Abstract
Here, we investigated whether titanium dioxide (TiO2) nanoparticles affect in vivo tumor growth through the modulation of mononuclear leukocytes. In vitro lymphocyte proliferation by lipopolysaccharide (LPS) or concanavalin A (ConA) was reduced by < 25 nm TiO2 with a dose-dependent manner. Similarly, TiO2 nanoparticles inhibited nitric oxide (NO) production from bone marrow-derived macrophages obtained from naïve mice. When mice were intraperitoneally (IP) injected with < 25 or < 100 nm TiO2 once a day for 7 days, total cell number of splenocytes was reduced in the spleen of TiO2 nanoparticle-exposed mice. Both CD4+ and CD8+ T-lymphocyte numbers were significantly decreased and B-lymphocyte development was retarded by host exposure to the TiO2 nanoparticles. LPS-stimulated spleen cell proliferation was significantly reduced by host exposure to < 25 or < 100 nm TiO2, but no changes were detected in ConA-stimulated spleen cell proliferation. Further, LPS-stimulated cytokine production by peritoneal macrophages and the percentage of NK1.1+ natural killer cells among splenocytes was reduced by the host exposures to the TiO2 nanoparticles. When mice were IP injected with TiO2 nanoparticles once a day for 28 days prior to the subcutaneous implantation of B16F10 melanoma cells, tumor growth was subsequently significantly increased. Collectively, these results show that TiO2 nanoparticles may damage the development and proliferation of B- and T-lymphocytes, reduce the activity of macrophages, and decrease natural killer (NK) cell population levels, outcomes that appear to lead to an increase in tumor growth in situ. These studies allow us to suggest that TiO2 nanoparticles might have the potential to enhance tumor growth through immunomodulation of B- and T-lymphocytes, macrophages, and NK cells.
| Abbreviations: | ||
| TiO2, | = | Titanium dioxide; |
| FBS, | = | fetal bovine serum; |
| RBC, | = | red blood cells; |
| ROS, | = | reactive oxygen species; |
| LPS, | = | lipopolysaccharide; |
| ConA, | = | concanavalin A; |
| FITC, | = | fluorescein isothiocyanate; |
| PE, | = | phycoerythrin; |
| IL, | = | interleukin. |
Introduction
Titanium dioxide (TiO2) nanoparticles have been widely used as a photocatalyst in air and water cleaning, cosmetics, and pharmaceuticals (Gélis et al., Citation2003; Sun et al., Citation2004). Thus, TiO2 nanoparticles have the potential to impact upon our daily life. However, before the use of any TiO2 nanoparticles for direct human exposure could be approved, both their beneficial and harmful effects on the human population need to be addressed; such information can be gleaned by understanding the effects of these agents on biological systems (Dobrovolskaia and McNeil, Citation2007). Among the many potential health effects that could evolve from exposure, nanoparticles have been shown to possess an immunomodulatory potential, as they can stimulate or suppress the immune system (Dobrovolskaia and McNeil, Citation2007; Dwivedi et al., Citation2009). However, as both conditions are undesirable, any nanoparticle-based therapeutics should avoid immunostimulatory or immunosuppressive reactions once the nanomaterials have been administered into the body. Because one additional effect of the modification by nanoparticles of the immune system is that this, in turn, alters the bioavailability and prolongs host exposure time to the particles themselves (Dwivedi et al., Citation2009), more data about the impact of these agents needs to be generated via in vivo studies.
Nanoparticles could induce host responses, i.e., changes in reactive oxygen species (ROS) generation, formation/release of inflammatory cytokines, and matrix metalloproteinase (MMP) release, that each/together can play a major role in tissue destruction and remodeling (Wan et al., Citation2008). It is already known that TiO2 nanoparticles can induce pulmonary emphysema, macrophage accumulation, extended disruption of alveolar septa, Type II pneumocyte hyperplasia, and epithelial cell apoptosis (Chen et al., Citation2006). Following inhalation of TiO2 nanoparticles, local macrophages were found to be activated (Chen et al., Citation2006; Geiser et al., Citation2008); within these cells, there was the induced differential expression of hundreds of genes involved in cell cycle regulation, apoptosis, chemokine formation, and activation of the complement cascade (Chen et al., Citation2006). When mice were exposed to TiO2 nanoparticles (by inhalation) acutely or subacutely, histopathologic changes with a significant but moderate inflammatory response were detected in their lung tissues (Grassian et al., Citation2007). In in vitro studies, TiO2 nanoparticles induced both apoptotic and necrotic modifications in U937 cells (Vamanu et al., Citation2008). In another study, formation of interleukin (IL)-8 was induced in BEAS-2B cells (derived from human bronchial epithelial cells) by TiO2 nanoparticles via p38 mitogen-activated protein kinase (MAPK) and/or extracellular-signal-regulated kinase (ERK) pathways (Park et al., Citation2008).
Related to its effects on the immune system, TiO2 also has serious health implications with regard to cancer surveillance, especially in the lungs. The World Health Organization (i.e., its International Agency for Research on Cancer) has classified TiO2 as a Group 2B carcinogen, i.e., possible carcinogen to humans (IARC, Citation2006). In one study, treatment with TiO2 nanoparticles by intrapulmonary spraying for 9 days significantly increased the multiplicity of N-nitroso-bis-(2-hydroxypropyl)-amine-induced alveolar cell hyperplasias and adenomas in the lung and the multiplicity of mammary adenocarcinomas (Xu et al., Citation2010). TiO2 nanoparticles also have the potential to convert benign tumor cells to malignant ones via the generation of ROS in the target cells (Onuma et al., Citation2009). Nevertheless, little is known about effects of TiO2 nanoparticles on tumor growth in the context of modulated immunosurveillance and less so with regard to mechanistic bases for these outcomes; this is especially true with regard to long-term exposures in vivo.
In this study, to begin to clarify potential mechanisms for the above-noted effects on host tumor immunosurveillance, the effect of TiO2 nanoparticles upon both mononuclear leukocytes and solid tumor growth were investigated. Examinations of potential effects of the particles on lymphocyte development and the percentages of natural killer (NK) cells and CD11b+ cell populations in the exposed hosts were also performed. The data here revealed that TiO2 nanoparticles were immunosuppressive to mouse immune cells and led to an augmentation of solid tumor growth in mice that were intraperitoneally (IP) exposed daily to the particles prior to/after tumor implantation.
Materials and methods
Mice and reagents
C57BL/6J mice were obtained from Dae-Han Biolink (Chungju, South Korea). Animals were maintained in the pathogen-free authorized facility in Sejong University (Seoul, South Korea) where temperature was maintained at 20–22°C, relative humidity at 50–60%, and with 12-h dark/light cycles. All animals were provided water and mouse (rodent) chow (Purina Co., Seoul, South Korea) ad libitum. All animal procedures were conducted in accordance with the guidelines of the institutional Animal Care and Use Committee at Sejong University.
TiO2 nanoparticles sized at < 25 nm (Cat No. 637254) or < 100 nm (Cat No. 634662) were purchased from Sigma (St. Louis, MO). Fluorescein isothiocyanate (FITC)- or phycoerythrin (PE)-labeled antibodies were obtained from BD Biosciences (Franklin Lakes, NJ). Lipopolysaccharide (LPS) and concanavalin A (ConA) were purchased from Sigma-Aldrich and resuspended at 1 mg/ml serum-free RPMI 1640 and stored at−20°C. Except where indicated, all other materials were obtained from the Sigma.
In vivo exposures to TiO2 nanoparticles
TiO2 nanoparticles were suspended in normal saline to yield a stock 1.0 mg particle/ml solutions. Prior to use for injection, the suspensions underwent sonication to assure no significant agglomeration in the aqueous solution took place. Immediately after this step, C57BL/6J mice (n = 5 per group) were IP injected with ~200 µl of the suspension of < 25- or < 100-nm TiO2 nanoparticles such that a dose of 10 mg particle/kg was achieved. The animals were thereafter weighed daily and the amount of saline needed to dilute the stock solution (so as to prepare the 200 µl injection volume) adjusted accordingly based on these weight values.
For analysis of T- and B-lymphocyte development, mice were daily injected for 7 days prior to preparation of splenocyte, thymocyte, and peritoneal macrophage suspensions 1 day after the final injection. In addition, to assess effects of the nanoparticles on tumor growth, additional sets of mice were daily injected for 28 days prior to subcutaneous implantation of B16F10 mouse melanoma cells into their flank skin. These now-tumor-bearing mice continued to receive daily TiO2 nanoparticle injections until sacrifice 24 h after the final TiO2 exposure on Day 11 post-implantation; on Day 12 post-implantation, the tumor was excised for analyses.
Cell culture
B16F10 mouse melanoma cells (ATCC, CRL-6475) used for analyses of in situ tumor growth were obtained from the Korea Institute of Radiological and Medical Science (KIRMS) cell bank (Seoul, Korea). Cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS; Hyclone, Kansas City, MO), 2 mM l-glutamine, 100 U penicillin/ml, and 100 U streptomycin/ml in a 5% CO2 incubator maintained at 37°C.
Splenocytes prepared from TiO2-exposed and control mice, as well as from naïve mice (for in vitro studies), were cultured in RPMI 1640 supplemented with 10% FBS, 2 mM l-glutamine, 100 U penicillin/ml, and 100 U streptomycin/ml in a 5% CO2 incubator maintained at 37°C.
Cell preparations
After euthanization, spleens and thymuses were dissected from the control or TiO2 nanoparticle-injected mice and placed in RPMI 1640 supplemented with 2% FBS. Splenocytes and thymocytes were obtained by teasing the respective tissues gently with the plunger of a 3-ml syringe and then sieving the suspension through a 70-µm cell strainer (Falcon, BD Biosciences). Red blood cells (RBC) were then removed by incubating the cell mixture with RBC Lysis Buffer (Sigma) for 5 min at room temperature. Cells thus prepared were washed twice with RPMI 1640 and their numbers adjusted to appropriate concentrations for placement in 24- or 96-well plates (Corning Inc., Life Sciences, Lowell, MA) to be used in the various assays described below.
Generation of bone marrow-derived macrophages (BMDM)
BMDM were generated using marrow cells from the femurs and tibias of the naive mice used for isolation of splenocytes for the in vitro studies here. RBC were removed by incubating the cell mixture with RBC Lysis Buffer for 5 min at room temperature and then washed twice with serum-free RPMI 1640. The resultant cells were further incubated at 5 × 105 cell/well in RPMI 1640 supplemented with 20 ng M-CSF/ml (R&D System, Minneapolis, MN) for 3–4 days. Non-adherent cells were discarded on Day 3, and culture medium was replenished with fresh medium. On day 7, adherent cells were harvested for use in the experiments outlined below.
Cell proliferation assay after in vitro exposure to TiO2 nanoparticles
Splenocytes from the naïve mice were plated at 2 × 105 cells/well in a 96-well plate and cultured with various concentrations of TiO2 nanoparticles in the presence or absence of LPS (25 µg/ml) or ConA (2 µg/ml) for 48 h. As in the in vivo studies, the nanoparticles were suspended in saline to yield stock 1.0 mg particle/ml solutions. Prior to their use in treating the cells, each suspension underwent sonication to assure no agglomeration had taken place. Following the 48-h incubation period, the cells were then pulsed with 1 µCi [3H]-thymidine/well (113 Ci/nmol, NEN, Boston, MA) for 4–6 h before collection on nitrocellulose filters using an automated cell harvester (Inotech, Dottikon, Switzerland). Total [3H]-thymidine incorporated into cells was measured (as cpm) in a Microbeta scintillation counter (Wallac, Turku, Finland).
Nitrite formation after in vitro exposure to TiO2 nanoparticles
Accumulated nitrites were measured in the cell supernatant by the Griess reaction (Green et al., Citation1982). In brief, 100 µl of supernatant from each well was mixed with 100 µl Griess reagent (a mixture of 0.1% naphthylethylenediamine dihydrochloride and 1% sulfanilamide, in 2% phosphoric acid) in 96-well microtiter plates. The absorbance in each well was then read at 540 nm using an enzyme-linked immunosorbent assay (ELISA) reader (Molecular Devices, Sunnyvale, CA).
MTT assay
Ex vivo cell proliferation by splenocytes from naïve mice was quantified using a colorimetric assay that measures intracellular mitochondrial enzyme (succinate dehydrogenase) activity with an MTT [3(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide] substrate (Denizot and Lang, Citation1986; Moon, Citation2008). Briefly, isolated splenocytes were cultured with LPS (25 µg/ml) or ConA (2 µg/ml) for 48 h. The splenocytes were then received 50 µg MTT/ml and were incubated a further 2 h at 37°C. The formazan that had formed within the cells was then dissolved by addition of 100 μl dimethyl sulfoxide. Optical density (OD) in each well on the plate was then read at 540 nm using an ELISA plate reader (Molecular Devices).
Cell proliferation assay after in vivo exposure to TiO2 nanoparticles
Splenocytes from control mice or those injected daily for 7 days prior to harvesting of the cells were plated at 2 × 105 cells/well in 96-well plates and cultured in the presence or absence of LPS (25 µg/ml) or ConA (2 µg/ml) for 48 h. Proliferative activity was then assessed using the MTT assay described above.
ELISA
Peritoneal macrophages (PEM) from TiO2 nanoparticle-injected mice were plated at 1 × 106 cells/ml and stimulated with LPS (1 µg/ml) for 12 h. Culture supernatants were then collected and the concentrations of TNF-α, IL-6, and IL-1β determined by sandwich immunoassays using a protocol supplied by R&D Systems. Absorbance values in each well were measured at 450 nm using ELISA reader (Molecular Devices).
Flow cytometric analyses
Aliquots of 1.0 × 106 thymocytes or splenocytes (as separate populations) from control or TiO2-injected mice were suspended in 2% FCS containing RPMI 1640. Thymocytes were then treated with FITC-conjugated anti-CD4 and PE-conjugated anti-CD8 antibodies; splenocytes received FITC-conjugated anti-CD43, -CD21, -IgD antibodies, and/or PE-conjugated anti-IgM antibodies. Splenocytes were also treated with PE-conjugated anti-NK1.1 antibodies and FITC-conjugated anti-CD11b antibodies for analysis of NK cells. Each suspension was incubated with antibodies for 20 min on ice. Thereafter, the cells were washed with medium and analyzed by CELLQuest™ software in an FACScalibur™ system (Becton Dickinson, Franklin Lakes, NJ). A minimum of 10,000 events were analyzed/cell population of interest.
In vivo tumor growth
Mice were subcutaneously implanted with 2 × 105 B16F10 mouse melanoma cells. Tumor growth was monitored by the measurement of short axis and long axis of tumor mass with Vernier calipers (Mititoyo, Mizuyo, Japan). Tumor volume was calculated by the multiplication of the [(long axis) / 2] by the (short axis)2. Mice were sacrificed on the 12th day after the implantation of B16F10 cells and tumor mass was excised to measure its weight.
Statistical analyses
Experimental differences were tested for statistical significance using analysis of variance (ANOVA) and Student’s t-test. A P value of < 0.05 or < 0.01 was considered to be significant.
Results
In vivo TiO2 nanoparticle effects on B- and T-lymphocyte proliferation and BMDM nitric oxide formation
To investigate effects of TiO2 nanoparticles on immune cells, effects on their viability first had to be determined. To achieve this, the activity of mitochondrial succinate dehydrogenase in spleen cells from naïve mice was assessed following in vitro exposure to the TiO2 nanoparticles. Using an MTT assay (Moon, Citation2008) to quantify this activity, no changes in viability were seen to be induced by either size TiO2 nanoparticle at concentrations from 0.1 to 10 µg/ml (). Of note, enzyme activity was significantly increased by the nanoparticles at concentrations from 30 to 100 µg/ml. Taken together, these results indicate that the doses of TiO2 nanoparticle used here did not adversely impact on the viability of spleen cells treated ex vivo with either size particle. Future studies will utilize alternative measures (i.e., propidium iodide staining and flow cytometric analyses) to better clarify if there is really any shift in viability—especially at high particle doses—since the noted increases in MTT activity (alone suggestive of spontaneous induced proliferation) might be masking any concurrent increase in cell death.
Figure 1. Inhibition of splenocyte proliferation by TiO2 nanoparticles after in vitro exposures. Splenocytes were prepared from naive C57BL/6 mice. (A) Splenocytes were cultured with various concentrations of TiO2 nanoparticles and cell viability then measured using an MTT assay. (B and C) Splenocytes were cultured with various concentrations of TiO2 nanoparticles in the presence of (B) lipopolysaccharide (LPS, 25 µg/ml) or (C) concanavalin A (ConA, 2 µg/ml) for 48 h. Cells were then pulsed with 1 µCi [3H]-thymidine/well for 4–6 h before collection onto nitrocellulose filters. Total [3H]-thymidine incorporated into cells was measured (as cpm) via a scintillation counter. (D) Bone marrow-derived macrophages (BMDM) from naïve mice were stimulated with LPS in the presence of various concentrations of TiO2 nanoparticles for 48 h. Nitric oxide (NO) production in the supernatant was then measured by the Griess reaction. Each bar shown is a representative of three experiments performed. Data shown are in terms of mean ± SE. Values significantly (*P < 0.05, **P < 0.01) different from LPS- or ConA-stimulated control (receiving medium only) counterpart cultures [as applicable] are indicated.
![Figure 1. Inhibition of splenocyte proliferation by TiO2 nanoparticles after in vitro exposures. Splenocytes were prepared from naive C57BL/6 mice. (A) Splenocytes were cultured with various concentrations of TiO2 nanoparticles and cell viability then measured using an MTT assay. (B and C) Splenocytes were cultured with various concentrations of TiO2 nanoparticles in the presence of (B) lipopolysaccharide (LPS, 25 µg/ml) or (C) concanavalin A (ConA, 2 µg/ml) for 48 h. Cells were then pulsed with 1 µCi [3H]-thymidine/well for 4–6 h before collection onto nitrocellulose filters. Total [3H]-thymidine incorporated into cells was measured (as cpm) via a scintillation counter. (D) Bone marrow-derived macrophages (BMDM) from naïve mice were stimulated with LPS in the presence of various concentrations of TiO2 nanoparticles for 48 h. Nitric oxide (NO) production in the supernatant was then measured by the Griess reaction. Each bar shown is a representative of three experiments performed. Data shown are in terms of mean ± SE. Values significantly (*P < 0.05, **P < 0.01) different from LPS- or ConA-stimulated control (receiving medium only) counterpart cultures [as applicable] are indicated.](/cms/asset/0a5016f5-0c5a-4535-8372-3d0d3177534d/iimt_a_543995_f0001_b.gif)
Having seen that the doses of the nanoparticles used here did not overtly impact on cell viability, studies of the particles’ impact on lymphocyte proliferation were then performed. Specifically, splenocytes from naïve mice were stimulated in vitro with LPS or ConA as B- and T-lymphocyte mitogens, respectively, in the presence or absence of < 25- or < 100-nm TiO2 nanoparticles. As shown in , LPS-stimulated lymphocyte proliferation was reduced by the < 25-nm TiO2 nanoparticles with the concentrations ≥ 25 µg TiO2/ml. ConA-stimulated lymphocyte proliferation was decreased by these particles at concentrations ≥ 50 µg TiO2/ml (). In contrast, no changes in LPS- or ConA-stimulated lymphocyte proliferation were detected after exposures to the < 100-nm TiO2 nanoparticles. No changes in proliferation were induced by 1-µm (micron-sized) TiO2 (data not shown).
In the analyses of effects of the particles on NO formation by BMDM, whereas LPS-induced NO production by these derived cells was decreased by treatments with 10 µg TiO2 nanoparticle/ml—with particles of either size (), no effects were detected at concentrations of either particle at < 1 µg/ml.
In vivo exposure to TiO2 nanoparticles affected B- and T-lymphocyte development
Because B-lymphocytes pass through various stages of maturation (Ollila and Vihinen, Citation2005; Srivastava et al., Citation2005; Casola, Citation2007), the studies here also investigated whether TiO2 nanoparticles affected B-lymphocyte development in the spleen of hosts exposed daily (for 7 days) to the particles. Using various FITC- or PE-labeled antibodies for analyses of B-lymphocytes in various stages of development (maturation), no changes were detected in the size of the CD43+ IgM− pro-B-lymphocytes or CD21− IgM+ T1 B-lymphocyte populations due to the 7 days of TiO2 nanoparticle exposures (). In contrast, the sizes of the CD21+ IgM+ T2 B-lymphocyte, the mature IgD+IgM+, and the re-circulating IgD+IgM− B-lymphocyte populations were each decreased by the IP injections of TiO2 nanoparticles (of either diameter tested; ). No changes were induced by 1-µm (micron-sized) TiO2 particles in any of the B-lymphocyte populations (data not shown).
Figure 2. Inhibition of ex vivo lymphocyte development due to exposure to TiO2 nanoparticles. Mice were intraperitoneally (IP) injected with 10 mg TiO2 nanoparticles/kg once a day for 7 days. (A) Splenocytes and (B) thymocytes from each animal were prepared from organs harvested 24 h after the final TiO2 injection. One million cells from each organ were then incubated on ice for 30 min with fluorescent-labeled antibodies to the indicated markers. The cells were then washed twice with RPMI 1640-5% fetal bovine serum (FBS) and then analyzed via flow cytometry. Each population was determined and sizes compared between the control and TiO2 nanoparticle-injected groups. Each bar shown is a representative of three experiments performed. Data shown are in terms of mean ± SE. Value significantly (*P < 0.05, **P < 0.01) different from the control group.
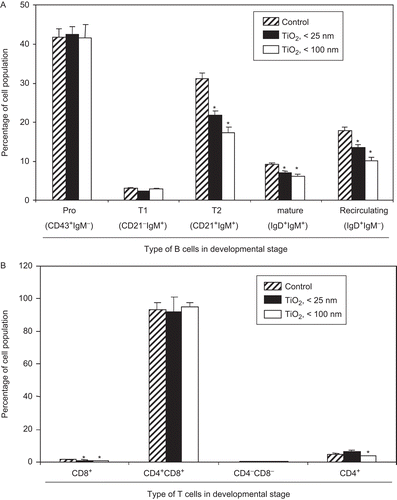
As T-lymphocytes develop in the thymus, CD4+ or CD8+ single positive (SP) cells mature from CD4+CD8+ double positive (DP) forms via positive and negative selection. The DP cells are themselves derived from CD4−CD8− double negative (DN) T-lymphocytes (Ellmeier et al., Citation1999; Sohn et al., Citation2003). The studies here also investigated whether TiO2 nanoparticles affected T-lymphocyte development in the thymus of hosts exposed daily (for 7 days) to the particles. Significant reductions in population sizes of the CD4+ or CD8+ SP T-lymphocytes were noted; with CD4+ SP cells, the effect was only evident with the < 100 nm particles, whereas each diameter particle caused significant reductions in CD8+ SP cell levels (). No significant changes were detected in levels of splenic CD4−CD8− DN and CD4+CD8+ DP T-lymphocytes.
In vivo exposure to TiO2 nanoparticles reduced B-lymphocyte proliferation, macrophage activity, and NK cell population size
To confirm that the in vitro effect of TiO2 nanoparticles on B- and T-lymphocyte proliferative activity also might take place in situ, splenocytes from hosts exposed daily to TiO2 nanoparticles for 7 days were incubated with LPS or ConA. To allow for better interpretation of any results pertaining to potential changes in proliferative activity, analyses of the isolated splenocytes (total numbers, individual cell types) were performed. As seen in , total spleen cell number was decreased by ≈30–50% relative to the control mice populations in both (i.e., different diameter particle) TiO2-exposed groups. Irrespective of these overall shifts in total population sizes, when equal populations (i.e., numbers) of splenocytes were examined by flow cytometry, no significant changes were noted in the percentages of cells that were apoptotic (). Similarly, the percentages of B220+ B- and CD3+ T-lymphocytes were relatively unchanged; values for each cell type were, respectively, ≈49–53% and ≈40–43%, regardless of the size TiO2 (control values were, respectively, 50 and 41% of total population analyzed). Nevertheless, when we apply these cell percentage values to the starting population sizes, it becomes apparent that the absolute cell counts for B- and T-lymphocyte populations would seem to be significantly lower (by ≈20–30% and ≈10–30%, respectively) in the spleens of these different diameter particle TiO2-exposed hosts than those in the organs of control hosts (). In this case, the 100 nm nanoparticles had the greater effect on both cell types. Taken together, although the overall size of the splenocyte populations and those of the individual cell types appeared to have been adversely impacted by the TiO2 treatments, these effects did not seem to be cell type-specific.
Figure 3. LPS-stimulated ex vivo splenocyte proliferation was reduced by host exposure to nanoparticle TiO2. Mice were IP injected with 10 mg TiO2 nanoparticles/kg once a day for 7 days. (A) Splenocytes from control and TiO2-injected mice were prepared from organs harvested 24 h after the final TiO2 injection and total spleen cell number determined. (B) One million isolated splenocytes/host were incubated with anti-B220 and anti-CD3 antibodies to permit flow cytometric determination, respectively, of B- and T-lymphocyte levels. Each population was determined and sizes compared between the control and nanoparticle-injected groups. Numbers inside each bar indicate percentage of each cell type in the total population analyzed. (C) Splenocytes were cultured with LPS (25 µg/ml) or ConA (2 µg/ml) for 48 h and cell proliferation then measured using an MTT assay. Each bar shown is a representative of three experiments performed. Data shown are in terms of mean ± SE. Value significantly (*P < 0.05, **P < 0.01) different from that of the control group.
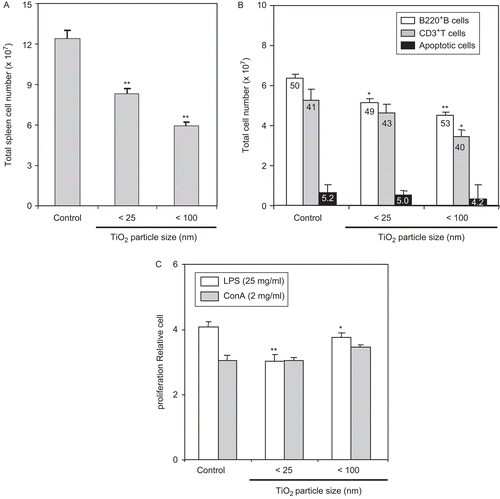
With respect to effects on the proliferative capacity of the splenic cells recovered from the nanoparticle-treated hosts, LPS-induced cell proliferation by splenocytes from the mice that received either diameter particle was reduced ≈10–25% (). In contrast, no changes were detected in ConA-stimulated proliferation by these cells. These outcomes are consistent with the data in and that demonstrated that the TiO2 nanoparticles (primarily those < 25 nm) impacted far more strongly upon B-lymphocytes.
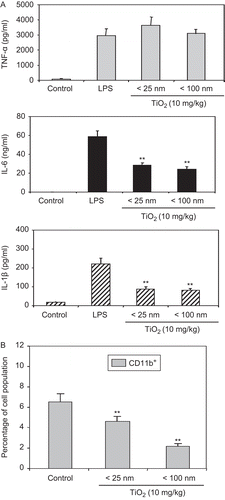
Given that CD11b+ including macrophages and NK cells can control innate immunity (Colucci et al., Citation2002; Moretta et al., Citation2002), any changes in CD11b+ cell function due to the particle exposures were examined. The activity of CD11b+ PEM was assessed via measures of their production of three select cytokines, for example, TNF-α, IL-6, and IL-1β. As shown in , production of IL-6 and IL-1β was decreased by ≈50% as a result of daily exposures for 7 days to < 25- or < 100-nm TiO2 particles. No changes in TNF-α production were detected. These latter results are in contrast with our in vitro data that showed that LPS-stimulated TNF-α production was decreased from BMDM (as compared to control cells) when they were co-treated with < 25- or < 100-nm TiO2 particles (data not shown). The percentages of CD11b+ cells in the mouse spleen were also examined to provide a numerical (as opposed to a functional) basis for any changes that were noted in the measured activities of the harvested PEM. As shown in , the percentage of CD11b+ cells was significantly reduced by host exposures to either diameter of TiO2 nanoparticle. It is interesting to note that whereas each diameter particle yielded an ≈50% reduction in the release of IL-6 and IL-1β, the total changes in population/cells available were much more reduced in mice that had received the larger diameter particles. This could suggest that the smaller particles were more toxic at a cytokine formation level than the larger forms (i.e., more cells available, yet still producing same amounts of cytokine as the smaller “< 100 nm population”).
Figure 4. Inhibitory effect of TiO2 nanoparticles on cytokine production. Mice were IP injected with 10 mg TiO2 nanoparticles/kg once a day for 7 days. Peritoneal cells were then collected from the peritoneal cavity of each mouse (with RPMI 1640 medium) 24 h following the final TiO2 injection. (A) Cells were stimulated with LPS for 48 h and culture supernatants were then collected for analyses of TNF-α (top), IL-6 (middle), and IL-1β (bottom) levels by enzyme-linked immunosorbent assay (ELISA). Data shown are in terms of mean ± SE. Value significantly (**P < 0.01) different from the control group. (B) Splenocytes from these control and TiO2 nanoparticle-injected mice were prepared and reacted with fluorescein isothiocyanate (FITC)-labeled anti-CD11b antibodies for 30 min. The cells were then washed twice with RPMI 1640-5% FBS and then analyzed via flow cytometry. Each population was determined and sizes compared between the control and TiO2 nanoparticle-injected groups. Each bar shown is a representative of three experiments performed. Data shown are in terms of mean ± SE. Value significantly (**P < 0.01) different from that of the control group.
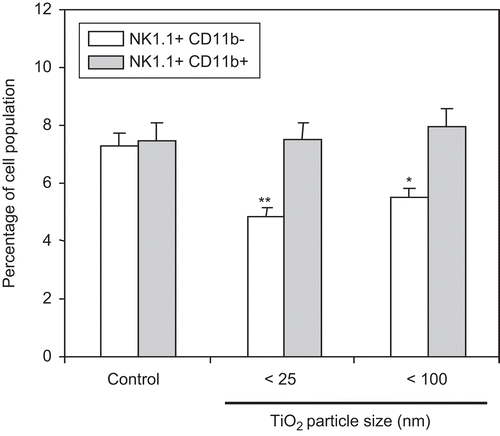
Given that NK cells can be activated by CD11b+ cells and play a key role in controlling the development of certain types of malignancies in humans (Trinchieri, Citation1989; Lotzová, Citation1991; Colucci et al., Citation2002; Moretta et al., Citation2002), this study also examined changes in NK cell population sizes as a result of the host daily exposures for 7 days to the TiO2 nanoparticles. As illustrated in , the percentage of NK1.1+CD11b− cells was reduced ≈30–40% by host exposures to either diameter TiO2 nanoparticle. No changes were detected in the percentage of NK1.1+CD11b+ cells.
Figure 5. Measurement of NK1.1+/CD11b+ cells in spleens of mice pre-exposed to TiO2 nanoparticles. Mice were IP injected with 10 mg TiO2 nanoparticles/kg once a day for 7 days. Splenocytes from control and TiO2-injected mice were prepared from organs harvested 24 h after the final TiO2 injection and reacted with FITC-labeled anti-CD11b or PE-labeled anti-NK1.1 antibodies for 30 min. The cells were then washed twice with RPMI 1640-5% FBS and each cell analyzed via flow cytometry. Each population was determined and sizes compared between the control and TiO2 nanoparticle-injected groups. Each bar shown is a representative of three experiments performed. Data shown are in terms of mean ± SE. Value significantly (*P < 0.05, **P < 0.01) different from that of the control group.
Solid tumor growth was increased by host pre-exposure to TiO2 nanoparticles
To confirm the immunosuppressive effect of TiO2 nanoparticles in vivo, we investigated whether changes in immunological function could also impact upon tumor growth (i.e., reflect an alteration in tumor immunosurveillance). Specifically, this study examined the effect of TiO2 nanoparticles on the growth of B16F10 mouse melanoma in situ. Mice were IP injected daily with 10 mg/kg TiO2 nanoparticles for 4 weeks and then implanted with B16F10 mouse melanoma cells. No significant changes in body weight of animals were detected as a result of TiO2 exposure prior to tumor implantation (). Such results imply there was likely to have been little “overt” toxicity (i.e., stress/induction of hypothalamus pituitary adrenocortical axis [HPA] system) as a result of the TiO2 exposures that clearly would have complicated the ability to discern relationship(s) between changes in tumor size/growth and tumor immunosurveillance.
Figure 6. Tumor growth was increased by host pre-tumor implantation exposure of TiO2 nanoparticles. Five mice per group were IP injected with 10 mg TiO2 nanoparticles/kg once a day for 28 days. B16F10 mouse melanoma cells were then subcutaneously implanted 24 h after the final nanoparticles injection; control mice were injected with vehicle only. Tumor size was then measured daily with Vernier calipers for 12 days. (A) Body weight of each animal was measured daily. (B) Tumor volume was calculated by the multiplication of the [(long axis)/2] × (short axis)2. The line graph shown is a representative of three experiments performed. Data shown are in terms of mean ± SE. Value significantly (*P < 0.05, **P < 0.01) different from that of the control group.
![Figure 6. Tumor growth was increased by host pre-tumor implantation exposure of TiO2 nanoparticles. Five mice per group were IP injected with 10 mg TiO2 nanoparticles/kg once a day for 28 days. B16F10 mouse melanoma cells were then subcutaneously implanted 24 h after the final nanoparticles injection; control mice were injected with vehicle only. Tumor size was then measured daily with Vernier calipers for 12 days. (A) Body weight of each animal was measured daily. (B) Tumor volume was calculated by the multiplication of the [(long axis)/2] × (short axis)2. The line graph shown is a representative of three experiments performed. Data shown are in terms of mean ± SE. Value significantly (*P < 0.05, **P < 0.01) different from that of the control group.](/cms/asset/1e5408f1-74f4-493f-9c86-b2412a15249f/iimt_a_543995_f0006_b.gif)
The now-tumor-bearing mice continued to receive daily TiO2 nanoparticle injections until they were sacrificed 24 h following the final TiO2 exposure on Day 11 post-implantation; on Day 12 post-implantation, the tumor was excised for analyses. As shown in , by Day 12 post-implantation, the tumor growth was ≈30% higher in TiO2-injected mice (regardless of particle diameter) as compared to among the control hosts. The difference in the ability of the injected cells to grow into tumors in the particle-exposed hosts was already evident by Day 8 post-implantation; these differences became significant by Day 10. Excised tumor size was also bigger in the TiO2 nanoparticle-injected mice than in their control counterparts (pictures not shown; available to readers upon request). Accordingly, tumor weights in both the < 25- and < 100-nm TiO2-injected mice groups were about twice that of the tumors in the control mice (data not shown). These outcomes allow for the suggestion to be made that the immunosuppressive effects of TiO2 nanoparticles noted herein may give rise to an increase in the incidence and/or growth of tumors in situ.
Discussion
For successful nanoparticle-based therapeutics/products to be developed for marketing, any undesirable immunomodulation by the particles must be defined and appropriately prevented (Dwivedi et al., Citation2009). Among the many nanoagents used in cosmetics and pharmaceuticals, TiO2 nanoparticles have a significant ongoing/increasing potential to impact on our daily lives. However, although many studies (most often related to occupational exposure impact) have documented the immunomodulatory effects of the parent TiO2 (see reviews of Di Gioacchino et al., Citation2007; Forte et al., Citation2008), in general these effects have yet to be clearly defined for nanoparticle forms. Further, whether effects on the immune response induced by TiO2 nanoparticles mitigate, exacerbate, or have no impact at all on tumor growth remains to be determined. This is because the hypothesis of tumor immunosurveillance suggests that though neoplasms arise frequently in the body, most are destroyed almost at inception by a rigorous immune response (Prehn, Citation2006; Bui and Schreiber, Citation2007).
Here, we investigated the effect of TiO2 nanoparticles on immune cells and solid tumor growth. We found that TiO2 nanoparticles reduced B-lymphocyte development, LPS-stimulated splenocyte proliferation, LPS-induced cytokine production from PEM, and the percentage of NK cells in the spleen. The data also revealed that TiO2 nanoparticle effects on B- and T-lymphocytes differed. Whereas the TiO2 nanoparticles impaired both B-lymphocyte development and proliferation, effects on T-lymphocyte development (more precisely, conversion to either SP form) appeared to be particle size-dependent. The data also illustrated that tumor growth in mice exposed to TiO2 nanoparticles for 4 weeks was increased as compared to that in control hosts. Based on our findings, it is plausible to suggest that TiO2 nanoparticles could enhance tumor growth via effects on host immune cells leading to altered tumor immunosurveillance. These results are in accordance with a previous report that nanocrystalline fullerene suspensions could potentiate tumor growth in vivo, possibly by causing an NO-dependent suppression of anticancer immune responses (Zogovic et al., Citation2009).
There are a few possible explanations to explain our data indicating decreases in immune cell function in conjunction with increases in tumor growth. First, it is possible that the TiO2 nanoparticles damaged immune cells (and thereby caused reductions in their levels) via apoptosis or necrosis. It has been shown previously in vitro that TiO2 nanoparticles induced both apoptotic and necrotic modifications in U937 cells (Vamanu et al., Citation2008). Even if the levels of certain immune cell types were not overtly altered by the nanoparticles, critical cell functions (including release of cytokines, chemokines, inflammatory agents) may have been impacted. It is well known that inhalation of TiO2 nanoparticles can induce a significant (local) inflammatory response in exposed animals (Grassian et al., Citation2007). In the case of respiratory epithelial cells (i.e., BEAS-2B cells), formation/release of inflammatory cytokines was induced by TiO2 nanoparticles through activation of p38 MAPK/ERK signal pathways (Park et al., Citation2008).
A second explanation might be that host macrophages had become differentially polarized by the TiO2 nanoparticles. Macrophages can promote specific immunity (including removal of cancer cells) by inducing granulocyte recruitment as well as T- and B-lymphocyte activation. Nevertheless, the presence of macrophages within the tumor itself/microenvironment has been associated with enhanced tumor progression (i.e., promoted cancer cell growth and spread, increased local angiogenesis, and tumor-driven immunosuppression). This paradoxical role of macrophages in cancer finds an explanation in functional plasticity (Sica et al., Citation2008). Depending on functional state, macrophages can be divided into fully polarized M1 and M2 subtypes (Mantovani, Citation2006). M1 forms are characterized, in part, by: production of large amounts of reactive oxygen/nitrogen intermediates (ROS/RNS) and inflammatory cytokines; an IL-12high, IL-10low phenotype; being part of the afferent and efferent limb of TH1 responses; and an ability to mediate resistance against tumors. M2 describes cells characterized, in part, by: exposure to IL-4, -13, or -10; an IL-12low, IL-10high phenotype; a general involvement in TH2 responses; and an ability to promote tissue repair, remodeling, and tumor progression (Mantovani et al., Citation2002).
Though there is no direct evidence here (or in the literature) regarding potential shifts in macrophage status due to TiO2 agents, there are suggestions these likely occurred in our nanoparticle-exposed hosts. Recent studies with similar nanoagents showed increasing shifts toward TH2-dominant responses (including promotion of allergic responses and inflammation in situ) in exposed hosts (Park et al., Citation2009; Larsen et al., Citation2010). Other studies using the parent TiO2 agent noted significant increases in many types of cytokines in the lungs (but not of IL-12; Hubbard et al., Citation2002) or increases in IL-10 levels released by cells isolated from peribronchial lymph nodes (de Haar et al., Citation2006). To provide clear evidence in support of this purported M1 →M2 status shift in the nanoparticle-exposed hosts, and then relate these changes to decreased tumor resistance in situ, additional studies (i.e., examination of relative numbers of macrophages expressing CD163 or CD206 marker [as M2 cells are CD163high/CD206high; Choi et al., Citation2010; Niino et al., Citation2010] vs. those expressing CD80 [Badylak et al., Citation2008]) of the macrophages from particle-exposed hosts are required.
A third explanation might be that downregulation (i.e., in either numbers or function) of CD11b+ cells in nanoparticle-exposed hosts could serve to negatively regulate host protection against tumor growth. CD11b (a portion of CD11b/CD18 integrin [i.e., Mac-1]) has been implicated in receptor cooperation with Fc receptors (FcR) and is normally expressed on monocytes/macrophages and NK cells. Activation of these cells by a variety of factors (i.e., cytokines, ROS, colony-stimulating factors) causes them to express more CD11b/CD18 on their surface (Mazzone and Ricevuti, Citation1995; Iwasaki, Citation2003). Such activation is critical in that expression of Mac-1 is essential for FcR-mediated cytotoxicity as well as leukocyte migration and adhesion—events critical to an anti-tumor cell response (Bühling et al., Citation2000; van Spriel et al., Citation2001). CD11b+ cells are also capable of causing activation of T-lymphocytes (Filippi et al., Citation2003; Iwasaki, Citation2003) that in turn contribute to increased activation of NK cells that can eliminate tumor cells (Colucci et al., Citation2002; Moretta et al., Citation2002). Because CD11b+ cells in the exposed hosts could readily non-specifically phagocytize the TiO2 nanoparticles, for the earlier-noted reasons, they might then also undergo apoptosis or necrosis (cytotoxic death). Thus, it should not then be unexpected that any decreases in the percentages of NK1.1 and/or CD11b+ cells would be mirrored by increase in tumor growth in so-affected hosts. As it is unclear what effects the nanoparticles might have on surface proteins on (these) cells, antibodies against additional markers (e.g., CD16, CD56, CD107a; Nagler et al., Citation2000; Alter et al., Citation2004) should be utilized for cell isolation to afford the greatest potential to catch “all”of the critical NK cells that might be present in the spleen of an exposed host.
In conclusion, the studies here showed that TiO2 nanoparticles could cause a decrease in lymphocyte proliferation/development, macrophage activity, and the size of NK cell populations. Further, host exposure to these agents caused significant increases in tumor growth—an outcome suggestive of decreased host anti-tumor immunosurveillance activity. We are mindful that because the experiments to see the effect of TiO2 nanoparticles following a 7-day exposure indicated significant immunosuppressive effects on immune cell proliferation and development, we surmised that a more long-term exposure might result in a more profound effect on the immune system, i.e., a change in immunosurveillance against cancer (that manifests as increase in [implanted] tumor growth). The ongoing exposure to TiO2 in the post-implantation period allowed us to maintain any ongoing exposure-related effects on host immune responses. In retrospect, we recognize a critical experiment (to be done in a follow-up study) would be one wherein we examine if the initial 7-day induced changes were long-lasting, i.e., no TiO2 treatments over the period of post-tumor cell implantation. This would let us address what immune parameters would be further impacted or even newly impacted by the additional 3 weeks of exposures.
The concentrations of TiO2 nanoparticles found in cosmetic/pharmaceutical products are not likely to ever be in the range of those used in this study. Nevertheless, after taking into account the potential for bioaccumulation in the body over time, the results here describing the potential for immunomodulatory effects on various immune cell types allow us to suggest that caution against chronic exposure to these agents is somewhat justified.
Acknowledgement
This work was supported by the Nanotoxicology Program (#08162KFDA545), Food and Drug Administration (KFDA) in 2008, and Grants from Mid-career Research Program (#R01-2008-000-20127-0) and National Nuclear R&D Program (#2010-0018545) through National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (MEST), Republic of Korea.
Declaration of interest
The authors report no conflicts of interest. The authors are alone responsible for the content and writing of the article.
References
- Alter, G., Malenfant, J. M., and Altfeld, M. 2004. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 294:15–22.
- Badylak, S. F., Valentin, J. E., Ravindra, A. K., McCabe, G. P., and Stewart-Akers, A. M. 2008. Macrophage phenotype as a determinant of biologic scaffold remodeling. Tissue Eng. Part A 14:1835–1842.
- Bühling, F., Ittenson, A., Kaiser, D., Thölert, G., Hoffmann, B., Reinhold, D., Ansorge, S., and Welte, T. 2000. MRP8/MRP14, CD11b and HLA-DR expression of alveolar macrophages in pneumonia. Immunol. Lett. 71:185–190.
- Bui, J. D., and Schreiber, R. D. 2007. Cancer immunosurveillance, immunoediting and inflammation: Independent or interdependent processes? Curr. Opin. Immunol. 19:203–208.
- Casola, S. 2007. Control of peripheral B-cell development. Curr. Opin. Immunol. 19:143–149.
- Chen, H. W., Su, S. F., Chien, C. T., Lin, W. H., Yu, S. L., Chou, C. C., Chen, J. J., and Yang, P. C. 2006. Titanium dioxide nanoparticles induce emphysema-like lung injury in mice. FASEB J. 20:2393–2395.
- Choi, K. M., Kashyap, P. C., Dutta, N., Stoltz, G. J., Ordog, T., Shea Donohue, T., Bauer, A. J., Linden, D. R., Szurszewski, J. H., Gibbons, S. J., and Farrugia, G. 2010. CD206-positive M2 macrophages that express heme oxygenase-1 protect against diabetic gastroparesis in mice. Gastroenterology 138:2399–409, 2409.e1.
- Colucci, F., Di Santo, J. P., and Leibson, P. J. 2002. Natural killer cell activation in mice and men: Different triggers for similar weapons? Nat. Immunol. 3:807–813.
- de Haar, C., Hassing, I., Bol, M., Bleumink, R., and Pieters, R. 2006. Ultrafine but not fine particulate matter causes airway inflammation and allergic airway sensitization to co-administered antigen in mice. Clin. Exp. Allergy 36:1469–1479.
- Denizot, F., and Lang, R. 1986. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods. 89:271–277.
- Di Gioacchino, M., Verna, N., Di Giampaolo, L., Di Claudio, F., Turi, M. C., Perrone, A., Petrarca, C., Mariani-Costantini, R., Sabbioni, E., and Boscolo, P. 2007. Immunotoxicity and sensitizing capacity of metal compounds depend on speciation. Int. J. Immunopathol. Pharmacol. 20:15–22.
- Dobrovolskaia, M. A., and McNeil, S. E. 2007. Immunological properties of engineered nanomaterials. Nat. Nanotechnol. 2:469–478.
- Dwivedi, P. D., Misra, A., Shanker, R., and Das, M. 2009. Are nanomaterials a threat to the immune system? Nanotoxicology 3:19–26.
- Ellmeier, W., Sawada, S., and Littman, D. R. 1999. The regulation of CD4 and CD8 co-receptor gene expression during T-cell development. Annu. Rev. Immunol. 17:523–554.
- Filippi, C., Hugues, S., Cazareth, J., Julia, V., Glaichenhaus, N., and Ugolini, S. 2003. CD4+ T-cell polarization in mice is modulated by strain-specific major histocompatibility complex-independent differences within dendritic cells. J. Exp. Med 198:201–209.
- Forte, G., Petrucci, F., and Bocca, B. 2008. Metal allergens of growing significance: epidemiology, immunotoxicology, strategies for testing and prevention. Inflamm. Allergy Drug Targets. 7:145–162.
- Geiser, M., Casaulta, M., Kupferschmid, B., Schulz, H., Semmler-Behnke, M., and Kreyling, W. 2008. The role of macrophages in the clearance of inhaled ultrafine titanium dioxide particles. Am. J. Respir. Cell Mol. Biol. 38:371–376.
- Gélis, C., Girard, S., Mavon, A., Delverdier, M., Paillous, N., and Vicendo, P. 2003. Assessment of the skin photoprotective capacities of an organo-mineral broad-spectrum sunblock on two ex vivo skin models. Photodermatol. Photoimmunol. Photomed. 19:242–253.
- Grassian, V. H., O’shaughnessy, P. T., Adamcakova-Dodd, A., Pettibone, J. M., and Thorne, P. S. 2007. Inhalation exposure study of titanium dioxide nanoparticles with a primary particle size of 2 to 5 nm. Environ. Health Perspect. 115:397–402.
- Green, L. C., Wagner, D. A., Glogowski, J., Skipper, P. L., Wishnok, J. S., and Tannenbaum, S. R. 1982. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem. 126:131–138.
- Hubbard, A. K., Timblin, C. R., Shukla, A., Rincón, M., and Mossman, B. T. 2002. Activation of NF-κB-dependent gene expression by silica in lungs of luciferase reporter mice. Am. J. Physiol. Lung Cell Mol. Physiol. 282:L968–L975.
- International Agency for Research on Cancer. 2006. Carbon Black, Titanium Dioxide, and Non-asbestiform Talc. In: IARC Monographs on the Evaluation of Carcinogenic Risks of Chemicals to Humans, Volume 93. Lyon: IARC, World Health Organization.
- Iwasaki, A. 2003. The importance of CD11b+ dendritic cells in CD4+ T-cell activation in vivo: With help from interleukin 1. J. Exp. Med. 198:185–190.
- Larsen, S. T., Roursgaard, M., Jensen, K. A., and Nielsen, G. D. 2010. Nano titanium dioxide particles promote allergic sensitization and lung inflammation in mice. Basic Clin. Pharmacol. Toxicol. 106:114–117.
- Lotzová, E. 1991. Natural killer cells: Immunobiology and clinical prospects. Cancer Invest. 9:173–184.
- Mantovani, A. 2006. Macrophage diversity and polarization: In vivo veritas. Blood 108:408–409.
- Mantovani, A., Sozzani, S., Locati, M., Allavena, P., and Sica, A. 2002. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23:549–555.
- Mazzone, A., and Ricevuti, G. 1995. Leukocyte CD11/CD18 integrins: Biological and clinical relevance. Haematologica 80:161–175.
- Moon, E. Y. 2008. Serum deprivation enhances apoptotic cell death by increasing mitochondrial enzyme activity. Biomol. Ther. 16:1–8.
- Moretta, A., Bottino, C., Mingari, M. C., Biassoni, R., and Moretta, L. 2002. What is a natural killer cell? Nat. Immunol. 3:6–8.
- Nagler, A., Rabinowitz, R., Rosengolts-Rat, J., Condiotti, R., and Schlesinger, M. 2000. Natural killer (NK) and T-cell-associated surface marker expression following allogeneic and autologous bone marrow transplantation (BMT). J. Hematother. Stem Cell Res. 9:63–75.
- Niino, D., Komohara, Y., Murayama, T., Aoki, R., Kimura, Y., Hashikawa, K., Kiyasu, J., Takeuchi, M., Suefuji, N., Sugita, Y., Takeya, M., and Ohshima, K. 2010. Ratio of M2 macrophage expression is closely associated with poor prognosis for Angioimmunoblastic T-cell lymphoma (AITL). Pathol. Int. 60:278–283.
- Ollila, J., and Vihinen, M. 2005. B-cells. Int. J. Biochem. Cell Biol. 37:518–523.
- Onuma, K., Sato, Y., Ogawara, S., Shirasawa, N., Kobayashi, M., Yoshitake, J., Yoshimura, T., Iigo, M., Fujii, J., and Okada, F. 2009. Nano-scaled particles of titanium dioxide convert benign mouse fibrosarcoma cells into aggressive tumor cells. Am. J. Pathol. 175:2171–2183.
- Park, E. J., Yi, J., Chung, K. H., Ryu, D. Y., Choi, J., and Park, K. 2008. Oxidative stress and apoptosis induced by titanium dioxide nanoparticles in cultured BEAS-2B cells. Toxicol. Lett. 180:222–229.
- Park, E. J., Yoon, J., Choi, K., Yi, J., and Park, K. 2009. Induction of chronic inflammation in mice treated with titanium dioxide nanoparticles by intratracheal instillation. Toxicology 260:37–46.
- Prehn, R. T. 2006. An immune reaction may be necessary for cancer development. Theor. Biol. Med. Model. 3:6.
- Sica, A., Larghi, P., Mancino, A., Rubino, L., Porta, C., Totaro, M. G., Rimoldi, M., Biswas, S. K., Allavena, P., and Mantovani, A. 2008. Macrophage polarization in tumour progression. Semin. Cancer Biol. 18:349–355.
- Sohn, S. J., Rajpal, A., and Winoto, A. 2003. Apoptosis during lymphoid development. Curr. Opin. Immunol. 15:209–216.
- Srivastava, B., Lindsley, R. C., Nikbakht, N., and Allman, D. 2005. Models for peripheral B-cell development and homeostasis. Semin. Immunol. 17:175–182.
- Sun, D., Meng, T. T., Loong, T. H., and Hwa, T. J. 2004. Removal of natural organic matter from water using a nano-structured photocatalyst coupled with filtration membrane. Water Sci. Technol. 49:103–110.
- Trinchieri, G. 1989. Biology of natural killer cells. Adv. Immunol. 47:187–376.
- Vamanu, C. I., Cimpan, M. R., Høl, P. J., Sørnes, S., Lie, S. A., and Gjerdet, N. R. 2008. Induction of cell death by TiO2 nanoparticles: studies on a human monoblastoid cell line. Toxicol. In Vitro 22:1689–1696.
- van Spriel, A. B., Leusen, J. H., van Egmond, M., Dijkman, H. B., Assmann, K. J., Mayadas, T. N., and van de Winkel, J. G. 2001. Mac-1 (CD11b/CD18) is essential for Fc receptor-mediated neutrophil cytotoxicity and immunologic synapse formation. Blood 97:2478–2486.
- Wan, R., Mo, Y., Zhang, X., Chien, S., Tollerud, D. J., and Zhang, Q. 2008. Matrix metalloproteinase-2 and -9 are induced differently by metal nanoparticles in human monocytes: The role of oxidative stress and protein tyrosine kinase activation. Toxicol. Appl. Pharmacol. 233:276–285.
- Xu, J., Futakuchi, M., Iigo, M., Fukamachi, K., Alexander, D. B., Shimizu, H., Sakai, Y., Tamano, S., Furukawa, F., Uchino, T., Tokunaga, H., Nishimura, T., Hirose, A., Kanno, J., and Tsuda, H. 2010. Involvement of macrophage inflammatory protein-1α (MIP1α) in promotion of rat lung and mammary carcinogenic activity of nanoscale titanium dioxide particles administered by intra-pulmonary spraying. Carcinogenesis 31:927–935.
- Zogovic, N. S., Nikolic, N. S., Vranjes-Djuric, S. D., Harhaji, L. M., Vucicevic, L. M., Janjetovic, K. D., Misirkic, M. S., Todorovic-Markovic, B. M., Markovic, Z. M., Milonjic, S. K., and Trajkovic, V. S. 2009. Opposite effects of nanocrystalline fullerene (C(60)) on tumour cell growth in vitro and in vivo and a possible role of immunosupression in the cancer-promoting activity of C(60). Biomaterials 30:6940–6946.