
Figure 2. Deletion of any of the extracellular domains of PTPρ disrupts cell-cell aggregation. Sf9 cells expressing deletion constructs of the individual extracellular domains of PTPρ were allowed to aggregate for 30 minutes prior to imaging. Scale bar equals 100 μm. The percentage of Sf9 cell-cell aggregation for each deletion protein was calculated from a minimum of three experiments. An asterisk indicates a statistically significant reduction in aggregation (p < .005).

Tumor-Derived PTPρ Mutations in the FNIII Repeats Result in Defective Cell-Cell Aggregation
We next tested the effects of human tumor-derived mutations in the FNIII repeats of PTPρ on PTPρ-mediated cell-cell aggregation. We recently demonstrated that tumor-derived mutations in the MAM and Ig domains of PTPρ are defective in cell-cell aggregation using the Sf9 aggregation assay (CitationYu et al. 2008), and given the above results, we hypothesized that mutations in the FNIII domains would likewise result in defective aggregation. Ten human tumor-derived mutations are located in the second, third and fourth FNIII repeats of PTPρ (CitationWang et al. 2004). To test whether these mutations also affect cell-cell aggregation, we engineered the 10 human mutations by site-directed mutagenesis using the full-length PTPρ baculoviral construct as a template (see ). The 10 mutations are expressed in Sf9 cells at the expected molecular weight and at similar levels to full-length human PTPρ (). All of these mutants are expressed at the cell surface (). When these mutant PTPρ proteins were expressed in Sf9 cells and tested in aggregation assays, we observed that all of the mutant PTPρ proteins resulted in fewer and smaller aggregates than wild-type PTPρ (). When the level of aggregation was quantified, we determined that all of the FNIII mutations significantly reduced cell-cell aggregation (16–43%) compared to cells expressing wild-type PTPρ (68% aggregation, p < .008; ).
Figure 3. PTPρ proteins containing tumor-derived mutations in their FNIII domains are expressed at the cell surface. Ten mutations in the FNIII domains of PTPρ were generated by site-directed mutagenesis and expressed in Sf9 cells. Total cellular and cell surface proteins were isolated and analyzed by immunoblotting with the SK18 antibody.
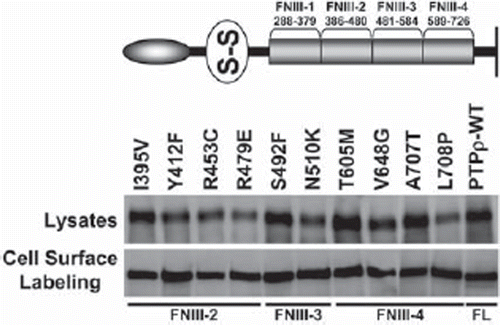
Figure 4. Tumor derived mutations in FNIII domains of PTPρ are defective in cell-cell aggregation. Sf9 cells expressing the mutated PTPρ proteins were allowed to aggregate for 30 min prior to imaging. Scale bar equals 100 μm. The percentage of Sf9 cell-cell aggregation for each mutant protein was calculated from a minimum of three experiments. An asterisk indicates a statistically significant reduction in aggregation (p < .008).
DISCUSSION
PTPRT/PTPρ is the most highly mutated tyrosine phosphatase in human colon carcinomas. We and others found that PTPRT/ PTPρ is also mutated in lung, gastric, and skin (melanoma) cancers (CitationForbes et al. 2008; CitationWang et al. 2004). The spectrum of mutations, which included nonsense mutations and frameshifts, suggested that these mutations were inactivating (CitationWang et al. 2004). Biochemical analyses demonstrated that missense mutations in the catalytic domains of PTPRT diminished its phosphatase activity and overexpression of PTPRT inhibited colon cancer cell growth. Moreover, we showed previously that the tumor-derived mutations in the MAM and Ig domains of PTPρ are defective in cell-cell adhesion (CitationYu et al. 2008). All these data suggest that PTPRT/PTPρ normally functions as a tumor suppressor gene. The experiments presented in this paper evaluate the contribution of the individual extracellular domains of PTPρ to PTPρ-mediated adhesion. We demonstrate that in addition to the MAM and Ig domains, the FNIII repeats are required for PTPρ-mediated adhesion. Importantly, we also show that PTPρ-mediated adhesion is impaired by human tumor-derived mutations in those FNIII repeats. These studies indicate that all the tumor-derived mutations located in the extracellular domain of PTPRT are loss-of-function mutations, thereby providing further evidence that PTPRT is a tumor suppressor. This notion is further supported by our recent study showing that PTPRT knockout mice are highly susceptible to the colon-specific carcinogen azoxymethane (AOM)-induced colon cancer (CitationZhao et al. 2010).
The entire extracellular domain of PTPμ is predicted to make a rigid conformation with extensive interfaces between each of the domains (CitationAricescu et al. 2006). The minimal adhesive unit for functional cell-cell aggregation is the MAM domain, the Ig domain, and the first two FNIII repeats (CitationAricescu et al. 2006; CitationBrady-Kalnay and Tonks 1994; CitationCismasiu et al. 2004; CitationZondag et al. 1995). However, the FNIII repeats appear to play a role in spacing the key homophilic interaction domain(s) a certain distance from the plasma membrane. Deletion of the FNIII repeats changed the intercellular spacing of two apposing membranes (CitationAricescu et al. 2007). Cell-cell aggregation assays of the FNIII deletion constructs were not performed in that study (CitationAricescu et al. 2007). The amino acid residues of the interfaces between the extracellular domains are highly conserved between members of the type IIB RPTP subfamily (CitationAricescu et al. 2006, Citation2007), therefore it is likely that the PTPρ adhesive interface resembles that of PTPμ. Together our data support the hypothesis that the essential regions for PTPρ-mediated adhesion resemble those of PTPμ.
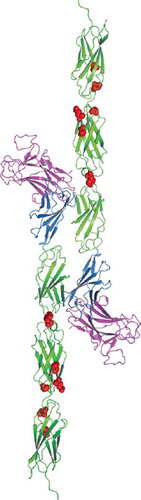
To gain insight into how the point mutations may alter the adhesive function of PTPρ, we modeled the human tumor derived PTPρ mutations onto the crystal structure of PTPμ (). The crystal structure of PTPμ includes only the first three FNIII repeats (CitationAricescu et al. 2007). Four of these mutations fall within the second FNIII (FNIII-2) repeat (I395V, Y412F, R453C, Q479E). It is important to note that FNIII-2 is required for efficient cell-cell adhesion (CitationAricescu et al. 2006). Two of the mutations fall in the third FNIII (FNIII-3) repeat (S492F, N510K). The mutation within the linker region between FNIII-1 and FNIII-2 (Y412F) could alter the flexibility or positioning between the two domains. The other mutations (I395V R453C, Q479E, S492F, N510K) appear to be clustered in surface exposed regions and/or near the linker regions between FNIII-2 and FNIII-3 that could alter flexibility, position of individual FNIII repeats, and/or protein-protein interactions. Although these mutations do not correspond to the adhesive interface hypothesized for PTPμ based upon a single static crystal structure (CitationAricescu et al. 2007), some of the mutations fall within the FNIII-2 repeat which is required for adhesion, and all of these surface modifications would likely alter either the three-dimensional topology and/or cis/trans interactions of PTPρ.
Figure 5. Homology model of tumor derived point mutations of PTPρ. The MAM domain (Magenta), Ig domain (Blue) and FNIII repeats (Green) and point mutations (Red) evaluated in this work were modeled onto the equivalent sites in the crystal structure of PTPμ (PDB ID: 2V5Y. The PTPρ mutations likely alter cell-cell aggregation either by altering the three-dimensional topology and/or cis/trans interactions of PTPρ.
Other type IIB RPTPs also function as tumor suppressor genes, by regulating adhesion and/or tyrosine phosphorylation of effector proteins. Expression of PTPμ is reduced in human glioblastomas (GBMs) and cell lines (CitationBurgoyne et al. 2009a). Reduction in full-length PTPμ expression with a concomitant increase in a proteolytically processed cytoplasmic fragment of PTPμ is observed in GBM cells and both are correlated with decreased adhesion and increased migration observed in the disease (CitationBurgoyne et al. 2009a, 2009b). Overexpression of the full-length form of PTPμ suppressed migration of GBM cells and reduces their survival (CitationBurgoyne et al. 2009b). Of note, at the same time that stable cell adhesion is reduced due to the loss of full-length PTPμ expression in GBM cells, a catalytically active cytoplasmic fragment of PTPμ is released and translocates to the nucleus (CitationBurgoyne et al. 2009b). Catalytic activity of this fragment is also necessary for cell migration and viability of GBM cells (CitationBurgoyne et al. 2009b). Therefore, both the tyrosine phosphatase activity of the cytoplasmic fragment of PTPμ and the loss of PTPμ-mediated adhesion may contribute to the invasiveness of GBM cells.
PTPκ is also implicated in tumor progression. PTPκ expression is reduced in melanoma cell lines and human tissue biopsies (CitationMcArdle et al. 2001), and in Hodgkin’s lymphoma cells (CitationFlavell et al. 2008). Overexpression of PTPκ in Hodgkin lymphoma cells reduces cellular proliferation and survival (CitationFlavell et al. 2008). Like PTPμ, PTPκ is proteolytically processed (CitationAnders et al. 2006), potentially as a result of aberrant glycosylation (CitationKim et al. 2006). It is not clear whether adhesion or catalytic activity of PTPκ is required for its tumor suppressor activity (CitationFlavell et al. 2008; CitationKim et al. 2006). Of note, the cleaved cytoplasmic domain fragment of PTPκ is catalytically active, translocates to the nucleus, and promotes β-catenin-mediated transcription (CitationAnders et al. 2006). Signaling via the PTPκ cytoplasmic fragment might augment the loss of stable cell-cell adhesion and may promote tumor progression.
Although speculative, it is interesting to postulate that tyrosine phosphatase activity and/or cleavage of PTPρ will also be an important element of its tumor suppressor activity, as has been demonstrated for PTPμ and PTPκ. Interestingly, we identified and validated STAT3 as a direct substrate of PTPRT (CitationZhang et al. 2007). STAT3 is latent transcription factor that translocates from the cytoplasm to the nucleus after being phosphorylated at the Y705 residue. It is possible that a cleaved intracellular fragment of PTPρ, which contains the phosphatase domains, translocates to nucleus and dephosphorylates STAT3. Besco and colleagues found that PTPRT associates with the adhesion molecule E-cadherin and its binding partner p120 catenin, which are PTPRT substrates (CitationBesco et al. 2006). p120 also translocates to the nucleus and associates with the Kaiso transcription factor (CitationDaniel and Reynolds 1999). Given the frequency and distribution of mutations in PTPRT/PTPρ in human colon and other cancers, a more thorough understanding of its biological function is warranted.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Anders L, Mertins P, Lammich S, Murgia M, Hartmann D, Saftig P, Haass C, Ullrich A (2006). Furin-, ADAM 10-, and gamma-secretase-mediated cleavage of a receptor tyrosine phosphatase and regulation of beta-catenin’s transcriptional activity. Mol Cell Biol. 26: 3917–3934.
- Aricescu AR, Hon WC, Siebold C, Lu W, van der Merwe PA, Jones EY (2006). Molecular analysis of receptor protein tyrosine phosphatase mu-mediated cell adhesion. EMBO J. 25: 701–712.
- Aricescu AR, Siebold C, Choudhuri K, Chang VT, Lu W, Davis SJ, van der Merwe PA, Jones EY (2007). Structure of a tyrosine phosphatase adhesive interaction reveals a spacer-clamp mechanism. Science. 317: 1217–1220.
- Aricescu AR, Siebold C, Jones EY (2008). Receptor protein tyrosine phosphatase mu: Measuring where to stick. Biochem Soc Trans. 36: 167–172.
- Bellosta P, Costa M, Lin DA, Basilico C (1995). The receptor tyrosine kinase ARK mediates cell aggregation by homophilic binding. Mol Cell Biol. 15: 614–625.
- Besco JA, Hooft van Huijsduijnen R, Frostholm A, Rotter A (2006). Intracellular substrates of brain-enriched receptor protein tyrosine phosphatase rho (RPTPrho/PTPRT). Brain Res. 1116: 50–57.
- Brady-Kalnay SM (2001). Protein tyrosine phosphatases. Cell Adhesion: Frontiers in Molecular Biology, Beckerle M. Oxford University Press, Oxford, UK. 217–258.
- Brady-Kalnay SM, Flint AJ, Tonks NK (1993). Homophilic binding of PTP mu, a receptor-type protein tyrosine phosphatase, can mediate cell-cell aggregation. J Cell Biol. 122: 961–972.
- Brady-Kalnay SM, Tonks NK (1994). Identification of the homophilic binding site of the receptor protein tyrosine phosphatase PTP mu. J Biol Chem. 269: 28472–28477.
- Burgoyne AM, Palomo JM, Phillips-Mason PJ, Burden-Gulley SM, Major DL, Zaremba A, Robinson S, Sloan AE, Vogelbaum MA, Miller RH, . (2009a). PTPmu suppresses glioma cell migration and dispersal. Neuro Oncol. 11: 767–778
- Burgoyne AM, Phillips-Mason PJ, Burden-Gulley SM, Robinson S, Sloan AE, Miller RH, Brady-Kalnay SM (2009b). Proteolytic cleavage of protein tyrosine phosphatase mu regulates glioblastoma cell migration. Cancer Res. 69: 6960–6968.
- Cheng J, Wu K, Armanini M, O’Rourke N, Dowbenko D, Lasky LA (1997). A novel protein-tyrosine phosphatase related to the homotypically adhering κ and μ receptors. J Biol Chem. 272: 7264–7277.
- Cismasiu VB, Denes SA, Reilander H, Michel H, Szedlacsek SE (2004). The MAM (meprin/A5-protein/PTPmu) domain is a homophilic binding site promoting the lateral dimerization of receptor-like protein-tyrosine phosphatase mu. J Biol Chem. 279: 26922–26931.
- Daniel JM, Reynolds AB (1999). The catenin p120(ctn) interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol Cell Biol. 19: 3614–3623.
- Del Vecchio RL, Tonks NK (2005). The conserved immunoglobulin domain controls the subcellular localization of the homophilic adhesion receptor protein-tyrosine phosphatase mu. J Biol Chem. 280: 1603–1612.
- Flavell JR, Baumforth KR, Wood VH, Davies GL, Wei W, Reynolds GM, Morgan S, Boyce A, Kelly GL, Young LS, . (2008). Down-regulation of the TGF-beta target gene, PTPRK, by the Epstein-Barr virus encoded EBNA1 contributes to the growth and survival of Hodgkin lymphoma cells. Blood. 111: 292–301.
- Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR (2008). The catalogue of somatic mutations in cancer (COSMIC). Current Protocols in Humum Genetics. New York: John Wiley & Sons, Chapter 10, Units 10, 11.
- Freiss G, Vignon F (2004). Protein tyrosine phosphatases and breast cancer. Crit Rev Oncol Hematol. 52: 9–17.
- Gebbink MF, Zondag GC, Wubbolts RW, Beijersbergen RL, van Etten I, Moolenaar WH (1993). Cell-cell adhesion mediated by a receptor-like protein tyrosine phosphatase. J Biol Chem. 268: 16101–16104.
- Gouveia RM, Gomes CM, Sousa M, Alves PM, Costa J (2008). Kinetic analysis of L1 homophilic interaction: Role of the first four immunoglobulin domains and implications on binding mechanism. J Biol Chem. 283: 28038–28047.
- Hortsch M, Wang YM, Marikar Y, Bieber AJ (1995). The cytoplasmic domain of the Drosophila cell adhesion molecule neuroglian is not essential for its homophilic adhesive properties in S2 cells. J Biol Chem. 270: 18809–18817.
- Inohara H, Raz A (1995). Functional evidence that cell surface galectin-3 mediates homotypic cell adhesion. Cancer Res. 55: 3267–3271.
- Kim YS, Kang HY, Kim JY, Oh S, Kim CH, Ryu CJ, Miyoshi E, Taniguchi N, Ko JH (2006). Identification of target proteins of N-acetylglucosaminyl transferase V in human colon cancer and implications of protein tyrosine phosphatase kappa in enhanced cancer cell migration. Proteomics. 6: 1187–1191.
- McArdle L, Rafferty M, Maelandsmo GM, Bergin O, Farr CJ, Dervan PA, O’Loughlin S, Herlyn M, Easty DJ (2001). Protein tyrosine phosphatase genes downregulated in melanoma. J Invest Dermatol. 117: 1255–1260.
- Nose A, Mahajan VB, Goodman CS (1992). Connectin: A homophilic cell adhesion molecule expressed on a subset of muscles and the motoneurons that innervate them in Drosophila. Cell. 70: 553–567.
- Ostman A, Hellberg C, Bohmer FD (2006). Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 6: 307–320.
- Paschos KA, Canovas D, Bird NC (2009). The role of cell adhesion molecules in the progression of colorectal cancer and the development of liver metastasis. Cell Signal. 21: 665–674.
- Phan D, Han E, Birrell G, Bonnal S, Duggan L, Esumi N, Gutstein H, Li R, Lopato S, Manogaran A, . (2001). Purification and characterization of human cell–cell adhesion molecule 1 (C-CAM1) expressed in insect cells. Protein Expr Purif. 21: 343–351.
- Pulido D, Campuzano S, Koda T, Modolell J, Barbacid M (1992). Dtrk, a Drosophila gene related to the trk family of neurotrophin receptors, encodes a novel class of neural cell adhesion molecule. EMBO J 11: 391–404.
- Sap J, Jiang YP, Friedlander D, Grumet M, Schlessinger J (1994). Receptor tyrosine phosphatase R-PTP-κ mediates homophilic binding. Mol Cell Biol. 14: 1–9.
- Shinza-Kameda M, Takasu E, Sakurai K, Hayashi S, Nose A (2006). Regulation of layer-specific targeting by reciprocal expression of a cell adhesion molecule, capricious. Neuron 49: 205–213.
- Snow PM, Bieber AJ, Goodman CS (1989). Fasciclin III: A novel homophilic adhesion molecule in Drosophila. Cell. 59: 313–323.
- Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, Ptak J, Silliman N, Peters BA, van der Heijden MS, . (2004). Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 304: 1164–1166.
- Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, Johnson KR (2008). Cadherin switching. J Cell Sci. 121: 727–735.
- Yan HX, Yang W, Zhang R, Chen L, Tang L, Zhai B, Liu SQ, Cao HF, Man XB, Wu HP, . (2006). Protein-tyrosine phosphatase PCP-2 inhibits beta-catenin signaling and increases E-cadherin-dependent cell adhesion. J Biol Chem. 281: 15423–15433.
- Yu J, Becka S, Zhang P, Zhang X, Brady-Kalnay SM, Wang Z (2008). Tumor-derived extracellular mutations of PTPRT /PTPrho are defective in cell adhesion. Mol Cancer Res. 6: 1106–1113.
- Zhang X, Guo A, Yu J, Possemato A, Chen Y, Zheng W, Polakiewicz RD, Kinzler KW, Vogelstein B, Velculescu VE, . (2007). Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc Natl Acad Sci U S A. 104: 4060–4064.
- Zhao Y, Zhang X, Guda K, Lawrence E, Sun Q, Watanabe T, Iwakura Y, Asano M, Wei L, Yang Z, . (2010). Identification and functional characterization of paxillin as a target of protein tyrosine phosphatase receptor T. Proc Natl Acad Sci U S A. 107:2592-2597.
- Zondag GC, Koningstein GM, Jiang YP, Sap J, Moolenaar WH, Gebbink MF (1995). Homophilic interactions mediated by receptor tyrosine phosphatases mu and kappa. A critical role for the novel extracellular MAM domain. J Biol Chem. 270: 14247–14250.