Abstract
The potential for adverse health effects of using tungsten and its alloys in military munitions are an important concern to both civilians and the US military. The toxicological implications of exposure to tungsten, its alloys, and the soluble tungstate (Na2WO4) are currently under investigation. To examine tungstate toxicity, a series of experiments to determine its in vitro effects on cells of the immune system were performed. We identified alterations in isolated human peripheral blood lymphocytes (PBL) treated in vitro with sodium tungstate (0.01, 0.1, 1.0, and 10 mM). Analyses of apoptosis with annexin V and propidium iodide revealed a dose- and time-dependent increase in the quantity of cells in early apoptosis after tungstate exposure. Reductions in the number of cells entering into the cell cycle were also noted. Exposure of PBL to tungstate (1 mM) and Concanavalin A (ConA) for 72 h reduced the number of cells in S and G2/M phases of the cell cycle. There were alterations in the numbers of cells in G0/G1, S, and G2/M phases of the cell cycle in long-term THP-1 (acute leukemic monocytes) cultures treated with tungstate (0.01, 0.1, 1.0, and 10 mM). Gel electrophoresis, silver staining, and LC-MS/MS showed the cytoplasmic presence of histone H1b and H1d after 72 h of tungstate exposure. The addition of tungstate to cultures resulted in significant reductions in the quantity of interleukin-10 (IL-10), tumor necrosis factor-α (TNF-α), and IL-6 produced by stimulated [CD3/CD28, ConA, or lipopolysaccharide (LPS)] and tungstate-treated lymphocytes. Taken together, these data indicate that tungstate increases apoptosis of PBL, alters cell cycle progression, reduces cytokine production, and therefore warrants further investigation.
Keywords::
Introduction
The heavy metal tungsten (W) is used in many applications worldwide due to its high melting point and strength when alloyed with other metals. Tungsten and its alloys were generally considered inert and therefore posed little threat to the environment and biological systems (Koutsospyros et al., Citation2006). Consequently, in the late 1990s, the US military replaced lead (Pb) and depleted uranium (DU) in kinetic energy penetrators and small ammunitions with tungsten and its alloys. The tungsten containing munitions were thought to be safe and were termed “green bullets” in the media. However, recent findings suggest that tungsten may have detrimental effects on biological systems.
In Fallon, Churchill County, NV, an acute lymphoblastic leukemia (ALL) cluster was identified in the children of this area (Steinberg et al., Citation2007). Sierra Vista, AZ and Elk Grove, CA have also reported leukemic clusters associated with tungstate exposure (Koutsospyros et al., Citation2006; Fastje et al., Citation2009). These locations have high levels of environmental tungsten and high background levels of tungstate in the drinking water (Haneke, Citation2003; ATSDR, Citation2005). While suggestive, the question of tungsten as the causative agent in these leukemic clusters remains open (CDC, Citation2003). In animal systems, intramuscularly-embedded tungsten alloy resulted in the development of metastatic cancer in rats (Kalinich et al., Citation2005). However, the presence of nickel and cobalt in the implanted alloy makes it difficult to identify tungsten as the sole source of carcinogenesis. Studies on rat L6 and mouse C2C12 skeletal muscle cell lines show that these cells have different susceptibilities to tungstate and other soluble metals (i.e., Co, Ni, Fe) (Kane et al., Citation2009).
Tungsten, in aqueous environments, can be converted to the soluble tungstate ion (WO4)2-. In mammalian systems, tungstate is a competitive inhibitor of molybdenum-containing enzymes such as xanthine oxidoreductase (XOR) and is reported to function as a phosphatase inhibitor (Stankiewicz and Gresser, Citation1988; Foster et al., Citation1998; Vorbach et al., Citation2003). Alterations in XOR function and potential changes to intracellular signaling networks due to tungstate-dependent phosphatase inhibition suggest that tungstate could have effects on biological responses. Rats exposed to sodium tungstate in their drinking water demonstrated tungstate-dependent neurobehavioral changes (McInturf et al., Citation2008). Xanthine oxidase is involved in innate immunity (Vorbach et al., Citation2003) and in antigen presentation by Kupffer cells (Maemura et al., Citation2005). XOR is also up-regulated during infection or sepsis (Chinnaiyan et al., Citation2001), suggesting that inhibition of this enzyme may be associated with reduced host defense against microbial challenge (Deitch et al., Citation1992; Kahl and Elsasser Citation2004). As a phosphatase inhibitor, tungstate has been reported to alter phosphorylation of p38 and PI3K in MIN6β cells (Piquer et al., Citation2007), GSK3β in cultured neural cells (Gomez-Ramos et al., Citation2006), ERK1/2 in Leydig cells and cultured hepatocytes (Dominguez et al., Citation2003; Ballester et al., Citation2005), and Stat1 in HeLa cells (Haque et al., Citation1995). Oral tungstate treatment is shown to regulate insulin sensitivity and glucose metabolism in diabetic models (Munoz et al., Citation2001; Heidari et al., Citation2008) and block adipocyte differentiation (Carmona et al., Citation2009). Taken together, these data suggest that tungstate can interact with a large number of cellular regulatory components.
The effects of tungstate on immune function have only been investigated to a limited degree. In this study, we describe an examination of the in vitro effects of tungstate on isolated leukocytes. Tungstate exposure altered apoptosis, cell cycle, and cytokine production. These findings suggest that tungstate exposure in vivo may have immunological consequences for an exposed individual.
Materials and methods
Reagents
Sodium tungstate dihydrate (Na2WO4·2H2O; CAS 10213-10-2), Histopaque 1077, and the T-cell mitogen Concanavalin A (ConA) were purchased from Sigma (St Louis, MO). Annexin V–APC and propidium iodide (PI) solutions were obtained from BD Biosciences (San Jose, CA). Lipopolysaccharide (LPS; Type O111:B4) was purchased from List Biological Laboratories (Campbell, CA). CD3/CD28 Dynabeads were obtained from Invitrogen (Carlsbad, CA). Interleukin-10 (IL-10), tumor necrosis factor-α (TNF-α), and IL-6 ELISAs and recombinant IL-2 were obtained from eBioscience (San Diego, CA). THP-1 cells were purchased from American Type Culture Collection (ATCC; Manassas, VA).
Isolation of human peripheral blood lymphocytes
Studies were conducted with the University of Cincinnati Institutional Review Board-approved protocols (#84-04-25-03). Whole blood was obtained from healthy volunteers by venipuncture into K2EDTA tubes. Blood was layered on Histopaque 1077 and centrifuged at 300 x g at room temperature for 30 min. Peripheral blood mononuclear cells (PBMC) were removed from the gradient interface and washed twice with RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT). Peripheral blood lymphocytes (PBL) were isolated by allowing PBMC to adhere twice onto cell culture dishes for 1 h at 37°C with 5% CO2. Non-adherent lymphocytes were gently washed and collected. Determinations of cell number were made on a Hemavet 950 analyzer (Drew Scientific Inc., Dallas, TX). For the studies here, each experiment included at least one female and two male donors. The donor pool consisted of three men (aged 42, 40, and 38) and two women (aged 47 and 22). Previous work in the literature has documented that an n = 3 is suitable to assess the in vitro immunotoxicity of various compounds (Berek et al., Citation2001; Alves et al., Citation2008; Eke and Celik, Citation2008).
Flow cytometry
Flow cytometry was performed at the Shriners Hospital for Children in Cincinnati Flow Cytometry Core Facility. All cytometry, with the exception of DNA cell cycle determinations, was performed on an LSRII flow cytometer (BD Biosciences). The LSRII is equipped with 488 and 633 nm lasers; and fluorescence was collected with the appropriate filters. Data were analyzed using Diva Software v5.0.2 (BD Bioscience). DNA measurements were made using a Coulter Epics XL system (Beckman-Coulter, Fullerton, CA) with 488 nm excitation. Data were collected with System II software (v3) as described previously (Knauf et al., Citation2006). Listmode files were analyzed using ModFit v3.1 (Verity Software House, Inc., Topsham, ME) to determine quantities of cells in G0/G1, S, and G2/M phases of the cell cycle. For all cytometry, at least 10,000 cells were measured per sample.
Measurement of apoptosis PBL with annexin V and PI
Isolated human PBL were treated with sodium tungstate (0, 0.1, 1, 10 mM) with or without ConA (2.5 μg/mL) for 24, 48, and 72 h. After treatment, cells were harvested and washed twice with ice-cold wash buffer [1X PBS (phosphate-buffered saline; pH 7.2), 0.5% BSA (bovine serum albumin), 7 mM CaCl2]. Cells were then re-suspended in 1X Annexin V–binding buffer [0.01 M HEPES (pH 7.4), 0.14 M NaCl, 2.5 mM CaCl2]. Annexin V–APC and PI (1 μg/mL) were added and the cells were incubated in the dark, on ice, for 15 min. Cells were then immediately analyzed by using flow cytometry (Holder et al., Citation2000).
DNA cell cycle and EdU analysis of leukocytes isolated from whole blood
PBL were isolated as described previously and cultures seeded at 1 × 106 cells/well in RPMI supplemented with 10% FBS. Sodium tungstate (0.01, 0.1, 1.0, 10 mM) and/or ConA (0, 1, 2.5 µg/mL) were added to appropriate wells. After 24, 48, and 72 h, cells were washed with 1X PBS and fixed in ice-cold 75% isopropanol for at least 30 min. Cells were then washed twice in buffer (1X PBS, 0.5% BSA, 7 mM CaCl2, 0.1% sodium azide) followed by staining with PI solution (50 µg PI/mL, 0.1% Triton X-100, 25 µg RNase A, 1X PBS). Click-iT EdU Alexa Fluor 488 Cell Proliferation Assay Kit (Invitrogen, Carlsbad, CA) staining of PBL was performed according to the manufacturer’s directions.
THP-1 cells were grown in triplicate plates with doses of tungstate (0, 0.01, 0.1, 1, or 10 mM). At each sampling interval, an aliquot of cells was removed from the culture and processed for DNA determinations as mentioned earlier. When cultures were split due to cell density, new medium was added as well as an appropriate quantity of sodium tungstate to amend tungstate concentrations.
Cytokine expression of PBL after treatment with tungstate
Isolated PBL were seeded at 1 × 106 cells/mL and treated with sodium tungstate (0, 0.1, 1 mM), with or without ConA (2.5 µg/mL), LPS (100 ng/mL), CD3/CD28 Dynabeads (1:1 beads to cells and IL-2 [1 ng/mL]), for 24, 48, and 72 h. Culture supernatants were harvested at each timepoint. Each timepoint represents total cytokine production of 24, 48, and 72 h (i.e., 0–24, 0–48, and 0–72 h of culture production). The ELISAs for IL-6, TNF-α, and IL-10 were performed in triplicate, according to the manufacturer’s instructions. Absorbance at 590 nm was measured using a SpectraMax Plus 384 system (Molecular Devices, Sunnyvale, CA).
Isolation of soluble and insoluble cellular fractions
Protein preparations were made, with slight modifications, according to Yan et al. (Citation1995) by the addition of CSK [25 mM PIPES (pH 6.9), 4 M glycerol, 1 mM MgCl2, 0.2% Triton X-100, 1 mM EGTA, 5 µg pepstatin/mL, 5 µg leupeptin/mL, 1 mM PMSF, 1 mM EDTA, 1 mM dithiothreitol (DTT), 100 μM sodium orthovanadate, and 10 μM phenylarsine oxide]. Cells were incubated for 20 min on ice and subsequently centrifuged at 12,000 x g at 4°C for 15 min. The non-detergent–resistant membrane fraction (non-DRM) supernatant was removed and the detergent-resistant pellet was solubilized by the addition of SDS buffer [2% sodium dodecyl sulfate, 80 mM Tris (pH 6.8), 10% glycerol, 2.5% 2-mercaptoethanol), followed by a 5-sec sonication. This fraction was centrifuged at 12,000 g at 4°C for 15 min. The DRM fraction was then recovered and the pellet was discarded. SDS–PAGE gel electrophoresis was then performed using pre-cast 12% Tris/glycine gels (BioRad, Hercules, CA), according to the standard procedures. The gel was visualized with Silver Stain Plus (BioRad). Bands were excised and analyzed by LC-MS/MS mass spectrometry in the Proteomics Laboratory at the University of Cincinnati’s Genome Research Institute.
Data analysis
Data were analyzed using Systat v11.00.01 (Systat Sofware Inc., Chicago, IL). Statistical significance was assessed by two-way ANOVA, followed by specific contrasts for post hoc comparisons. Results were considered statistically significant when P <0.05.
Results
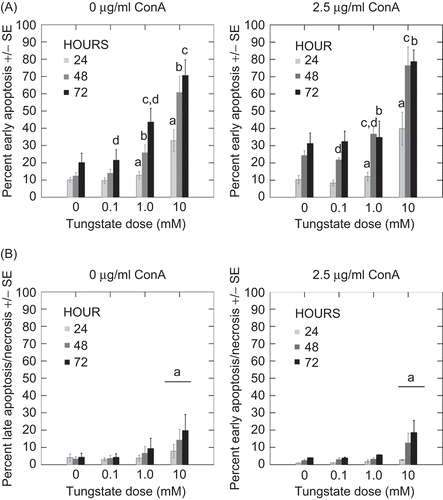
To determine whether tungstate treatment increased apoptosis in human peripheral leukocytes (PBL), these cells were treated in vitro with tungstate (0.1, 1.0, 10.0 mM) and ConA for 24, 48, and 72 h. The data in (upper panels) depicts increases in early apoptosis (annexin V+/PI−) in tungstate-treated PBL. The 0 and 0.1 mM tungstate doses resulted in no statistically significant increases in early apoptosis with either the 0 or 2.5 µg/mL ConA treatments even though there was a trend of increased early apoptosis as a function of culture length. Activating PBL with the T-cell mitogen ConA (2.5 µg/mL) increased the overall level of early apoptosis (as compared with controls) at 0 and 0.1 mM tungstate. At 72 h, in the absence of ConA, significant increases in early apoptosis were observed at 1.0 mM versus 0.1 mM (P <0.001) tungstate-treated PBL. When ConA was added to these conditions, statistically significant differences were not noted. The PBL cultured in 10 mM, with and without ConA, had significantly higher percentages of cells in early apoptosis (compared with all other groups) at all timepoints. At 24 h, the 10 mM sample had a more than two-fold increase in early apoptosis levels as compared with cells that received 1 mM tungstate (P < 0.007); the increases in early apoptosis was observed following 48 and 72 h of 10 mM tungstate treatment were also highly significant (P <0.001 in both cases). A similar pattern of increases in apoptosis was shown in the ConA-treated samples. However, there was a trend toward increased early apoptosis at 48 h. Quantities of late apoptotic (annexin V+/PI+) and/or necrotic cells (annexin V−/PI+ cells) were not significantly increased except with 10 mM tungstate (P < 0.02) (, lower panels). Unlike with early apoptosis, only the 10 mM tungstate caused an increase in the number of late apoptotic cells as a function of culture time. The osmolarity of the tungstate-supplemented RPMI-1640 was determined and found not to be significantly different from the tungstate-free media (data not shown).
Figure 1. (A) Early apoptosis with sodium tungstate and ConA. Isolated PBL were treated with sodium tungstate and ConA (2.5 μg/mL) for 24, 48, and 72 h. Early apoptosis was measured as Annexin V+ and PI− cells. Data represent the mean and standard error of three individuals. Statistical differences between groups were determined by specific post hoc contrasts. (B) Late apoptosis/necrosis with sodium tungstate and ConA. Isolated PBL were treated with sodium tungstate and ConA (2.5 μg/mL) for 24, 48, and 72 h. Late apoptosis/necrosis was measured as Annexin V+/− and PI+ cells. Data represent the mean and standard error of three individuals. Statistical differences between groups were determined by specific post hoc contrasts. At each timepoint, bars with identical letters indicate statistically significant differences were present between the groups.
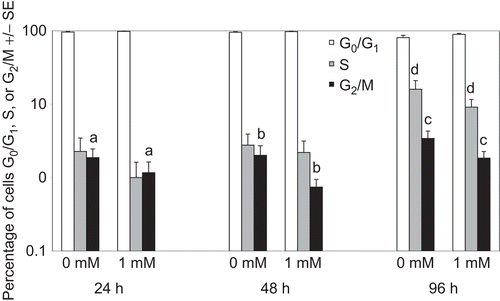
The effects of tungstate on cell cycle progression of PBL were examined following stimulation with ConA after 24, 48 and 96 h of culture with (or without) 1 mM tungstate (). The addition of 1 mM tungstate to ConA (2.5 µg/mL)-treated PBL resulted in a significant decrease in the number of cells in G2/M phase at 24, 48, and 96 h as compared with among control cells (P < 0.001, P < 0.007, P < 0.021, respectively). In addition, there was a trend toward fewer cells in S phase in 1 mM–treated PBL at all timepoints; however, this change only reached statistical significance at 96 h of culture (P < 0.05).
Figure 2. Cell cycle of PBL treated with sodium tungstate (1 mM) and ConA for 24, 48, and 96 h. Determinations of cell cycle were made with PBL stained with PI. Data represent the mean and standard error of three individuals. Bars with identical letters indicate statistically significant differences were present between the groups.
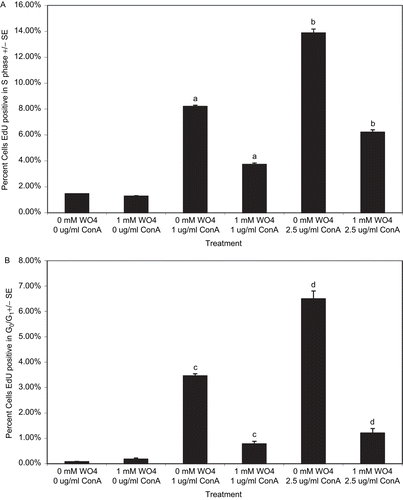
To expand upon the effect of tungstate on cell cycle progression of PBL, DNA synthesis was measured by EdU (5-ethynyl-2′-deoxyuridine) incorporation. PBL were treated with 1 mM tungstate and 1.0 or 2.5 µg/mL of ConA for 72 h, with EdU added during the last 24 h of culture. The data in indicates that PBL treated with ConA and 1 mM tungstate had a >50% reduction in the percentage of EdU+ cells relative to control cell values (at each ConA level, P < 0.001). Tungstate also significantly decreased the percentage of EdU+ cells in G0/G1 compared with controls () (at each ConA level, P < 0.001). These data suggest that tungstate reduced the number of cells capable of entering the cycle.
Figure 3. EdU staining in PBL treated with sodium tungstate for 72 h. Isolated PBL were treated with sodium tungstate (1 mM) and ConA (1.0 and 2.5 μg/mL) for 72 h. At 24 h before analysis, cells were incubated with EdU. (A) PBL that are EdU+ and in S phase of the cell cycle. (B) PBL that are EdU+ in G0/G1. Data represent the mean and standard error of values from three individuals. Bars with identical letters indicate statistically significant differences were present between the groups.
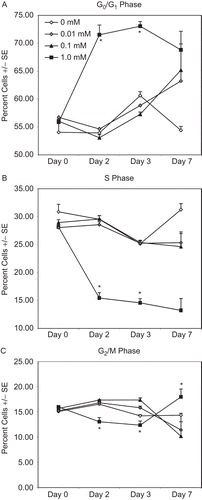
We also sought to determine whether tungstate-dependent effects on cell cycle could be observed in cells that continuously proliferate without the addition of an external agonist. Continuously proliferating cells have the further advantage of having reproducible cell cycle profiles, which make perturbations in the cycle easier to detect. To investigate this, THP-1 (an immortalized monocytic leukemia line) cells were treated with tungstate (0.01, 0.1, 1.0, 10 mM) for up to 7 days. The highest dose of tungstate (10 mM) resulted in complete loss of viable cells in culture after 72 h. These cultures were predominantly in G0/G1 before the depletion of viable cells due to the tungstate exposure (data not shown). The data indicate that 1 mM tungstate caused a significant increase in the number of cells in G0/G1 (P < 0.001 vs. each other treatment group; ) and a significant decrease in cells in S phase () and G2/M phase () at Days 2 and 3 (P < 0.001 vs. each other treatment group in 4B and 4C) of the culture. By Day 7, the percentages of cells in G0/G1 and S phase were different compared with control cultures, but the differences were not statistically significant. The quantity of cells in G2/M, after tungstate (1 mM) treatment was now greater compared with the controls (P < 0.001). Treatments of 0.1 mM and 0.01 mM did not appear to have an impact on cell cycle in THP-1 cells.
Figure 4. Cell cycle of THP-1 cells treated with sodium tungstate for 7 days. Cell cycle in THP-1 monocytes was determined by PI staining. Cells were kept in culture with appropriate quantities (0.01–10 mM) of tungstate for up to 7 days. Data represent the mean and standard error of three independent culture wells. The 10 mM tungstate treatment resulted in the loss of culture viability after 72 h; for clarity, the data from these cultures is not shown.
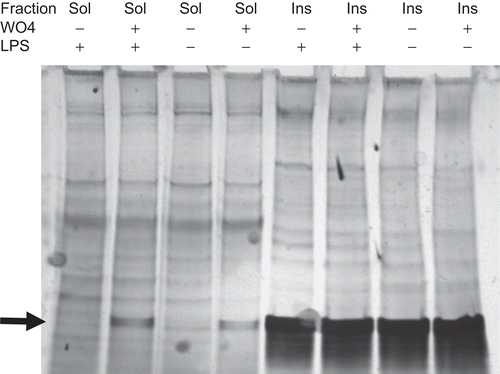
To determine whether tungstate induced changes in the expression of cellular proteins, detergent-soluble (non-DRM) and detergent-insoluble fractions (DRM) were prepared from PBL treated with tungstate (1 mM) for 72 h. Analysis of the soluble and insoluble fractions by silver staining revealed the presence of a low molecular weight band (∼20–30 kDa) that was present only in the soluble fractions from tungstate-treated cells and in all of the insoluble fractions (, indicated by arrow). This band was excised and identified by GC-MS/MS mass spectrometry as histone H1b and histone H1d.
Figure 5. Silver stain of PBL soluble and insoluble fractions after treatment with tungstate. Soluble (non-DRM) and insoluble (DRM) fractions were prepared from PBL treated with tungstate for 72 h. Soluble and insoluble fractions were loaded at 12.5 and 6 µg protein per lane, respectively. Protein bands were visualized by silver staining.
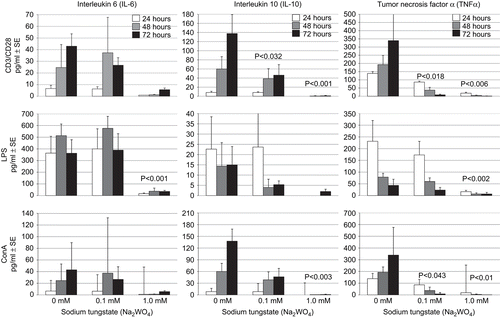
Production of interleukin-6 (IL-6), IL-10, and TNF-α was measured in culture supernatants from PBL stimulated with CD3/CD28 beads, LPS, and ConA. As shown in , 1 mM treatment with tungstate resulted in significant effects upon PBL cytokine production. For IL-6, there was a significant reduction (P < 0.001 vs. control; all timepoints) in the release of this cytokine in the LPS-stimulated cultures. Determinations of monocytes in PBL isolations were not made and a portion of IL-6 production in LPS-stimulated cultures is likely due to these monocytes. At this same dose, the reduction in IL-6 synthesis by ConA- and CD3/CD28-stimulated cultures was not significant. IL-10 formation was significantly reduced with 1 mM tungstate in the CD3/CD28− and the ConA-stimulated cultures (P < 0.001 and P < 0.003, respectively, vs. control; all timepoints). In addition, 0.1 mM tungstate resulted in a significant reduction (P < 0.032 vs. control; 48 and 72 h timepoints) in IL-10 production among CD3/CD28-stimulated cells. TNF-α synthesis was reduced in ConA-treated samples that received 0.1 mM or 1 mM tungstate (P < 0.043 and P < 0.010, respectively, vs. control; all timepoints). In addition, in LPS-exposed PBL, the 1 mM tungstate dose resulted in cytokine levels that were near baseline (P < 0.002 vs. control; all timepoints). In CD3/CD28-treated PBL, the 0.1 mM and 1 mM tungstate doses again resulted in significant reductions in TNF-α production (P < 0.018 and P < 0.006, respectively, vs. control; all timepoints). Statistical analyses of these data revealed that, generally, the tungstate dose was more important in terms of cytokine production than was the culture length. In addition, no statistically significant interactions between the tungstate dose and culture time were noted. Therefore, the P-values in indicate the level of significance of a tungstate dose compared with its appropriate control, across culture timeframes.
Figure 6. Cytokine production of IL-10, IL-6, and TNF-α by PBL treated with tungstate and CD3/CD28, ConA, or LPS for 24, 48, and 72 h. PBL were treated with LPS, ConA, or CD3/CD28 beads and culture supernatants subsequently collected for cytokine determinations. Cytokine production was measured from PBL of three individuals. Bars represent the mean and standard error. Note that y-axis scale bars vary depending on cytokine. P-values shown indicate level of significance of the difference between the value at a given dose of tungstate compared with the value of the control; generally, statistical analyses showed that dose was more important than culture time and so these P-values reflect the overall impact of the indicated dose across the three timeframes cited.
Discussion
Tungsten is used in many industrial, medical, and military applications throughout the world. Relatively little is known about its toxicological profile with respect to human and animal environmental exposures. Our studies have focused on the immunotoxicological effects the soluble tungstate ion (WO42−) has on human leukocytes in vitro, and was intended as a starting point for understanding the potential mechanistic effects of exposure rather than to reflect a particular exposure scenario. In the context of this study, the 1 mM tungstate dose represents the exposure in which significant toxicity (i.e., cell cycle effects, apoptosis, etc.) occurs in all of the assays. With that being said, the 1 mM (240 ppm of tungstate ion), 0.1 mM (24 ppm), and 0.01 mM (2.4 ppm) treatments represent doses that are likely higher than environmental exposures. However, in animal studies in which mice were exposed to tungstate for 28 days, we have found immunological impairments and tungsten concentrations in the spleen and bone ranging from 2 to 70 ppm (unpublished data). Furthermore, the use of tungstate as a therapeutic agent for the treatment of obesity is under examination, and therapeutic levels of tungstate may exceed those of environmental exposure. In addition, tungstate follows a two-compartment pharmacokinetic model and organs such as spleen, bone, or kidneys may have local tungstate concentrations that are significant (Leggett, Citation1997).
In the current study, we show that there were increases in tungstate-dependent apoptosis in PBL and reductions in the number of cells progressing through the cell cycle after tungstate treatment. There are also significant reductions in the number of tungstate-treated cells synthesizing DNA. The presence of histones H1b/H1d in the cytoplasm of only the tungstate-treated PBL is suggestive as a sign of early apoptosis. Finally, we observed that there were significant reductions in the quantity of cytokines produced after CD3/CD28, LPS, or ConA stimulation of tungstate-treated PBL. Taken together, these data are consistent with tungstate-dependent changes in the in vitro function of cells of the immune system.
In unstimulated PBL, 10 mM tungstate increased the percentage of cells in early apoptosis at all timepoints compared with lower tungstate doses. At 1 mM of tungstate, there were significantly greater numbers of cells undergoing apoptosis at 72 h of culture. In ConA-stimulated cultures, there was an overall increase in the quantity of cells in early apoptosis at 48 and 72 h, as well as significant differences in quantities of apoptotic leukocytes between the 0.1 and 1.0 mM tungstate doses. This would suggest that the action of tungstate is more pronounced when PBL are responding to mitogenic stimuli. Cell cycle determinations indicated that there were significantly reduced quantities of tungstate-treated PBL in G2/M versus control cells. In addition, there were fewer cells with synthesizing DNA in tungstate-treated samples when EdU incorporation was measured. Consistent with this, there were significantly reduced numbers of tungstate-treated cells that successfully passed through G2/M to G0/G1 (see ). However, the effects on PBL cell cycle could be attributed to tungstate-dependent inhibition of ConA-induced proliferation. To determine whether cell cycle alterations were present in cells that proliferate without the requirement for agonist, we used THP-1 cell culture monocytes. These cells are inherently proliferative and, therefore, perturbations in cell cycle kinetics can be measured. Our data () shows that there was a considerable increase in G0/G1 at 1 mM tungstate and no effect at lower doses. These outcomes suggest that 1 mM tungstate affects cell cycle kinetics; however, the mechanism for this is still unclear. Studies in our laboratory (data not shown) and by others showed that tungstate alters phosphorylation of cellular proteins (Gomez-Ramos et al., Citation2006; Piquer et al., Citation2007); this could account for the altered cytokine profiles observed. Potentially, tungstate-dependent phosphatase inhibition could alter phosphorylation of cyclins and cell cycle regulatory machinery.
Proteomic analysis of a band present in soluble protein preparations from tungstate-treated PBL was identified as histone 1 family members H1b (H1.5, H1F5) and H1d (H1.3, H1F3). The identification of these histones in the soluble fraction is consistent with the cytoplasmic distribution of histone H1. It would appear unlikely that contamination from the insoluble fraction is responsible, as there is little to no appearance of this band in the fractions isolated from tungstate-untreated preparations. The histone 1 family is predominantly involved in chromatin regulation through nucleosomal linker functions (Happel and Doenecke, Citation2009). Recent evidence suggests that H1 histones may functionally connect nuclear double-strand breaks (DSB), in a p53-dependent manner, with mitochondrial apoptotic machinery (Konishi et al., Citation2003).
Of the histone 1 family members, only histone H1c (i.e., H1.2, H1F2) has been directly linked to the induction of apoptosis. Histone H1.2 is released into the cytoplasm after apoptotic induction (Konishi et al., Citation2003; Okamura et al., Citation2008). However, H1.5 appears to be modified in HL-60 cells induced to undergo apoptosis by topotecan treatment (Kratzmeier et al., Citation1999). Cytoplasmic pools in histone H1 in murine hepatocytes have also previously been identified (Zlatanova et al., Citation1990), as well as the presence of cytoplasmic H1 in intestinal villi cells undergoing apoptosis (Rose et al., Citation1998). While a functional connection between the cytoplasmic localization of H1b and/or H1d and apoptosis has not been described, it would seem to be consistent with an increase in early apoptotic signaling (Bree et al., Citation2004). The histone H1 translocations described in the literature suggest that DNA DSB are the initiator of histone translocation. DSB in the context of tungstate exposure have not previously been described.
In terms of functional changes, tungstate-treated PBL produce significantly less TNF-α, IL-10, and IL-6 after stimulation with agonists. Some reduction in cytokine production can likely be attributed to increased apoptosis after tungstate exposure of the PBL. However, the cytokine production in PBL at the highest dose of tungstate (i.e., 1 mM), in which approximately half of the PBL were either early or late apoptotic/necrotic, is far less than this could account for. Fewer cells would produce cytokines; however, the reduction in total cytokine production is larger than the portion of cells undergoing apoptosis. Others have shown, for example, that lineage-specific NF-κB inhibition decreases cellular proliferation and cytokine production, as well as increases in apoptosis in T-cells after mitogen stimulation (Ferreira et al., Citation1999). However, with 0.1 mM tungstate, there was only a marginal increase in apoptosis between 0 and 2.5 µg/mL ConA (), while there was a significant reduction in the quantity of TNF-α (P < 0.043) and IL-10 (P < 0.032) produced. This suggests that there is an additional mechanism besides apoptosis responsible for reduction in cytokine production.
The data indicating changes in apoptosis, cell cycle, and cytokine production suggests that the effects of tungstate are related to its physiological functions rather than physical effects of the ion. At the doses used in this study, addition of tungstate to culture media did not significantly alter the osmolarity of the media. Tungstate is reported to replace the molybdenum reaction center in xanthine oxidase (Johnson et al., Citation1974). Xanthine oxidase (XO) contributes to reactive oxygen species production and is associated with tissue damage in certain situations (Victorino et al., Citation2008). Tungstate would reduce oxidant production by XO. This would suggest that excess oxidants are not responsible for apoptosis induction in the tungstate-treated samples. The observed effects of tungstate could be due to its inhibition of phosphatase. These data suggest that tungstate can alter important phosphorylation of several intracellular signaling molecules (Dominguez et al., Citation2003; Gomez-Ramos et al., Citation2006; Piquer et al., Citation2007). At lower doses of tungstate, apoptosis and cell cycle effects are not as pronounced. However, given potential phosphorylation changes, tungstate may make cells more susceptible to other insults. From these studies, we conclude that exposure to tungsten in the ionic form has pronounced effects on biological systems and warrants further study.
Acknowledgements
We would like to thank CDR Gail Chapman for her assistance.
Declaration of interest
The authors report no conflicts of interest. The authors are alone responsible for the content and writing of the paper.
References
- Alves, R.J., Jotz, G.P., do Amaral, V.S., Montes, T.M., Menezes, H.S. and de Andrade, H.H. 2008. The evaluation of maté (Ilex paraguariensis) genetic toxicity in human lymphocytes by the cytokinesis-block in the micronucleus assay. Toxicol. In Vitro 22:695–698.
- ATSDR (Agency for Toxic Substances and Disease Registry). 2005. Toxicological Profile for Tungsten. Atlanta: US-DHHS.
- Ballester, J., Domínguez, J., Muñoz, M.C., Sensat, M., Rigau, T., Guinovart, J.J. and Rodríguez-Gil, J.E. 2005. Tungstate treatment improves Leydig cell function in streptozotocin-diabetic rats. J. Androl. 26:706–715.
- Berek, L., Petri, I.B., Mesterházy, A., Téren, J. and Molnár, J. 2001. Effects of mycotoxins on human immune functions in vitro. Toxicol. In Vitro 15:25–30.
- Bree, R.T., Neary, C., Samali, A. and Lowndes, N.F. 2004. The switch from survival responses to apoptosis after chromosomal breaks. DNA Repair (Amst.) 3:989–995.
- Carmona, M.C., Amigó, M., Barceló-Batllori, S., Julià, M., Esteban, Y., Moreno, S. and Gomis, R. 2009. Dual effects of sodium tungstate on adipocyte biology: inhibition of adipogenesis and stimulation of cellular oxygen consumption. Int. J. Obes. (Lond). 33:534–540.
- CDC (Centers for Disease Control). 2003. Cross-sectional Exposure Assessment of Environmental Contaminants in Churchill County. Nevada, Atlanta: CDC.
- Chinnaiyan, A.M., Huber-Lang, M., Kumar-Sinha, C., Barrette, T.R., Shankar-Sinha, S., Sarma, V.J., Padgaonkar, V.A. and Ward, P.A. 2001. Molecular signatures of sepsis: multiorgan gene expression profiles of systemic inflammation. Am. J. Pathol. 159:1199–1209.
- Deitch, E.A., Kemper, A.C., Specian, R.D. and Berg, R.D. 1992. A study of the relationship among survival, gut-origin sepsis, and bacterial translocation in a model of systemic inflammation. J. Trauma 32:141–147.
- Domínguez, J.E., Muñoz, M.C., Zafra, D., Sanchez-Perez, I., Baqué, S., Caron, M., Mercurio, C., Barberà, A., Perona, R., Gomis, R. and Guinovart, J.J. 2003. The antidiabetic agent sodium tungstate activates glycogen synthesis through an insulin receptor-independent pathway. J. Biol. Chem. 278:42785–42794.
- Eke, D. and Celik, A. 2008. Genotoxicity of thimerosal in cultured human lymphocytes with and without metabolic activation sister chromatid exchange analysis proliferation index and mitotic index. Toxicol. In Vitro 22:927–934.
- Fastje, C.D., Kim, L., Sun, N.N., Wong, S.S., Sheppard, P.R. and Witte, M.L. 2009. Prenatal exposure of mice to tungstate is associated with decreased transcriptome-expression of the putative tumor suppressor gene, DMBT1: Implications for childhood leukemia. Land Contam. Reclam. 17:169–178.
- Ferreira, V., Sidénius, N., Tarantino, N., Hubert, P., Chatenoud, L., Blasi, F. and Körner, M. 1999. In vivo inhibition of NF-kappa B in T-lineage cells leads to a dramatic decrease in cell proliferation and cytokine production and to increased cell apoptosis in response to mitogenic stimuli, but not to abnormal thymopoiesis. J. Immunol. 162:6442–6450.
- Foster, J.D., Young, S.E., Brandt, T.D. and Nordlie, R.C. 1998. Tungstate: A potent inhibitor of multifunctional glucose-6-phosphatase. Arch. Biochem. Biophys. 354:125–132.
- Gómez-Ramos, A., Domínguez, J., Zafra, D., Corominola, H., Gomis, R., Guinovart, J.J. and Avila, J. 2006. Sodium tungstate decreases the phosphorylation of tau through GSK3 inactivation. J. Neurosci. Res. 83:264–273.
- Haneke, K. 2003. Tungsten and Selected Tungsten Compounds. Edited by NIOEH Sciences.
- Happel, N. and Doenecke, D. 2009. Histone H1 and its isoforms: Contribution to chromatin structure and function. Gene 431:1–12.
- Haque, S.J., Flati, V., Deb, A. and Williams, B.R. 1995. Roles of protein-tyrosine phosphatases in Stat1 a-mediated cell signaling. J. Biol. Chem. 270:25709–25714.
- Heidari, Z., Harati, M., Mahmoudzadeh-Sagheb, H.R. and Moudi, B. 2008. Beta cell protective effects of sodium tungstate in streptozotocin-induced diabetic rats: glycemic control, blockage of oxidative stress and beta cell histochemistry. Iran. Biomed. J. 12:143–152.
- Holder, I.A., Frede, E.A., Neely, A.N. and Babcock, G.F. 2000. Reduction of neutrophil apoptosis with Pseudomonas aeruginosa immune serum. In: 5th World Congress on Trauma, Shock, Inflammation and Sepsis (Faist, E., Ed.), Bologna, Italy: Monduzzi Editore.
- Johnson, J.L., Rajagopalan, K.V. and Cohen, H.J. 1974. Molecular basis of the biological function of molybdenum. Effect of tungsten on xanthine oxidase and sulfite oxidase in the rat. J. Biol. Chem. 249:859–866.
- Kahl, S. and Elsasser, T.H. 2004. Endotoxin challenge increases xanthine oxidase activity in cattle: Effect of growth hormone and Vitamin E treatment. Domest. Anim. Endocrinol. 26:315–328.
- Kalinich, J.F., Emond, C.A., Dalton, T.K., Mog, S.R., Coleman, G.D., Kordell, J.E., Miller, A.C. and McClain, D.E. 2005. Embedded weapons-grade tungsten alloy shrapnel rapidly induces metastatic high-grade rhabdomyosarcomas in F344 rats. Environ. Health Perspect. 113:729–734.
- Kane, M.A., Kasper, C.E. and Kalinich, J.F. 2009. The use of established skeletal muscle cell lines to assess potential toxicity from embedded metal fragments. Toxicol. In Vitro 23:356–359.
- Knauf, J.A., Ouyang, B., Knudsen, E.S., Fukasawa, K., Babcock, G. and Fagin, J.A. 2006. Oncogenic RAS induces accelerated transition through G2/M and promotes defects in the G2 DNA damage and mitotic spindle checkpoints. J. Biol. Chem. 281:3800–3809.
- Konishi, A., Shimizu, S., Hirota, J., Takao, T., Fan, Y., Matsuoka, Y., Zhang, L., Yoneda, Y., Fujii, Y., Skoultchi, A.I. and Tsujimoto, Y. 2003. Involvement of histone H1.2 in apoptosis induced by DNA double-strand breaks. Cell. 114:673–688.
- Koutsospyros, A., Braida, W., Christodoulatos, C., Dermatas, D. and Strigul, N. 2006. A review of tungsten: From environmental obscurity to scrutiny. J. Hazard. Mater. 136:1–19.
- Kratzmeier, M., Albig, W., Meergans, T. and Doenecke, D. 1999. Changes in the protein pattern of H1 histones associated with apoptotic DNA fragmentation. Biochem. J. 337(Pt 2):319–327.
- Leggett, R.W. 1997. A model of the distribution and retention of tungsten in the human body. Sci. Total Environ. 206:147–165.
- Maemura, K., Zheng, Q., Wada, T., Ozaki, M., Takao, S., Aikou, T., Bulkley, G.B., Klein, A.S. and Sun, Z. 2005. Reactive oxygen species are essential mediators in antigen presentation by Kupffer cells. Immunol. Cell Biol. 83:336–343.
- McInturf, S.M., Bekkedal, M.Y., Wilfong, E., Arfsten, D., Gunasekar, P.G. and Chapman, G.D. 2008. Neurobehavioral effects of sodium tungstate exposure on rats and their progeny. Neurotoxicol. Teratol. 30:455–461.
- Muñoz, M.C., Barberà, A., Domínguez, J., Fernàndez-Alvarez, J., Gomis, R. and Guinovart, J.J. 2001. Effects of tungstate, a new potential oral anti-diabetic agent, in Zucker diabetic fatty rats. Diabetes 50:131–138.
- Okamura, H., Yoshida, K., Amorim, B.R. and Haneji, T. 2008. Histone H1.2 is translocated to mitochondria and associates with Bak in bleomycin-induced apoptotic cells. J. Cell. Biochem. 103:1488–1496.
- Piquer, S., Barceló-Batllori, S., Julià, M., Marzo, N., Nadal, B., Guinovart, J.J. and Gomis, R. 2007. Phosphorylation events implicating p38 and PI3K mediate tungstate-effects in MIN6b cells. Biochem. Biophys. Res. Commun. 358:385–391.
- Rose, F.R., Bailey, K., Keyte, J.W., Chan, W.C., Greenwood, D. and Mahida, Y.R. 1998. Potential role of epithelial cell-derived histone H1 proteins in innate antimicrobial defense in the human gastrointestinal tract. Infect. Immun. 66:3255–3263.
- Stankiewicz, P.J. and Gresser, M.J. 1988. Inhibition of phosphatase and sulfatase by transition-state analogues. Biochemistry. 27:206–212.
- Steinberg, K.K., Relling, M.V., Gallagher, M.L., Greene, C.N., Rubin, C.S., French, D., Holmes, A.K., Carroll, W.L., Koontz, D.A., Sampson, E.J. and Satten, G.A. 2007. Genetic studies of a cluster of acute lymphoblastic leukemia cases in Churchill County, Nevada. Environ. Health Perspect. 115:158–164.
- Victorino, G.P., Ramirez, R.M., Chong, T.J., Curran, B. and Sadjadi, J. 2008. Ischemia-reperfusion injury in rats affects hydraulic conductivity in two phases that are temporally and mechanistically separate. Am. J. Physiol. Heart Circ. Physiol. 295:H2164–H2171.
- Vorbach, C., Harrison, R. and Capecchi, M.R. 2003. Xanthine oxidoreductase is central to the evolution and function of the innate immune system. Trends Immunol. 24:512–517.
- Yan, S. R., Fumagalli, L., Dusi, S. and Berton, G. 1995. Tumor necrosis factor triggers redistribution to a Triton X-100-insoluble, cytoskeletal fraction of beta 2 integrins, NADPH oxidase components, tyrosine phosphorylated proteins, and the protein tyrosine kinase p58fgr in human neutrophils adherent to fibrinogen. J Leukoc Biol. 58 (5):596–606.
- Zlatanova, J.S., Srebreva, L.N., Banchev, T.B., Tasheva, B.T. and Tsanev, R.G. 1990. Cytoplasmic pool of histone H1 in mammalian cells. J. Cell. Sci. 96 (Pt 3):461–468.