Abstract
Owing to increased obesity, non-alcoholic fatty liver disease (NAFLD) is now the most prevalent liver disease in the United States. NAFLD is considered a component of metabolic syndrome, a cluster of disorders that also includes diabetes mellitus, dyslipidemia, arteriosclerosis, and hypertension. Exposure to ambient air particulate matter with aerodynamic diameters < 2.5 μm (PM2.5) is a risk factor for arteriosclerosis and lung disease, but its effect on NAFLD is unknown. PM2.5 induces pulmonary dysfunction via Toll-like receptor (TLR) activation on alveolar macrophages. TLR activation of Kupffer cells, resident hepatic macrophages, and subsequent pro-inflammatory cytokine production have been shown to play a key role in NAFLD progression. We hypothesized that PM2.5 exposure is a significant risk factor for the progression of NAFLD. Thus, following exposure of male C57BL/6 mice fed high fat chow (HFC) to concentrated air particulate matter (CAPs) or filtered air for 6 weeks, progression of NAFLD was evaluated by standardized histological assessment of hepatic inflammation and fibrosis. In mice fed HFC, the hepatic inflammatory grade (3.00 ± 0.00 vs. 1.50 ± 0.71, P < 0.001) and fibrosis stage (1.00 ± 0.00 vs. 0.60 ± 0.52, P = 0.023) were both significantly higher in mice exposed to CAPs versus filtered air, respectively. Increased numbers of Kupffer cells contained PM in CAPs-exposed mice scores of (2.00 ± 0.94 vs. 0.20 ± 0.42, respectively, P < 0.001). PM exposure increased IL-6 secretion up to seven-fold in a dose-dependent manner by isolated wild-type but not TLR4−/− Kupffer cells (P < 0.050). In conclusion, ambient PM2.5 exposure may be a significant risk factor for NAFLD progression.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease in the United States (McCullough, Citation2004), but its pathogenesis is poorly understood. Indeed, the condition was not recognized as a cause of cirrhosis (end-stage liver disease) until fairly recently. NAFLD ranges from simple fatty liver (hepatic steatosis) to cirrhosis with intervening steatohepatitis characterized by progressive amounts of inflammation and fibrosis. It is unclear why steatohepatitis develops in only 10% of those with hepatic steatosis, but increased release of pro-inflammatory cytokines is likely essential (McCullough, Citation2006). Increased hepatic cytokine release appears to be related to Toll-like receptor (TLR) activation on Kupffer cells (Isogawa et al., Citation2005; Yohe et al., Citation2006). Identification of factors that induce hepatic pro-inflammatory cytokine production is an important area of research, since prevention of progression from simple steatosis to steatohepatitis may be sufficient to prevent the development of cirrhosis in NAFLD.
Epidemiological studies have demonstrated that exposure to airborne fine particulate matter (PM2.5) is positively associated with increases in the morbidity and mortality caused by cardiovascular disease (Mar et al., Citation2000; Schwartz et al., Citation2001; Sun et al., Citation2005) and pulmonary disease (Pope and Kanner, Citation1993; Sunyer et al., Citation2000). The main constituents of PM2.5 are organic carbon, elemental carbon, sulfates, and nitrates (Fuentes et al., Citation2006). Little data exists on the relationship between air particulate matter exposure and liver disease. Air pollution exposure related to toxic waste sites has been associated with an increased prevalence of autoimmune liver disease (Ala et al., Citation2006; Stanca et al., Citation2008). Two recent animal studies suggest that airborne pollutants may also play a role in the pathogenesis of NAFLD (Folkmann et al., Citation2007; Tomaru et al., Citation2007).
The mechanisms by which PM2.5 might affect liver disease are unclear. PM2.5 induces pulmonary damage at least partly through induction of cytokine release (Kumagai et al., Citation1997) that is dependent on TLR activation (Becker et al., Citation2005). A similar effect of PM2.5 in the liver is possible since activation of TLR4 on Kupffer cells results in the release of pro-inflammatory cytokines (Su et al., Citation2000; Uesugi et al., Citation2001). TLR4 signaling also enhances tumor-growth factor β responsiveness in hepatic stellate cells leading to their increased synthesis of collagen and fibrosis (Seki et al., Citation2007). Studies were performed in vivo and in vitro to determine whether air particulate matter exposure enhances hepatic inflammation in NAFLD and induces Kupffer cell activation.
Materials and methods
Mice
Six-week-old C57BL/6 male mice (Taconic Europe, Denmark) were enrolled and housed two to a cage in an Association for Assessment and Accreditation of Laboratory Animal Care–accredited animal housing facility. They were fed either with high fat chow (HFC) (n = 10; Adjusted Calories Diet, TD 88137; Harlan, Indianapolis, IN) or with normal chow (NC) (n = 10; Ralston Purina Co., Chicago, IL) during exposure to PM2.5 or filtered air. Assignments to HFC versus NC and PM2.5 versus filtered air were random. The Committees on Use and Care of Animals from New York University and Mount Sinai School of Medicine approved all experimental procedures.
For studies in vitro, cells were isolated from male wild-type (WT) and TLR4−/− C57BL/6 mice fed NC obtained from Dr. Maria T. Abreau (Adachi et al., Citation1998; Hoshino et al., Citation1999).
PM2.5 inhalation
Mice were exposed from October 2006 through December 2006 to concentrated PM2.5 (CAPs) composed of the northeastern regional background at the AJ Lanza Laboratory of the Department of Environmental Medicine of New York University (NYU), which is located within Sterling Forest State Park in Tuxedo, NY, 40 miles northwest of Manhattan, where the PM2.5 is largely attributable to long-range transport. Sterling Forest is a largely undeveloped woodland park. The NYU laboratory is located near the center of the park, on a relatively lightly traveled two-lane road that bisects the park. There are no large power generators or industrial operations within 20 miles of the site. The ambient fine PM in Sterling Forest is, therefore, representative of regional background PM2.5 aerosol of the megalopolis that extends from Virginia to Maine. The PM in Sterling Forest has been characterized previously (Maciejczyk et al., Citation2005).
The CAPs were collected and concentrated by a versatile aerosol concentration enrichment system (Sioutas et al., Citation2005), which was modified for long-term exposures (Chen and Nadziejko, Citation2005). The mice were exposed to PM2.5 (n = 10) at an average concentration of 85 μg/ m3 for 6 hr/d, 5 d/wk, for 6 wks. The filtered air (FA; n = 10) control mice in the experiment were exposed to an identical protocol with the exception that a high efficiency particulate air filter (HEPA; Pall Life Sciences, East Hills, NY) was positioned in the inlet valve to the exposure system to remove all of the PM2.5 from the air stream. For the measurement of CAPs in the exposure chambers, PM2.5 samples were collected on Teflon filters (Gelman “Teflon” 37 mm, 0.2 μm pore) and the filters were weighed before and after sampling in a temperature- and humidity-controlled weighing room. The long-term average exposure that the mice received over this period (15 μg/ m3) corresponds to the current EPA ambient air quality standard and the level of exposure for many U.S. residents. At the completion of the exposure period, mice were euthanized following anesthesia with ketamine/xylacine (100 mg/kg, intraperitoneally [IP]). Livers were then removed and fixed in 10% formalin.
Injection with PM
SRM 1469a, atmospheric particulate matter collected in the Washington, DC area in 1976–1977, was obtained from the National Institute of Standards and Technology (NIST; Gaithersburg, MD). SRM 1469a (500 μg) in phosphate-buffered saline (PBS; n = 3) or PBS alone (n = 3) was injected into the retinal vein of WT C57BL/6 male mice. After 24 hr, the mice were euthanized following anesthetization with ketamine/xylacine (100 mg/ kg, IP). Livers were removed and fixed in 10% formalin. Blinded, semi-quantitative scoring of the number of cells containing PM in ten high-power fields was conducted as follows: no cells = 0; one to two cells = 1; three to five cells = 2; over five cells = 3.
Histological staining
Formalin-fixed liver specimens were paraffin-embedded. Five-μm sections were prepared and stained either with hematoxylin and eosin (H&E) or with 0.1% Sirius red F3B (Sigma, St Louis, MO) in saturated picric acid. The H&E sections were scored for inflammation and fibrosis using the modified Brunt classification system for fatty liver disease (Brunt et al., Citation1999) and for the presence of particulate matter by a liver histopathologist blinded as to sample source. Relative fibrosis area (expressed as % of total liver area) was assessed by analyzing 40 Sirius red-stained liver sections per animal. Each field was acquired at 10X magnification and then analyzed with a computerized Bioquant morphometry system (Bioquant, Nashville, TN). Subtraction of the vascular luminal area from the total field area yielded the net field area (Horani et al., Citation2007). To evaluate the relative fibrosis area, the measured collagen area (red) was divided by the net field area and then multiplied by 100.
Immunohistochemistry and electron microscopy
Formalin-fixed liver sections underwent sequential dehydration in ethanol, peroxide blocking with 0.03% hydrogen peroxide, and antigen retrieval with citrate buffer (pH 7.6) in a microwave for 20 min. Sections were stained with horseradish peroxidase–conjugated primary antibody or isotype control antibody for 1 hr. DAB substrate (3,3′-diaminobenzidine; Vector, Burlingame, CA) was used for detection. Anti-mouse F4/80 (Abcam, Cambridge, MA) and isotype control antibody (Jackson Immunoresearch, West Grove, PA) were purchased for macrophage detection. Blinded, semi-quantitative scoring of the number of cells in 20 high-power fields with positive staining was conducted as follows: < 10 positive cells = 0; 10–49 cells = 1; 50–99 cells = 2; 100 or more cells = 3. Glutaraldehyde-fixed sections from PM-exposed (n = 3) and FA-exposed (n = 3) mice for electron microscopy were prepared, imaged, and scanned as described previously (Phelps et al., Citation1989; Sicari et al., Citation1999).
Cells
The RAW 264.7 murine macrophage cell line was a gift from Dr. J. Unkeless (Mount Sinai School of Medicine, New York, NY). Cell viability was measured using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) cell viability kit and following the manufacturer’s instructions (Biotum Inc., Hayward, CA). Kupffer cells were isolated from 2-month-old WT (n = 5) and TLR4−/− (n = 5) C57BL/6 mice. Following anesthetization with ketamine/xylacine, a medial laparotomy was performed and the portal vein was catheterized. Livers were perfused through the portal vein in situ with 10 mL of Hanks’ Balanced Salt Solution (SAFC Biosciences, Lenexa, KA) followed by 10 mL of 0.02% collagenase IV (Sigma). The liver was removed and digested for another 30 min in 0.02% collagenase IV. The digested liver was minced and strained through a steel mesh (70 μm). The dispersed liver cells were collected by centrifugation at 50 × g for 10 min at 4°C. Cells were washed twice in PBS. Kupffer cells were then isolated by Percoll (GE Healthcare, Pittsburg, PA) gradient centrifugation at room temperature. The cells were washed again in PBS twice. Cells were counted and viability was checked by Trypan blue exclusion (Smedsrod et al., Citation1985). Only isolations with > 95% cell viability were used. Cells were seeded in 24-well plates at 2 × 105 in RPMI with 10% heat-inactivated fetal bovine serum (FBS) for 72 hr before exposure to particulate matter. Stellate cells from WT and TLR4−/− C57BL/6 mice were immortalized as described previously (Guo et al., Citation2009).
Incubation of cells in vitro with PM
DMEM media supplemented with 10% heat-inactivated FBS, 1% l-glutamine, and 1% penicillin–streptomycin was used for all in vitro studies. All cells were cultured at 37°C and in a 5% CO2 atmosphere. Macrophages (RAW 264.7) were seeded at 8 × 105 cells/well; mouse stellate cells (WT and TLR4−/−) were seeded at 4 × 105 cells/well; and, primary Kupffer cells isolated from mice were seeded at 1 × 106 cells/well. All wells were at least 70% confluent after 24 hr. Media were completely replaced at 24 hr with media containing PM (SRM 1649a at 0–200 μg/mL). The doses of PM are based on prior studies of alveolar cells (Hetland et al., Citation2005; Imrich et al., Citation2007). TLR-blocking antibody (Ebioscience, San Diego, CA) or isotype control antibody (Jackson Immunoresearch, West Grove, PA) was added to specified wells at 20 μg/mL. The amount of bacterial lipopolysaccharide (LPS) contamination in SRM 1469a preparations was measured using the Chromogenic Limulus Amebocyte Lysate Test Kit (Cambrex, Walkersville, MD), according to the manufacturer’s instructions. LPS concentrations in SRM 1469a were < 125 pg/mL, well below the level known to stimulate macrophage cytokine secretion. In some experiments, conditioned media from macrophages exposed to particulate matter (0, 100, or 200 μg/mL) for 24 hr was employed. Stellate cells were then incubated in a 1:4 mixture of conditioned macrophage media and fresh media (final concentration of PM was 0, 20, or 40 μg/mL). Control stellate cells were likewise exposed to 0, 20, or 40 μg/mL of PM in fresh media. Cell culture supernatants and cells were retrieved at various timepoints, and mRNA and protein expression levels were assayed by qRT-PCR and enzyme-linked immunosorbent assay (according to manufacturer’s directions; R&D Systems, Minneapolis, MN), respectively. Measurements were performed in triplicate.
Statistical analysis
An unpaired Mann–Whitney U-test was used for non-parametric comparisons of histological staining. Fisher exact two-tailed test was used to compare the presence of hepatic PM in CAPs- versus FA-exposed mice. A one-way ANOVA test was used for parametric comparisons. A P-value < 0.050 was considered statistically significant for each test.
Results
Inhalation of PM2.5 increased hepatic inflammation and fibrosis
The livers of WT C57BL/6 mice exposed to either filtered air (FA) or concentrated air particles (CAPs) were examined histologically at the end of 6 wks. The average CAPs exposure that experimental mice received over this period (15 μg/m3) corresponds to the current EPA ambient air quality standard and the level of exposure for many U.S. residents. During exposure periods, the average CAPs concentration was 85 μg/m3 for the CAPs-exposed mice and 0 μg/m3 for the FA-exposed mice. In mice fed NC, no histological abnormalities were noted regardless of exposure to FA or CAPs ( and ). In contrast, increased hepatic steatosis was evident in both groups of mice fed HFC (). Lobular inflammation characterized by leukocyte infiltration appeared significantly increased along with hepatocyte ballooning and Mallory bodies in mice exposed to CAPs and fed HFC. Macrophage staining ( and ) was significantly increased in the CAPs-exposed mice compared with in the FA-exposed mice fed HFC (2.4 ± 1.1 vs. 1.1 ± 1.2 cells/hpf, P = 0.045). Bioquant analysis of Sirius red staining suggested a trend toward increased collagen staining ( and ) in CAPs-exposed versus FA-exposed mice (5.9 ± 2.7% vs. 4.5 ± 2.1%, respectively, P = 0.334). The collagen staining had the typical “chicken wire” pattern seen in human NAFLD.
Figure 1. Histological analysis of formalin-fixed sections from C57BL/6 mice exposed to FA or CAPs. H&E staining demonstrated that mice fed NC did not develop fatty livers or hepatic inflammation regardless of exposure to FA (A) or PM (B). Mice receiving HFC and exposed to FA had fatty livers with minimal hepatic inflammation (C); whereas CAPs-exposed mice on HFC developed fatty livers with increased hepatic inflammation (as noted by arrows) (D). F4/80 macrophage staining was less significant in HFC mice exposed to FA (E) versus CAPs (F). Sirius red staining of collagen deposition in mice fed HFC was slightly less in mice exposed to FA (G) versus CAPs (H). For each panel, a representative image is shown. Bars represent 10 μm.
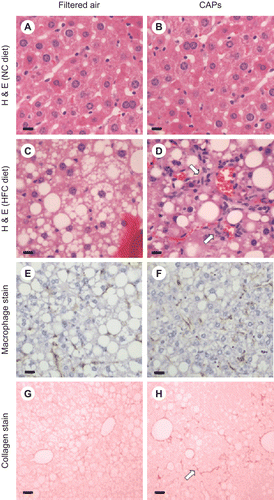
Liver sections were quantitatively scored in a blinded fashion using the modified Brunt scoring system to assess steatohepatitis (Brunt et al., Citation1999). CAPs exposure did not significantly alter the amount of hepatic fat compared to FA exposure for mice receiving HFC. However, the overall mean steatohepatitis grade (inflammation) was significantly (P < 0.001) higher in the CAPs- versus FA-exposed mice (). Increases in lobular inflammation, hepatocyte ballooning, and Mallory bodies were primarily responsible for the higher inflammatory grade in the CAPs-exposed mice. The mean fibrosis stage was also statistically significantly higher in the CAPs- versus FA-exposed mice (P = 0.023) after just 6 wks of exposure. Increased fibrosis typically follows increased inflammation in NAFLD (Jou et al., Citation2008). Similarly, the NAFLD activity score (Kleiner et al., Citation2005) based on steatosis, lobular inflammation, and hepatocyte ballooning was also significantly higher in the CAPs versus the FA group (7.10 ± 0.88 vs. 4.50 ± 1.90, respectively, P = 0.002).
Table 1. NAFLD grading and staging in C57BL/6 mice fed high fat chow.
PM localizes predominantly to Kupffer cells in the liver
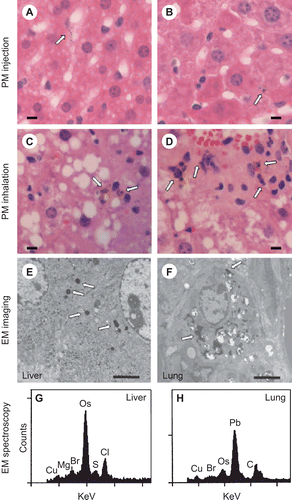
It is controversial whether PM reaches the liver following exposure to ambient CAPs. Before analyzing the liver PM content in the ambient CAPs- and FA-exposed mice, standard PM (SRM 1649a) suspended in PBS or PBS alone was injected intravenously into mice to determine whether circulating PM is taken up by the liver. Twenty-four hours after injection, H&E-stained liver sections were prepared for review. Dark intracytoplasmic PM of < 2 μm was detected only in the livers of PM injected mice (n = 3), mainly within Kupffer cells ( and ), and not in PBS-injected mice (n = 3) (data not shown). Larger particles were not observed in the livers of PM- or PBS-injected mice. Similar dark, intracytoplasmic PM (< 2 μm) was present in the livers of 10 out of 10 mice following ambient CAPs exposure ( and ) and only 2 out of 10 mice exposed to FA (data not shown) after a blinded analysis of H&E-stained liver sections (P < 0.001). PM was again observed predominantly in Kupffer cells. Semiquantitative scoring (0–3) of the percentage of liver cells containing PM indicated significantly more cells contained PM in CAPs- versus FA-exposed mice (2.00 ± 0.94 vs. 0.2 ± 0.42, respectively, P < 0.001). Prussian blue did not stain these particles, indicating little iron was in the PM (data not shown).
Electron microscope imaging of liver sections from the CAPs-exposed mice revealed dense, intracytoplasmic PM in both the liver and lungs ( and ). The PM in the lung cells appeared more heterogeneous in size and morphology than in the liver cells. Spectroscopic analysis of PM in liver cells identified sulfur, a major constituent of PM2.5 known to affect cytokine production () (Fuentes et al., Citation2006; Lipfert et al., Citation2006; Duvall et al., Citation2008). Bromine and chlorine, common PM2.5 constituents, were present in the liver particles as well. The spectroscopic analysis of PM in aorta plaques identified similar elements as found in liver cell PM (data not shown). Lead, which is known to partition poorly from the lungs into the circulation (Wallenborn et al., Citation2007), was present only in the lung particles ().
Figure 2. Examination of PM in liver sections. Following injection of PM, fine particles < 2 μm are observed in the cytosol of hepatic macrophages, primarily Kupffer cells as seen under high power (40X objective) in H&E-stained formalin-fixed sections (A and B; arrows mark PM; bar represents 10 μm). Similar PM was observed in hepatic macrophages following CAPs exposure (C and D; arrows mark PM; bar represents 10 μm). Imaging of glutaraldehyde-fixed sections by electron microscopy also identified electron dense fine PM in both livers (E) and lungs (F) of mice following exposure to ambient CAPs (arrows mark PM; bar represents 2 μm). Electron dispersive spectroscopy of the electron dense intracellular particles revealed sulfur (S), chlorine (Cl), and bromine (Br) in the liver (G) and lead (Pb) and bromine (Br) in the lung (H). The osmium (Os) peaks indicate deposition of osmium tetroxide, which is used to fix membranes. Copper (Cu) peaks are from the copper grid used to mount the tissue section. (Counts = electron counts, KeV = kilovolts.) For each panel, a representative image is shown.
PM exposure increased macrophage and Kupffer cell pro-inflammatory cytokine production in a TLR4-dependent manner
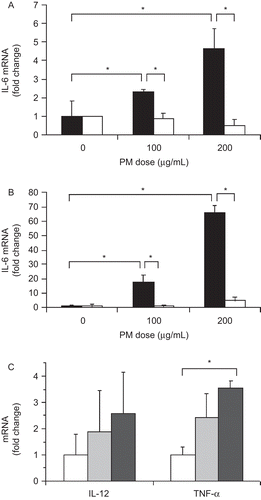
Studies were performed using cultured cells to determine the potential mechanism of any direct effect of PM exposure on inflammatory cytokine and collagen production by liver cells. Standard ambient PM (SRM 1469a) was diluted in media for these studies. The LPS contamination in the PM preparation was minimal, < 125 pg/mL. Since PM accumulated mostly in Kupffer cells (resident hepatic macrophages) following exposure to ambient CAPs, a macrophage cell line was exposed to PM (SRM 1469a) in vitro to assess changes in pro-inflammatory cytokine production (interleukin [IL]-6, IL-12, and tumor necrosis factor [TNF]-α). IL-6 mRNA levels were significantly increased in macrophages in a dose- and time-dependent manner. As early as 8 hr after the addition of PM, macrophage IL-6 mRNA levels were increased 2- to 5-fold relative to unexposed control cells (both P < 0.050) for the 100 μg/mL dose and the 200 μg/mL dose, respectively (, black columns). After 24 hr of exposure to PM (, black columns), the fold-increases in IL-6 mRNA levels were 17.65 ± 5.05 and 65.87 ± 4.93, respectively (both P < 0.010). Pre-incubation with inhibitory anti-TLR4 antibodies (white columns), as opposed to isotype control antibodies (black columns), completely blocked the increase in macrophage IL-6 mRNA levels due to PM exposure at both timepoints ( and ). To a lesser extent, IL-12 and TNFα mRNA levels were increased 24 hr following PM exposure relative to control macrophages, but only the increase in TNFα mRNA after exposure to 200 μg/mL PM was statistically significant (P>0.050) ().
Figure 3. PM effect on cytokine mRNA levels in cultured macrophages. IL-6 mRNA levels were increased in murine RAW macrophages (black bars) at both 8 hr (A) and 24 hr (B) after PM addition. Antibody blockade of TLR4 (white bars) inhibited the effect of PM on IL-6 mRNA levels. Isotype control antibody was present in other cultures (same black bars). Macrophage IL-12 and TNFα mRNA levels were more modestly increased by PM exposure after 24 hr (C). White bars, no PM, light gray bars, 100 μg/ml PM, dark gray bars, 200 μg/ml PM. The results are based on at least three independent experiments. *Statistically significant differences (P < 0.050).
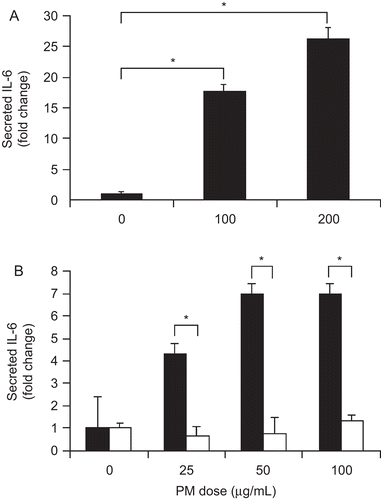
Changes due to PM exposure were also detectable at the protein level. Twenty-four hours after PM addition to the macrophage cell line, IL-6 secretion increased 17-fold for the 100 μg/mL dose and 27-fold for the 200 μg/mL dose (both P < 0.010) compared with control macrophages (). The effect of PM on IL-6 cytokine secretion was also examined in Kupffer cells isolated from both WT and TLR4−/− mouse livers. There was a significant increase in IL-6 secretion in PM-exposed, WT Kupffer cells (25–100 μg/mL PM) compared with unexposed WT Kupffer cells (all P < 0.050) (, black columns). The increase in IL-6 secretion plateaued at a lower dose of PM in Kupffer cells than in the RAW macrophage cell line. At each dose of PM, IL-6 secretion was significantly less in TLR4−/− Kupffer cells (, white columns) compared with WT Kupffer cells (black columns) (all P < 0.050).
Figure 4. PM effect on cultured macrophage and Kupffer cell IL-6 protein secretion. PM addition significantly increased mouse macrophage cell line IL-6 secretion after 24 hr (A). PM stimulated IL-6 secretion by isolated WT Kupffer cells (black bars) but not TLR4−/− Kupffer cells (white bars) after 24 hr (B). The results are based on at least three independent experiments. *Statistically significant differences (P < 0.050).
Hepatic stellate cell collagen expression was increased by conditioned media from PM-exposed macrophages, but not by direct PM exposure
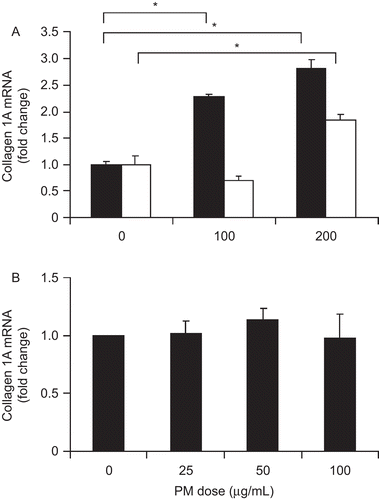
Pro-inflammatory cytokines released by Kupffer cells may enhance fibrosis by increasing collagen 1A production by hepatic stellate cells which are in close proximity to Kupffer cells in the hepatic sinusoids. Therefore, mouse hepatic stellate cells were incubated with four-fold diluted, conditioned media from macrophages exposed to 0, 100, and 200 μg/mL PM for 24 hr. In the presence of conditioned media from PM-exposed macrophages, collagen 1A mRNA expression increased significantly in WT (black columns) stellate cells (both P < 0.050) compared with WT stellate cells cultured in conditioned media from unexposed macrophages (). In TLR4−/− stellate cells (white columns), a significant increase in collagen 1A mRNA expression was observed in the presence of conditioned media from macrophages exposed to 200 μg/mL PM (P = 0.005). Exposure of stellate cells to PM in the absence of macrophage-conditioned media did not increase collagen 1A mRNA expression ().
Figure 5. PM effect on cultured stellate cells. Collagen 1A mRNA levels in both WT (black bars) and TLR4−/− (white bars) hepatic stellate cells were increased by exposure to conditioned media from macrophages exposed to PM (A). Direct PM exposure had no significant effect on collagen 1A mRNA expression (B). The results are based on at least three independent experiments. *Statistically significant differences (P < 0.050).
Discussion
Since NAFLD is the most common chronic liver disease, identification of risk factors responsible for NAFLD progression will significantly help reduce morbidity and mortality from chronic liver disease. The need for liver transplantation would be reduced as well. Limited data is available concerning environmental risk factors associated with NAFLD progression. Our results suggest that chronic inhalation of low levels of ambient air PM is an important risk factor for the progression of NAFLD. The minimal hepatic inflammation and fibrosis in mice exposed only to filtered air indicates that reductions in ambient air PM levels or specific PM components would reduce morbidity and mortality due to NAFLD.
To what extent reducing PM exposure will decrease the progression of benign fatty liver to steatohepatitis is uncertain. Even if only a small percentage of those with NAFLD are affected by PM exposure, millions might benefit from reduced PM exposure since a large epidemiologic study suggested NAFLD affects up to 30% of the U.S. population (as assessed by hepatic triglyceride content; Browning et al., 2004). Hispanics (45%) were most likely to have NAFLD, and the prevalence of insulin resistance (58%) and metabolic syndrome (30%) was increased three-fold in those with NAFLD. Of all factors analyzed, body mass index (i.e., obesity) had the closest positive correlation with hepatic triglyceride content. For now, the prevalence of NAFLD is less in the pediatric population; however, a four-fold increase in pediatric cases of NAFLD has been noted in the past two decades (Koebnick et al., Citation2009). These data suggest NAFLD will be even more prevalent among adults in the near future. Epidemiologic studies utilizing U.S. Environmental Protection Agency data on local PM levels may be helpful in assessing the importance of PM exposure in the progression of NAFLD.
Our animal exposure study was physiologic in that it utilized ambient air PM at doses mimicking naturally occurring levels, daily exposure, natural inhalation, and diet-induced hepatic steatosis. Previous studies using less physiologic models have examined the effect of air pollutant exposure on the liver (Folkmann et al., Citation2007; Gong et al., Citation2007; Tomaru et al., Citation2007). Tomaru et al. demonstrated that obese diabetic mice injected intratracheally bi-weekly for 4 mo with diesel exhaust particles developed increased hepatic steatosis and lipid peroxidation compared with mice exposed to vehicle only (Tomaru et al., Citation2007). Diesel exhaust particles are a major component of ambient PM in urban areas. Araujo et al. (Citation2008) also observed increased hepatic lipid peroxidation in ApoE knockout mice fed NC and exposed 3 d/wk for 6 wks to ambient air CAPs collected 300 m from a Los Angeles freeway. Hepatic lipid peroxidation is indicative of oxidative stress, commonly associated with the transition from simple fatty liver to a profibrotic inflammatory state (steatohepatitis) (Wortham et al., Citation2008). Unlike our study, the effect of air pollutant exposure on hepatic inflammation and fibrosis was not analyzed in prior studies.
Along with oxidative stress, Kupffer cell/macrophage activation and cytokine/chemokine production play a central role in the progression of NAFLD (McCullough, Citation2006). PM was primarily observed in the cytosol of Kupffer cells and to a lesser extent in portal macrophages. Our studies in vitro indicate that ambient PM that reaches the liver has the potential to induce Kupffer cell cytokine secretion in vivo. Kupffer cell IL-6 secretion was greatly increased following PM2.5 exposure. IL-6 levels are significantly elevated in human NAFLD (Wieckowska et al., Citation2008) and are significantly higher in those with steatohepatitis as opposed to simple fatty liver (Wieckowska et al., Citation2008). Kupffer cell activation by PM was TLR4-dependent, consistent with recent studies indicating that macrophage TLR4 expression is pivotal for the progression of NAFLD (Rivera et al., Citation2007). Similar to PM, bacterial LPS activation of hepatic macrophages via TLR4 may also play a role in NAFLD progression (Becker et al., Citation2002). Though LPS may be bound to ambient PM, the ambient PM used in our studies in vitro contained only trace amounts of LPS (< 125 pg/mL) insufficient for the activation of macrophages.
It is uncertain whether the particles noted in the liver represent intact ambient PM that translocated from the lungs into the circulation. The particles we observed in liver may be soluble components of PM that were released from their insoluble carbon backbone. Water- or acid-soluble metals have been shown to translocate to the systemic circulation and deposit in the rat liver following acute or chronic exposure to PM derived from incomplete oil or coal combustion, respectively (Mani et al., Citation2007; Wallenborn et al., Citation2007). In the latter study, increased hepatic inflammation was noted, but the exposure concentration was a 1000-fold higher than in the current study. Seasonal and geographic variability in ambient PM concentrations and components alter its effect on pulmonary (Nadadur and Kodavanti, Citation2002; Prophete et al., Citation2006) and cardiovascular disease (Steerenberg et al., Citation2006). Further studies are needed to determine the route and extent to which inhaled particulate matter reaches the liver.
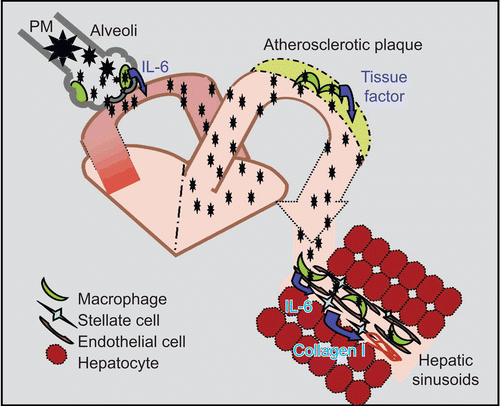
Our working model () illustrates how inhaled fine PM may reach the liver by crossing the alveolar membranes to reach the circulation. Circulating fine PM may then accumulate in both atherosclerotic plaques (Sun et al., Citation2005) and hepatic Kupffer cells. TLR4-dependent activation of cytokine release by Kupffer cells may then trigger inflammation and hepatic stellate cell collagen synthesis. A better understanding of the impact of ambient PM exposure on NAFLD progression may require studies utilizing a variety of ambient PM sources. Finally, the increased inflammation and fibrosis induced by ambient PM exposure in this animal model may facilitate the evaluation of treatments to slow fatty liver disease progression.
Figure 6. Dissemination of ambient PM2.5 and activation of tissue macrophages. Ambient PM2.5 may stimulate alveolar macrophage activation and IL-6 production. Soluble PM2.5 may then enter the circulation and enhance tissue macrophage production of pro-inflammatory cytokines and chemokines. In arteries, this process may enhance the progression of atherosclerosis while in liver the progression of NAFLD may be accelerated.
Acknowledgements
C. E. Alvarez’ technical support for mRNA analysis and Kupffer cell isolation were invaluable. C. E. Jackson’s helpful review and suggestions during manuscript preparation are much appreciated. Qinghua Sun is supported by NIH KO1ES016588
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Adachi, O., Kawai, T., Takeda, K., Matsumoto, M., Tsutsui, H., Sakagami, M., Nakanishi, K., and Akira, S. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143–150.
- Ala, A., Stanca, C.M., Bu-Ghanim, M., Ahmado, I., Branch, A.D., Schiano, T.D., Odin, J. A., and Bach, N. 2006. Increased prevalence of primary biliary cirrhosis near Superfund toxic waste sites. Hepatology 43:525–531.
- Araujo, J.A., Barajas, B., Kleinman, M., Wang, X., Bennett, B.J., Gong, K.W., Navab, M., Harkema, J., Sioutas, C., Lusis, A.J., and Nel, A.E. 2008. Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ. Res. 102:589–596.
- Becker, S., Fenton, M.J., and Soukup, J.M. 2002. Involvement of microbial components and Toll-like receptors 2 and 4 in cytokine responses to air pollution particles. Am. J. Respir. Cell. Mol. Biol. 27:611–618.
- Becker, S., Dailey, L.A., Soukup, J.M., Grambow, S.C., Devlin, R.B., and Huang, Y.C. 2005. Seasonal variations in air pollution particle-induced inflammatory mediator release and oxidative stress. Environ. Health Perspect. 113:1032–1038.
- Browning, J.D., Szczepaniak, L.S., Dobbins, R., Nuremberg, P., Horton, J.D., Cohen, J.C., Grundy, S.M., Hobbset, H.H. 2004. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 40:1387–1395.
- Brunt, E.M., Janney, C.G., Di Bisceglie, A.M., Neuschwander-Tetri, B.A., and Bacon, B. R. 1999. Non-alcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 94: 2467–2474.
- Chen, L.C., and Nadziejko, C. 2005. Effects of subchronic exposures to concentrated ambient particles (CAPs) in mice. V. CAPs exacerbate aortic plaque development in hyperlipidemic mice. Inhal. Toxicol. 17:217–224.
- Duvall, R.M., Norris, G.A., Dailey, L.A., Burke, J.M., McGee, J.K., Gilmour, M.I., Gordon, T., and Devlin, R.B. 2008. Source apportionment of particulate matter in the U.S. and associations with lung inflammatory markers. Inhal. Toxicol. 20:671–683.
- Folkmann, J.K., Risom, L., Hansen, C.S., Loft, S., and Moller, P. 2007. Oxidatively damaged DNA and inflammation in the liver of dyslipidemic ApoE−/− mice exposed to diesel exhaust particles. Toxicology 237:134–144.
- Fuentes, M., Song, H. R., Ghosh, S. K., Holland, D. M., and Davis, J. M. 2006. Spatial association between speciated fine particles and mortality. Biometrics 62:855–863.
- Gong, K. W., Zhao, W., Li, N., Barajas, B., Kleinman, M., Sioutas, C., Horvath, S., Lusis, A.J., Nel, A., and Araujo, J.A. 2007. Air-pollutant chemicals and oxidized lipids exhibit genome-wide synergistic effects on endothelial cells. Genome Biol. 8:R149.
- Guo, J., Loke, J., Zheng, F., Hong, F., Yea, S., Fukata, M., Tarocchi, M., Abar, O.T., Huang, H., Sninsky, J.J., and Friedman, S.L. 2009. Functional linkage of cirrhosis-predictive single nucleotide polymorphisms of Toll-like receptor 4 to hepatic stellate cell responses. Hepatology 49:960–968.
- Hetland, R.B., Cassee, F.R., Lag, M., Refsnes, M., Dybing, E., and Schwarze, P.E. 2005. Cytokine release from alveolar macrophages exposed to ambient particulate matter: Heterogeneity in relation to size, city and season. Part. Fibre Toxicol. 2:4.
- Horani, A., Muhanna, N., Pappo, O., Melhem, A., Alvarez, C.E., Doron, S., Wehbi, W., Dimitrios, K., Friedman, S.L., and Safadi, R. 2007. Beneficial effect of glatiramer acetate (Copaxone) on immune modulation of experimental hepatic fibrosis. Am. J. Physiol. 292:G628–G638.
- Hoshino, K., Takeuchi, O., Kawai, T., Sanjo, H., Ogawa, T., Takeda, Y., Takeda, K., and Akira, S. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J. Immunol. 162:3749–3752.
- Imrich, A., Ning, Y., Lawrence, J., Coull, B., Gitin, E., Knutson, M., and Kobzik, L. 2007. Alveolar macrophage cytokine response to air pollution particles: Oxidant mechanisms. Toxicol. Appl. Pharmacol. 218:256–264.
- Isogawa, M., Robek, M.D., Furuichi, Y., and Chisari, F.V. 2005. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J. Virol. 79:7269–7272.
- Jou, J., Choi, S.S., and Diehl, A.M. 2008. Mechanisms of disease progression in non-alcoholic fatty liver disease. Semin. Liver Dis. 28:370–379.
- Kleiner, D.E., Brunt, E.M., Van Natta, M., Behling, C., Contos, M.J., Cummings, O.W., Ferrell, L.D., Liu, Y.C., Torbenson, M.S., Unalp-Arida, A., Yeh, M., McCullough, A.J., and Sanyal, A.J. 2005. Design and validation of a histological scoring system for non-alcoholic fatty liver disease. Hepatology 41:1313–1321.
- Koebnick, C., Getahun, D., Reynolds, K., Coleman, K.J., Porter, A.H., Lawrence, J.M., Punyanitya, M., Quinn, V.P., and Jacobsen, S.J. 2009. Trends in non-alcoholic fatty liver disease-related hospitalizations in U.S. children, adolescents, and young adults. J Pediatr. Gastroenterol. Nutr. 48(5):597–603.
- Kumagai, Y., Arimoto, T., Shinyashiki, M., Shimojo, N., Nakai, Y., Yoshikawa, T., and Sagai, M. 1997. Generation of reactive oxygen species during interaction of diesel exhaust particle components with NADPH-cytochrome P450 reductase and involvement of the bioactivation in the DNA damage. Free Rad. Biol. Med. 22:479–487.
- Lipfert, F.W., Baty, J.D., Miller, J.P., and Wyzga, R.E. 2006. PM2.5 constituents and related air quality variables as predictors of survival in a cohort of U.S. military veterans. Inhal. Toxicol. 18:645–657.
- Maciejczyk, P., Zhong, M., Li, Q., Xiong, J., Nadziejko, C., and Chen, L.C. 2005. Effects of subchronic exposures to concentrated ambient particles (CAPs) in mice. II. The design of a CAPs exposure system for biometric telemetry monitoring. Inhal. Toxicol. 17:189–197.
- Mani, U., Prasad, A.K., Suresh Kumar, V., Lal, K., Kanojia, R.K., Chaudhari, B.P., and Murthy, R.C. 2007. Effect of fly ash inhalation on biochemical and histomorphological changes in rat liver. Ecotoxicol. Environ. Safety 68:126–133.
- Mar, T.F., Norris, G.A., Koenig, J.Q., and Larson, T.V. 2000. Associations between air pollution and mortality in Phoenix, 1995–1997. Environ. Health Perspect. 108:347–353.
- McCullough, A. J. 2004. The clinical features, diagnosis and natural history of non-alcoholic fatty liver disease. Clin. Liver. Dis. 8:521–533, viii.
- McCullough, A.J. 2006. Pathophysiology of non-alcoholic steatohepatitis. J. Clin. Gastroenterol. 40:S17–S29.
- Nadadur, S. S., and Kodavanti, U. P. 2002. Altered gene expression profiles of rat lung in response to an emission particulate and its metal constituents. J. Toxicol. Environ. Health A 65:1333–1350.
- Phelps, R.G., Bernstein, L.E., Harpaz, N., Gordon, R.E., Cruickshank, F.A., and Schwartz, E. 1989. Characterization of a dermal derived malignant mesenchymal tumor arising in ultraviolet irradiated mice. Am. J. Pathol. 135:149–159.
- Pope, C.A. III, and Kanner, R.E. 1993. Acute effects of PM10 pollution on pulmonary function of smokers with mild to moderate chronic obstructive pulmonary disease. Am. Rev. Respir. Dis. 147:1336–1340.
- Prophete, C., Maciejczyk, P., Salnikow, K., Gould, T., Larson, T., Koenig, J., Jaques, P., Sioutas, C., Lippmann, M., and Cohen, M. 2006. Effects of select PM-associated metals on alveolar macrophage phosphorylated ERK1 and -2 and iNOS expression during ongoing alteration in iron homeostasis. J. Toxicol. Environ. Health A 69:935–951.
- Rivera, C.A., Adegboyega, P., van Rooijen, N., Tagalicud, A., Allman, M., and Wallace, M. 2007. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 47:571–579.
- Schwartz, J., Ballester, F., Saez, M., Perez-Hoyos, S., Bellido, J., Cambra, K., Arribas, F., Canada, A., Perez-Boillos, M.J., and Sunyer, J. 2001. The concentration-response relation between air pollution and daily deaths. Environ. Health Perspect. 109:1001–1006.
- Seki, E., De Minicis, S., Osterreicher, C.H., Kluwe, J., Osawa, Y., Brenner, D.A., and Schwabe, R.F. 2007. TLR4 enhances TGFβ signaling and hepatic fibrosis. Nat. Med. 13:1324–1332.
- Sicari, M.C., Lebwohl, M., Baral, J., Wexler, P., Gordon, R.E., and Phelps, R.G. 1999. Photo-induced dermal pigmentation in patients taking tricyclic antidepressants: Histology, electron microscopy, and energy dispersive spectroscopy. J. Am. Acad. Dermatol. 40:290–293.
- Sioutas, C., Delfino, R.J., and Singh, M. 2005. Exposure assessment for atmospheric ultrafine particles (UFPs) and implications in epidemiologic research. Environ. Health Perspect. 113:947–955.
- Smedsrod, B., Pertoft, H., Eggertsen, G., and Sundstrom, C. 1985. Functional and morphological characterization of cultures of Kupffer cells and liver endothelial cells prepared by means of density separation in Percoll, and selective substrate adherence. Cell Tissue Res. 241:639–649.
- Stanca, C.M., Babar, J., Singal, V., Ozdenerol, E., and Odin, J.A. 2008. Pathogenic role of environmental toxins in immune-mediated liver diseases. J. Immunotoxicol. 5:59–68.
- Steerenberg, P.A., van Amelsvoort, L., Lovik, M., Hetland, R.B., Alberg, T., Halatek, T., Bloemen, H.J., Rydzynski, K., Swaen, G., Schwarze, P., Dybing, E., and Cassee, F.R. 2006. Relation between sources of particulate air pollution and biological effect parameters in samples from four European cities: An exploratory study. Inhal. Toxicol. 18:333–346.
- Su, G.L., Klein, R.D., Aminlari, A., Zhang, H.Y., Steinstraesser, L., Alarcon, W.H., Remick, D.G., and Wang, S.C. 2000. Kupffer cell activation by lipopolysaccharide in rats: Role for lipopolysaccharide binding protein and Toll-like receptor 4. Hepatology 31:932–936.
- Sun, Q., Wang, A., Jin, X., Natanzon, A., Duquaine, D., Brook, R.D., Aguinaldo, J.G., Fayad, Z.A., Fuster, V., Lippmann, M., Chen, L.C., and Rajagopalan, S. 2005. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA 294:3003–3010.
- Sunyer, J., Schwartz, J., Tobias, A., Macfarlane, D., Garcia, J., and Anto, J.M. 2000. Patients with chronic obstructive pulmonary disease are at increased risk of death associated with urban particle air pollution: A case-crossover analysis. Am. J. Epidemiol. 151:50–56.
- Tomaru, M., Takano, H., Inoue, K., Yanagisawa, R., Osakabe, N., Yasuda, A., Shimada, A., Kato, Y., and Uematsu, H. 2007. Pulmonary exposure to diesel exhaust particles enhances fatty change of the liver in obese diabetic mice. Int. J. Mol. Med. 19:17–22.
- Uesugi, T., Froh, M., Arteel, G.E., Bradford, B.U., and Thurman, R.G. 2001. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 34:101–108.
- Wallenborn, J.G., McGee, J.K., Schladweiler, M.C., Ledbetter, A.D., and Kodavanti, U.P. 2007. Systemic translocation of particulate matter-associated metals following a single intratracheal instillation in rats. Toxicol. Sci. 98:231–239.
- Wieckowska, A., Papouchado, B.G., Li, Z., Lopez, R., Zein, N.N., and Feldstein, A. E. 2008. Increased hepatic and circulating interleukin-6 levels in human non-alcoholic steatohepatitis. Am. J. Gastroenterol. 103:1372–1379.
- Wortham, M., He, L., Gyamfi, M., Copple, B.L., and Wan, Y.J. 2008. The transition from fatty liver to NASH associates with SAMe depletion in db/db mice fed a methionine choline-deficient diet. Dig. Dis. Sci. 53:2761–2774.
- Yohe, H.C., O’Hara, K.A., Hunt, J.A., Kitzmiller, T.J., Wood, S.G., Bement, J.L., Bement, W.J., Szakacs, J.G., Wrighton, S.A., Jacobs, J.M., Kostrubsky, V., Sinclair, P.R., and Sinclair, J.F. 2006. Involvement of Toll-like receptor 4 in acetaminophen hepatotoxicity. Am. J. Physiol. 290:G1269–G1279.