Abstract
Chronic obstructive pulmonary disease (COPD) is currently the fifth leading cause of death worldwide. Exposure to cigarette smoke (CS) is the primary factor associated with the COPD development. CS activates epithelial cells to secrete chemokines such as interleukin-8 (IL-8) and monocyte chemotactic protein-1 (MCP-1) that recruit neutrophils and macrophages to the lung. These inflammatory cells then release additional chemokines and cytokines leading to chronic inflammation that initiates apoptosis in epithelial and endothelial cells and destruction of alveolar structure. Pulmonary epithelium responds to oxidative stress mediated by CS through activating NRF2-dependent pathways, leading to an increased expression of antioxidant and cytoprotective enzymes thereby providing a protective response against CS-induced lung injury. We hypothesized that activating NRF2-dependent cytoprotective gene expression with sulforaphane (SFN) affords protection against CS-induced lung damage by inhibiting chemokine production. Results indicate that in the human BEAS-2B epithelial cell line, 5 μM SFN activated NRF2-dependent gene expression by triggering the translocation of NRF2 to the nucleus and significantly increased the expression of NRF2-dependent genes such as NADPH quinone oxidoreductase-1, heme oxygenase-1, and glutamate cysteine ligase modulatory subunit. Cigarette smoke extract (CSE) exposure of BEAS-2B cells significantly increased production of both IL-8 and MCP-1. Production of both chemokines was significantly reduced with SFN given prior to CSE; SFN inhibited IL-8 and MCP-1 gene expression at the transcription level. Our results indicate that activating NRF2 pathways with SFN inhibits CSE-induced chemokine production in human epithelial cells. However, the mechanism by which the production of chemokines is inhibited through SFN still remains to be elucidated. SFN may enhance NRF2 transcriptional activity resulting in the inhibition of proinflammatory pathways such as NF-κB.
Introduction
Chronic obstructive pulmonary disease (COPD) is currently the fifth leading cause of death worldwide and affects >210 million people (Viegi et al., Citation2007). The primary risk factor for developing COPD is exposure to cigarette smoke (CS) (Rabe et al., Citation2007). COPD is characterized by abnormal inflammation leading to irreversible airflow limitation in the lung. As COPD progresses, pulmonary inflammation (as characterized by increases in lung levels of neutrophils, macrophages, and lymphocytes) increases in tandem (Hogg et al., Citation2004). Currently, there are few effective therapies for this disease that reduce pulmonary inflammation to modify the progression of airflow obstruction.
Interleukin-8 (IL-8) and monocyte chemotactic protein-1 (MCP-1) are two major mediators in the inflammatory response induced by CS. IL-8 acts as a potent chemoattractant for neutrophils and monocytes to the lung (Barnes, Citation2009). IL-8 levels are elevated in induced sputum samples from patients with COPD (Keatings et al., Citation1996) and correlate with the degree of airflow limitation (Yamamoto et al., Citation1997). The importance of IL-8 levels in COPD disease progression is supported by its potential as a clinical biomarker (Chung, Citation2006) and the ability of IL-8 receptor antagonists to block pulmonary neutrophil and monocyte recruitment in COPD patients (Barnes, Citation2008).
MCP-1, also known as CCL2, enables the recruitment of monocytes, macrophages, and lymphocytes to sites of tissue injury. In smokers with chronic bronchitis, MCP-1 levels are increased in the bronchoalveolar lavage (BAL) fluid compared with the levels in BAL from smokers without the disease (Capelli et al., Citation2005). MCP-1 levels are significantly increased in sputum from patients with COPD and correlate with the number of inflammatory cells and with the degree of airflow limitation in these patients (Traves et al., Citation2002). Elevated mRNA and protein expression of both IL-8 and MCP-1 are observed in bronchiolar epithelium harvested from smokers with COPD compared with cells from patients without COPD (de Boer et al., Citation2000). Taken together, these data indicate that IL-8 and MCP-1 are increased in COPD patients and are involved in the inflammatory response, a major contributing factor in the pathogenesis of COPD.
Nuclear factor erythroid 2 p45-related factor (NRF2) is a basic leucine zipper transcription factor that regulates the expression of numerous antioxidant and cytoprotective genes (Kensler et al., Citation2007). Under normal conditions, NRF2 is maintained at low basal levels in cells by binding to its inhibitor protein, KEAP1 (Itoh et al., Citation1999). KEAP1 targets NRF2 for ubiquitination leading to its constitutive degradation (Kobayashi et al., Citation2004). Upon exposure to oxidative stress, NRF2 translocates to the nucleus (Itoh et al., Citation2003; Kobayashi et al., Citation2006) where it forms heterodimers with other transcription regulators and binds antioxidant response elements (ARE) to induce expression of detoxification enzymes such as NADPH quinone oxidoreductase-1 (NQO1), heme oxygenase-1 (HO-1), and enzymes important in GSH biosynthesis such as glutamate cysteine ligase modulatory (GCLM) subunit (Thimmulappa et al., Citation2002).
NRF2-dependent gene expression is a central mediator in several pulmonary inflammatory diseases such as sepsis, asthma, and emphysema (Rangasamy et al., Citation2004, 2005; Iizuka et al., Citation2005; Thimmulappa et al., Citation2006a). These studies are characterized by increased pulmonary inflammation in mice with a disruption of Nrf2 (Nrf2−/− mice) compared with wild-type mice (Nrf2+/+ mice), resulting in more severe pulmonary disease. For example, chronic CS exposure in Nrf2−/− mice resulted in increased inflammation and oxidative stress in the lungs, which was correlated with an earlier onset and more severe emphysema (Rangasamy et al., Citation2004). Emphysema in Nrf2−/− mice was characterized by more extensive alveolar destruction and increased bronchoalveolar inflammation of macrophages. The importance of the NRF2 pathway in protecting the lung from CS-induced lung destruction was evident through microarray analysis that indicated nearly 50 NRF2-dependent genes were enhanced during CS exposure. NRF2 pathway activation has also been shown to be important in COPD patients. Lung samples from COPD patients have a significantly reduced NRF2 activity that correlates to disease severity (Goven et al., Citation2008; Malhotra et al., Citation2008; Suzuki et al., Citation2008). Therefore, if deficiencies in the NRF2 pathways are associated with COPD pathogenesis, therapies directed toward enhancing the expression of NRF2-regulated antioxidants may be a novel strategy for attenuating the effects of inflammation in the pathogenesis of COPD.
Sulforaphane (SFN), an isothiocyanate agent derived from cruciferous vegetables such as broccoli, potently up-regulates expression of cytoprotective and antioxidant genes (Fahey and Talalay, Citation1999). SFN enhances NRF2-mediated transcriptional activity by modifying redox-sensitive sulfhydryl groups on the KEAP1 protein (Dinkova-Kostova et al., Citation2002). SFN has been shown to increase the expression of >70 cytoprotective and antioxidant genes in an NRF2-dependent manner (Thimmulappa et al., Citation2002). Therefore, we hypothesize that sustained activation of NRF2 with SFN will increase antioxidant and cytoprotective pathways to inhibit chemokine production and protect against pulmonary inflammation caused by CS.
Materials and methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), phosphate-buffered saline (PBS), dimethyl sulfoxide (DMSO), penicillin/streptomycin, and 0.5% trypsin–EDTA were purchased from Invitrogen (Carlsbad, CA). R-SFN was procured from LKT Laboratories (St. Paul, MN). All antibodies were purchased from Santa Cruz Biotechnology (Santo Cruz, CA). The human BEAS-2B lung epithelial cells were obtained from American Type Culture Collection (ATCC; Manassas, VA). Cigarette smoke extract (CSE) was purchased from Murty Pharmaceuticals Inc. (Lexington, KY). The CSE was prepared by smoking University of Kentucky 3R4F Standard Research Cigarettes on an FTC Smoke Machine. The total particulate matter (TPM) on the filter was calculated by the weight gain of the filter. From the TPM, the amount of DMSO to be used for extraction to prepare a 4% stock solution (40 mg/mL) was calculated. The condensate was extracted with DMSO by soaking and sonication.
Determination of cytotoxicity
BEAS-2B cells were plated at a density of 0.2 × 105 cells per well in 96-well plates, allowed to recover for 12 h, and then exposed for 24 h to 5, 10, 50, or 250 µM SFN in DMEM with 1% FBS. Cells were then washed once in PBS and cytotoxicity evaluated by an MTT assay. In brief, 10 µL of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) reagent was added to each well, the cells were incubated at 37°C for 2 h, and then the absorbance in each well was measured at 490 nm in an Infinite® 200 PRO series plate reader (Tecan Systems Inc., San Jose, CA). The experiment was repeated three times. Each data point represented a mean (±SEM) and was normalized to the value of the corresponding control cells.
Cell culture and treatments
BEAS-2B cells were grown and maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. For chemokine quantification, BEAS-2B cells in the exponential growth phase were plated at a density of 0.2 × 106 cells/mL and grown for 12 h. Cells were then treated with 5 µM SFN (or DMSO as a control) for 6 h in DMEM with 1% FBS. The optimal concentration of SFN was determined to be 5 µM because this concentration was non-cytotoxic in BEAS-2B cells as determined by the MTT assay described above. Cells were then washed once in PBS and treated with 200 µg CSE/mL or DMSO (as a control) for 15 min as previously described (Laan et al., Citation2004). Cells were then washed once in PBS and incubated with DMEM with 1% FBS for 5 h. Thereafter, the media was collected, centrifuged to clear cellular debris, and frozen at −80°C until chemokine levels were assayed.
RNA isolation and quantification of gene expression
BEAS-2B cells were treated with 5 µM SFN as indicated, washed with PBS, and exposed to 200 µg/mL CSE for 15 min. Cells were washed with PBS and maintained in DMEM with 1% FBS after CSE exposure. Total RNA was isolated using the RNeasy Mini Kit according to the manufacturer’s recommended protocol (Qiagen Inc., Valencia, CA). The cDNA reaction was completed using the high-capacity cDNA reverse transcriptase kit (Applied Biosystems, Foster City, CA). Gene expression was measured using assays on demand probe sets (Applied Biosystems). Reactions were completed and analyzed using the StepOne real-time PCR system. Actin was used for normalization. The cycle threshold (CT) value indicates the number of PCR cycles that are necessary for the detection of a fluorescence signal exceeding a fixed threshold. The level of fold change was calculated using the following formulas: ΔCT = CT (target gene)− CT (actin) and FC = 2−(ΔCT2 − ΔCT1), in which ΔCT1 represents the average ΔCT value among control samples and ΔCT2 represents the value of a particular sample. Control samples for the in vitro studies included mock-treated control BEAS-2B cells exposed to DMSO. The experiment was repeated twice. All results were expressed as mean (±SEM).
Immunoblotting
Cytoplasmic and nuclear fractions were obtained using the NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo Scientific, Rockford, IL). Total protein content was determined by the BCA protein assay (Pierce, Rockford, IL). Equal amounts of total protein (50 µg) were resolved by SDS-PAGE and were transferred to nitrocellulose membranes (Millipore, Marl-borough, MA). Non-specific binding sites were blocked by incubation for 1 h at room temperature with PBS containing 0.1% Tween-20 (PBST) and 5% non-fat dry milk. Membranes were then exposed to rabbit anti-NRF2 polyclonal antibody, goat anti-NQO1 polyclonal antibody, rabbit anti-HDAC2 polyclonal antibody, or goat anti-GAPDH polyclonal antibody in PBST–0.5% milk overnight at 4°C, followed by incubation with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare UK Ltd., Buckinghamshire, England). Immunoreactivity was visualized by chemiluminescence substrate according to the manufacturer’s instructions (Amersham Biosciences, Piscataway, NJ).
Quantification of IL-8 and MCP-1 chemokines
BEAS-2B cells were treated with 5 µM SFN for 6 h, washed with PBS, and then exposed to 200 µg CSE/mL for 15 min. Cells were then washed with PBS and conditioned medium was collected 5 h after CSE exposure. MCP-1 protein levels were quantified using a human CCL2 (MCP-1) ELISA according to the manufacturer’s recommended protocol (eBioscience, San Diego, CA). IL-8 protein levels were quantified using a human IL-8/NAP-1 ELISA (Bender MedSystems, Vienna, Austria) according to the manufacturer’s recommended protocol. IL-8 and MCP-1 levels were quantified in three separate experiments.
Statistical analysis
Data are presented as mean ± SEM. Data comparing two group means were analyzed by independent samples t-test. Significance was noted at P < 0.05. Outliers were detected through a Grubb’s test from GraphPad Software (GraphPad, La Jolle, CA).
Results
SFN significantly increases gene expression of well-characterized NRF2-dependent genes and enables nuclear translocation of NRF2
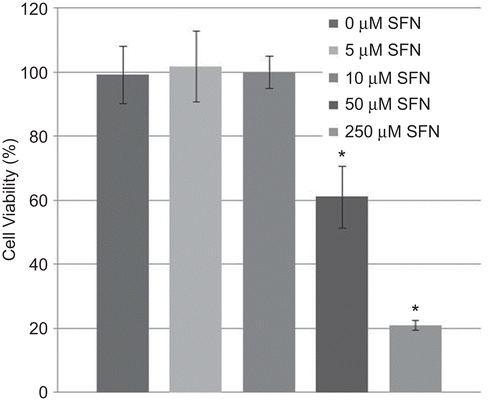
SFN, a naturally occurring component of cruciferous vegetables, induces numerous NRF2-dependent antioxidant and cytoprotective pathways (Thimmulappa et al., Citation2002). However, SFN has also been shown to be a potential anticancer agent by inducing apoptosis in both prostate and colon cancer cells (Clarke et al., Citation2008). Therefore, in order to select an appropriate SFN concentration that does not induce apoptosis, human epithelial cells (BEAS-2B cells) were exposed to four different concentrations of SFN (i.e. 5, 10, 50, or 250 µM) for 24 h. Cell viability was then determined using a standardized MTT assay. SFN had no effect on cell viability at low concentrations (5 and 10 µM); however, higher concentrations (50 and 250 µM) significantly reduced viability after 24 h (). Based on these results, 5 µM SFN was chosen as the optimal concentration to be used in all subsequent experiments.
Figure 1. Cell viability in BEAS-2B cells after treatment with sulforaphane (SFN). BEAS-2B cells were treated with different concentrations of SFN for 24 h. Viability was then quantified using an MTT assay. Data presented are mean percent viability (±SEM, n = 5). *Value is significantly different compared with control cells (P < 0.05).
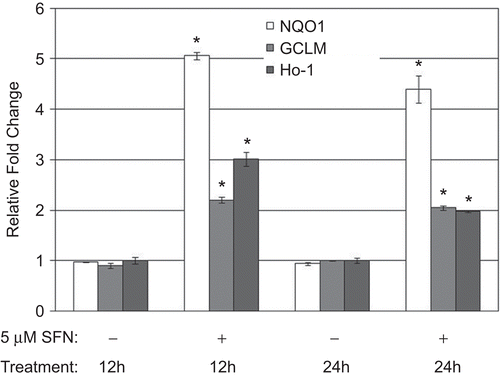
To determine if SFN enhanced NRF2-dependent gene expression in human epithelial cells, the BEAS-2B cells were treated with 5 µM SFN for 12 and 24 h, and the expression of several NRF2 target genes then quantified through real-time PCR (). NRF2-dependent gene expression of NQO1, GCLM, and HO-1 was significantly increased (>2-fold) in the cells after 12 and 24 h (indicated by asterisks in ). Gene expression was not altered in control DMSO-treated cells.
Figure 2. Messenger RNA levels of NQO1, GCLM, and HO-1 in BEAS-2B cells after 12 and 24 h treatment with DMSO (as a control) or 5 µM SFN. Fold change was calculated using the formula described in the Materials and methods. Data presented are mean fold-change (±SEM, n = 3). *Value is significantly different compared with control cells (P < 0.05).
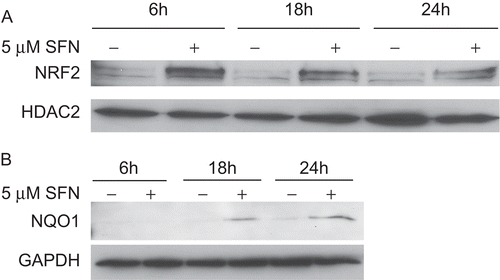
To provide additional support that NRF2 was activated by SFN at the protein level, the BEAS-2B cells were treated with SFN for 6, 18, and 24 h and then fractionated into cytosolic and nuclear components. Protein lysates were then analyzed via immunoblotting. Nuclear extracts were normalized to HDAC2 levels to ensure equal loading and cytosolic extracts were normalized to GAPDH levels to ensure equal loading. Treatment with the SFN resulted in observable nuclear translocation of NRF2 after 6 h compared with DMSO-treated control cells (). Nuclear accumulation of NRF2 was also enhanced 18 to 24 h after SFN treatment compared with DMSO-treated controls. Moreover, SFN treatment resulted in enhanced protein expression of NQO1 (a well-characterized NRF2-dependent target gene) after 18 and 24 h (). These results provide support that SFN enhances NRF2 transcriptional activity by allowing NRF2 to gain access to the nucleus, thereby increasing gene expression and protein synthesis.
Figure 3. Immunoblot analysis of NRF2 and NQO1 in BEAS-2B cells treated with DMSO (as a control) or 5 µM SFN for various lengths of time. (A) Immunoblots of the nuclear extracts from the treated cells were probed with an anti-human NRF2 antibody, stripped, and re-probed with an anti-human HDAC2 antibody. (B) Immunoblots of the cytosolic extracts were probed with an anti-human NQO1 antibody, stripped, and re-probed with an anti-human GAPDH antibody.
SFN inhibits IL-8 and MCP-1 chemokine production induced by CSE in epithelial cells
SFN has been shown to have anti-inflammatory properties in response to lipopolysaccharide (LPS) (Lin et al., Citation2008). Moreover, enhancing NRF2-dependent gene expression with other small molecular activators (CDDO-Im) has been shown to inhibit pulmonary diseases (e.g. emphysema and sepsis) driven by inflammation (Thimmulappa et al., Citation2006b; Sussan et al., Citation2009). However, it is unclear whether activating NRF2 with SFN will decrease the inflammatory processes due to CS in human epithelial cells. To this end, BEAS-2B cells were treated with either 5 µM SFN or DMSO (as a control) for 6 h prior to exposure to CSE. After SFN treatment, the cells were washed with PBS and then exposed to DMSO (as a control) or 200 µg CSE/mL for 15 min to mimic exposure to CS in vivo (Laan et al., Citation2004). Thereafter, the cells were washed with PBS, the medium was replaced with DMEM + 1% FBS, and the cells cultured a further 5 h before culture supernatants were collected to permit analyses of post-CSE exposure chemokine levels.
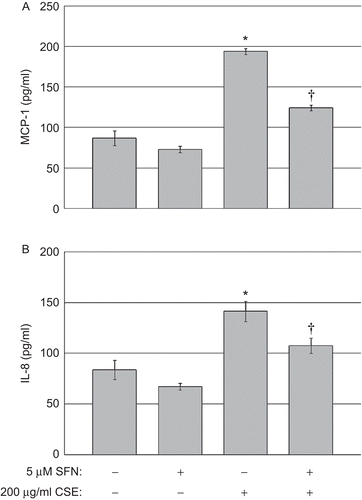
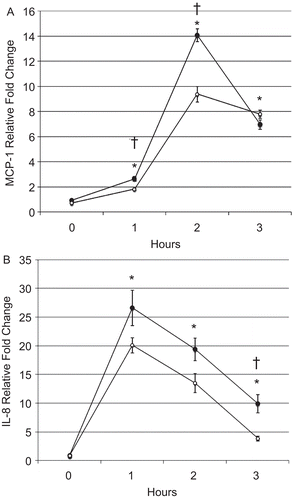
Exposure to CSE significantly increased levels of both IL-8 and MCP-1 in the cell culture supernatants 5 h after CSE exposure compared with after DMSO (control) exposure (indicated by asterisks in ). Treatment with 5 µM SFN prior to the CSE exposure significantly reduced the production of IL-8 and MCP-1 (indicated by asterisks in ). No differences in basal chemokine levels were observed between control cells treated with DMSO and cells treated with 5 µM SFN in the absence of CSE exposure. These results indicate that enhancing the transcriptional activity of NRF2 with SFN inhibited CSE-induced chemokine production by the human epithelial cells.
Figure 4. Chemokine quantification in human BEAS-2B epithelial cells exposed to CSE. (A) MCP-1 and (B) IL-8 levels in the culture supernatants of cells treated with DMSO or 5 µM SFN for 6 h, then treated with DMSO or 200 µg CSE/mL as described in the Materials and methods. Data presented are mean fold-change (±SEM, n = 3). *Value is significantly different compared with control cells not exposed to CSE (P < 0.05); †Value is significantly different compared with cells exposed to CSE alone (P < 0.05).
SFN inhibits de novo synthesis of IL-8 and MCP-1 due to CSE exposure by enhancing expression of NRF2-dependent genes
To determine whether SFN inhibits de novo synthesis of CSE-induced chemokines or inhibits the extracellular release of these chemokines from epithelial cells, mRNA levels of IL-8 and MCP-1 were quantified through real-time PCR. BEAS-2B cells were pretreated with either DMSO as a control or 5 µM SFN for 6 h prior and exposed to 200 µg/mL CSE as described above. Cells were collected and total RNA was isolated immediately before CSE exposure (0 h) and 1, 2, and 3 h after CSE exposure.
Messenger RNA expression of MCP-1 in the BEAS-2B cells was significantly increased 1, 2, and 3 h after CSE exposure compared with that in untreated control cells with and without SFN (indicated by asterisks in ). Compared with DMSO-treated control cells, SFN-treated cells displayed significantly decreased mRNA MCP-1 expression 1 and 2 h after CSE exposure (indicated by daggers in ). However, expression levels of MCP-1 in cells with and without SFN treatment were equivalent 3 h after the CSE exposure. CSE treatment also gave rise to significantly increased IL-8 mRNA expression 1, 2, and 3 h post-exposure compared with that in control cells, regardless of SFN treatment (indicated by asterisks in ). In comparison with DMSO-treated control cells, SFN-treated cells displayed decreased IL-8 gene expression levels 1 and 2 h after CSE exposure, albeit that statistical significance was not observed. However, the decrease in IL-8 mRNA levels was significant at 3 h post-exposure in cells treated with SFN compared with cells without SFN treatment (indicated by a dagger in ). These results indicate that SFN exerts its anti-inflammatory effect by suppressing transcription of CSE-induced chemokines and thereby diminishing de novo synthesis of MCP-1 and IL-8.
Figure 5. BEAS-2B cell (A) MCP-1 and (B) IL-8 messenger RNA levels after CSE treatment. Filled and open circles denote cells exposed, respectively, to DMSO or 5 µM SFN for 6 h, then exposed to CSE as described in the Materials and methods. Data presented are mean fold-change; fold-change was calculated using formula described in the Materials and methods. *Value is significantly different from treatment-matched control cells not exposed to CSE (0 h) (P < 0.05); †Value is significantly different between cells exposed to CSE treated with and without SFN (P < 0.05).
CS is a potent oxidant that contains greater than 1014 free radicals per puff (Pryor et al., Citation1983) and induces the transcriptional activity of NRF2, which is characterized by the expression of over 100 cytoprotective and antioxidant genes (Rangasamy et al., Citation2004). To test our central hypothesis that sustained activation of NRF2 will enhance the expression of cytoprotective genes and inhibit chemokine production, two important anti-inflammatory genes were quantified through real-time PCR; NQO1 and HO-1. Both of these genes are well-characterized NRF2 target genes (Venugopal and Jaiswal, Citation1996; Alam et al., Citation1999). BEAS-2B cells were treated with SFN or DMSO as a control for 6 h and then exposed to CSE. NQO1 expression levels were significantly increased at all time points in cells treated with SFN compared with the levels in DMSO-treated control cells (as indicated by daggers in ). Exposure to CSE significantly increased NQO1 gene expression 2 and 3 h after CSE exposure in cells that had been treated with SFN and in cells treated without SFN. NQO1 expression in SFN-treated epithelial cells continued to increase after CSE exposure indicting that expression of this gene can be enhanced in CSE-exposed cells.
Figure 6. BEAS-2B cell (A) NQO1 and (B) HO-1 messenger RNA levels after treatment with SFN and CSE. Fold change was calculated using the formula described in the section “Materials and methods.” Filled and open circles denote cells exposed, respectively, to DMSO or 5 µM SFN for 6 h, then exposed to CSE as described in the Materials and methods. Data presented are mean fold-change; fold-change was calculated using formula described in the Materials and methods. *Value is significantly different from treatment-matched control cells not exposed to CSE (0 h) (P < 0.05) †Value is significantly different between cells exposed to CSE treated with and without SFN (P < 0.05).
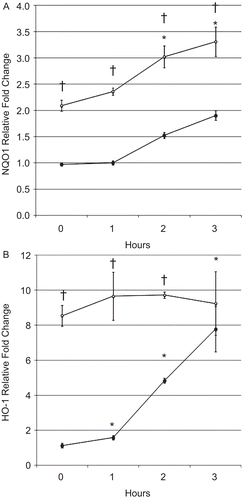
Exposure to CSE significantly increased HO-1 mRNA expression 1, 2, and 3 h after CSE exposure in control cells treated with DMSO (indicated by asterisks in ). Epithelial cells treated with SFN had significantly higher HO-1 expression compared with DMSO-treated control cells but in contrast to NQO1 expression no increase of HO-1 was observed after CSE exposure. HO-1 mRNA expression was equivalent 3 h after CSE exposure regardless of SFN treatment. These observations demonstrate that SFN enhances NRF2-dependent gene expression in human epithelial cells and inhibits CSE-induced chemokine production in vitro.
Discussion
Exposure to CS is the primary factor associated with the development of COPD. Although the exact mechanism leading to the development of COPD remains to be elucidated, mounting evidence indicates that the recruitment of inflammatory cells to the lung is a major factor that contributes to COPD pathogenesis and severity (Stockley, Citation2009). Current therapies for COPD, such as anti-inflammatory agents and bronchodilators, are generally ineffective because they do not target the underlying disease processes to suppress inflammation. Indeed, the majority of these therapies do not reduce the progression of COPD (Barnes, Citation2005). Current COPD therapies in clinical development are either inhibitors of a single pro-inflammatory mediator to reduce inflammation or individual antioxidants that suppress oxidative stress associated with COPD severity (Barnes and Hansel, Citation2004). However, administration of a single antioxidant or cytokine inhibitor may not alleviate the multiple disease processes involved in COPD pathogenesis (Rahman and Kilty, Citation2006; Barnes, Citation2009). An excellent alternative to administering a single compound that targets one disease component in COPD would be to utilize a small molecule, such as SFN, that activates a diverse array of antioxidant and detoxifying genes to inhibit the multiple inflammatory pathways generated by CS exposure to reduce the overall inflammation in the lung and modify the progression of the disease.
The augmentation of NRF2 and transcription of its target genes may act to protect against CS-induced emphysema to decrease lung inflammation and alveolar destruction. Enhancing the transcriptional activity of NRF2 has been shown to attenuate emphysema in an NRF2-dependent manner over a 6-month period of chronic CS exposure (Sussan et al., Citation2009). Antioxidant and cytoprotective genes were expressed at the highest levels in the lungs of Nrf2+/+ mice that were exposed to CS and given the small molecular activator, CDDO-Im. Nrf2+/+ mice exposed to CS and given CDDO-Im had significantly decreased levels of oxidative stress in the lung. However, the number of inflammatory cells in the lung was not decreased in Nrf2+/+ mice given the drug indicating that NRF2 activation did not alter pulmonary inflammation. In contrast, pulmonary inflammation due to acute CS exposure is significantly reduced in a genetic model of NRF2 activation (Blake et al., Citation2009). Conditional Keap1 knockout mice were generated by disrupting the Keap1 gene only in Clara cells of the lung. The Keap1 deletion resulted in the up-regulation of NRF2-dependent gene expression and increased glutathione levels in the lung. Conditional Keap1 knockout mice had a significantly reduced numbers of inflammatory macrophages in their BAL that was associated with decreased expression of MCP-1 in their lungs following CS exposure (Blake et al., Citation2009).
Activating NRF2 pathways has been shown to be important in reducing pulmonary inflammation in other experimental disease models. For example, in an experimental sepsis model utilizing LPS, Nrf2+/+ mice given CDDO-Im expressed NRF2-dependent genes at high levels and had reduced expression levels of pro-inflammatory molecules such as tumor necrosis factor (TNF)-α, MCP-1, and IL-6 compared with Nrf2−/− mice (Thimmulappa et al., Citation2006b). Moreover, only Nrf2+/+ mice given CDDO-Im were protected against LPS-induced mortality indicating the protective effects were dependent upon NRF2. SFN has also been shown to be protective in an NRF2-dependent manner. The anti-inflammatory effects of SFN on LPS-stimulated inflammation have been observed in Nrf2+/+ but not in Nrf2−/− macrophages leading to a significant reduction in TNF-α and IL-1β gene expression (Lin et al., Citation2008). SFN also significantly decreased production of IL-8, GM-CSF, and IL-1β in bronchial epithelial cells in response to diesel exhaust particles (Ritz et al., Citation2007). SFN has been shown to be protective in vivo against pulmonary inflammation induced by respiratory syncytial virus (RSV) infection. Only Nrf2+/+ mice given SFN, but not Nrf2−/− given SFN, had a significant decrease in the number of neutrophils and eosinophils recruited to the lung after RSV challenge (Cho et al., Citation2009). SFN has also been shown to protect arterial endothelial cells in vivo from expressing a pro-inflammatory state, which was dependent on NRF2 (Zakkar et al., Citation2009). Our results confirm previous reports that SFN is a potent anti-inflammatory compound that is able to exert its cellular effects in an NRF2-dependent manner. Future studies will be necessary to determine whether the suppression of MCP-1 and IL-8 requires the presence of NRF2.
The reduction in chemokine production by SFN is important with regard to COPD pathogenesis because both IL-8 and MCP-1 levels are increased in COPD patients and these agents can cause recruitment of additional inflammatory cells (e.g. macrophages, neutrophils, and lymphocytes) to the lungs, an outcome associated with increases in COPD disease severity. We believe SFN will be effective in inhibiting pulmonary inflammation induced by CS in vivo due to its ability to decrease the production of two important inflammatory chemokines concurrently. Moreover, SFN may exert its anti-inflammatory effects in other pulmonary cell types such as macrophages and Type II epithelial cells, thereby enhancing the anti-inflammatory effect in vivo.
SFN has been shown to regulate >70 genes in an NRF2-dependent manner (Thimmulappa et al., Citation2002). Therefore, understanding which antioxidant and cytoprotective genes, under the transcriptional control of NRF2, are most important to suppress pulmonary inflammation may be of therapeutic importance. One NRF2-dependent gene of interest is NQO1, which promotes reduction of quinones, thereby reducing generation of reactive oxygen species and depletion of thiol pools. Over-expression of NQO1 has been shown to inhibit IL-8 production due to diesel exhaust particles (Ritz et al., Citation2007). HO is a second NRF2-dependent gene whose product is able to degrade, via oxidation, heme into biliverdin, carbon monoxide, and free iron. Induction of HO-1 has been shown to inhibit MCP-1 expression in monocytes and endothelial cells (Sacerdoti et al., Citation2005; Shokawa et al., Citation2006). Expression of both NQO1 and HO-1 were increased in human epithelial cells treated with SFN prior to CSE exposure. Interestingly, HO-1 was expressed at a much higher level than NQO1; however, HO-1 expression levels did not increase in SFN-treated cells after CSE exposure. In contrast, NQO1 levels increased significantly after CSE exposure regardless of SFN treatment.
Since SFN treatment suppresses transcriptional expression of both MCP-1 and IL-8, we hypothesize that NRF2 activation may inhibit DNA binding or nuclear accumulation of the transcription factors involved in the expression of these chemokines. NRF2 has been shown to have direct and indirect inhibitory interactions with nuclear factor κ light-chain enhancer of B-cells (NF-κB) (Wakabayashi et al., Citation2010). Because two NF-κB binding sites have been identified in the MCP-1 promoter (Ueda et al., Citation1997) and IL-8 is also under the transcriptional control of NF-κB (Zhou et al., Citation2003), NRF2 may modify the transcriptional responsiveness of NF-κB. Alternatively, both MCP-1 and IL-8 are under transcriptional control of activator protein-1 (AP-1) (Martin et al., Citation1997; Zhou et al., Citation2003). Therefore, future studies will determine whether CSE-induced NF-κB or AP-1 activity is altered in human epithelial cells given SFN.
Acknowledgement
This work was supported by funds given to Dr. David Blake by Fort Lewis College.
Declaration of interest
The authors have no declarations of interest to disclose. The authors are alone responsible for the content and writing of the article.
References
- Alam, J., Stewart, D., Touchard, C., Boinapally, S., Choi, A. M. and Cook, J. L. 1999. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 274:26071–26078.
- Barnes, P. J. 2008. Emerging pharmacotherapies for COPD. Chest. 134:1278–1286.
- Barnes, P. J. 2005. Emerging targets for COPD therapy. Curr Drug Targets Inflamm Allergy. 4:675–683.
- Barnes, P. J. 2009. The cytokine network in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 41:631–638.
- Barnes, P. J. and Hansel, T. T. 2004. Prospects for new drugs for chronic obstructive pulmonary disease. Lancet. 364:985–996.
- Blake, D. J., Singh, A., Kombairaju, P., Malhotra, D., Mariani, T. J., Tuder, R. M., Gabrielson, E. and Biswal, S. 2009. Deletion of Keap1 in the lung attenuates acute cigarette smoke-induced oxidative stress and inflammation. Am J Respir Cell Mol Biol. 42:524–536.
- Capelli, A., Di Stefano, A., Gnemmi, I. and Donner, C. F. 2005. CCR5 expression and CC chemokine levels in idiopathic pulmonary fibrosis. Eur Respir J. 25:701–707.
- Cho, H. Y., Imani, F., Miller-DeGraff, L., Walters, D., Melendi, G. A., Yamamoto, M., Polack, F. P. and Kleeberger, S. R. 2009. Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. Am J Respir Crit Care Med. 179:138–150.
- Chung, K. F. 2006. Cytokines as targets in chronic obstructive pulmonary disease. Curr Drug Targets. 7:675–681.
- Clarke, J. D., Dashwood, R. H. and Ho, E. 2008. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 269:291–304.
- de Boer, W. I., Sont, J. K., van Schadewijk, A., Stolk, J., van Krieken, J. H. and Hiemstra, P. S. 2000. Monocyte chemoattractant protein-1, interleukin 8, and chronic airways inflammation in COPD. J Pathol. 190:619–626.
- Dinkova-Kostova, A. T., Holtzclaw, W. D., Cole, R. N., Itoh, K., Wakabayashi, N., Katoh, Y., Yamamoto, M. and Talalay, P. 2002. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of Phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA. 99:11908–11913.
- Fahey, J. W. and Talalay, P. 1999. Antioxidant functions of sulforaphane: A potent inducer of Phase II detoxication enzymes. Food Chem Toxicol. 37:973–979.
- Goven, D., Boutten, A., Leçon-Malas, V., Marchal-Sommé, J., Amara, N., Crestani, B., Fournier, M., Lesèche, G., Soler, P., Boczkowski, J. and Bonay, M. 2008. Altered Nrf2/Keap1-Bach1 equilibrium in pulmonary emphysema. Thorax. 63:916–924.
- Hogg, J. C., Chu, F., Utokaparch, S., Woods, R., Elliott, W. M., Buzatu, L., Cherniack, R. M., Rogers, R. M., Sciurba, F. C., Coxson, H. O. and Paré, P. D. 2004. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 350:2645–2653.
- Iizuka, T., Ishii, Y., Itoh, K., Kiwamoto, T., Kimura, T., Matsuno, Y., Morishima, Y., Hegab, A. E., Homma, S., Nomura, A., Sakamoto, T., Shimura, M., Yoshida, A., Yamamoto, M. and Sekizawa, K. 2005. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells. 10:1113–1125.
- Itoh, K., Wakabayashi, N., Katoh, Y., Ishii, T., Igarashi, K., Engel, J. D. and Yamamoto, M. 1999. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13:76–86.
- Itoh, K., Wakabayashi, N., Katoh, Y., Ishii, T., O’Connor, T. and Yamamoto, M. 2003. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 8:379–391.
- Keatings, V. M., Collins, P. D., Scott, D. M. and Barnes, P. J. 1996. Differences in interleukin-8 and tumor necrosis factor-α in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med. 153:530–534.
- Kensler, T. W., Wakabayashi, N. and Biswal, S. 2007. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 47:89–116.
- Kobayashi, A., Kang, M. I., Okawa, H., Ohtsuji, M., Zenke, Y., Chiba, T., Igarashi, K. and Yamamoto, M. 2004. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 24:7130–7139.
- Kobayashi, A., Kang, M. I., Watai, Y., Tong, K. I., Shibata, T., Uchida, K. and Yamamoto, M. 2006. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 26:221–229.
- Laan, M., Bozinovski, S. and Anderson, G. P. 2004. Cigarette smoke inhibits lipopolysaccharide-induced production of inflammatory cytokines by suppressing the activation of activator protein-1 in bronchial epithelial cells. J Immunol. 173:4164–4170.
- Lin, W., Wu, R. T., Wu, T., Khor, T. O., Wang, H. and Kong, A. N. 2008. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2-dependent pathway. Biochem Pharmacol. 76:967–973.
- Malhotra, D., Thimmulappa, R., Navas-Acien, A., Sandford, A., Elliott, M., Singh, A., Chen, L., Zhuang, X., Hogg, J., Pare, P., Tuder, R. M. and Biswal, S. 2008. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am J Respir Crit Care Med. 178:592–604.
- Martin, T., Cardarelli, P. M., Parry, G. C., Felts, K. A. and Cobb, R. R. 1997. Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-κ B and AP-1. Eur J Immunol. 27:1091–1097.
- Pryor, W. A., Prier, D. G. and Church, D. F. 1983. Electron-spin resonance study of mainstream and sidestream cigarette smoke: nature of the free radicals in gas-phase smoke and in cigarette tar. Environ Health Perspect. 47:345–355.
- Rabe, K. F., Hurd, S., Anzueto, A., Barnes, P. J., Buist, S. A., Calverley, P., Fukuchi, Y., Jenkins, C., Rodriguez-Roisin, R., van Weel, C. and Zielinski, J.; Global Initiative for Chronic Obstructive Lung Disease. 2007. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 176:532–555.
- Rahman, I. and Kilty, I. 2006. Antioxidant therapeutic targets in COPD. Curr Drug Targets. 7:707–720.
- Rangasamy, T., Cho, C. Y., Thimmulappa, R. K., Zhen, L., Srisuma, S. S., Kensler, T. W., Yamamoto, M., Petrache, I., Tuder, R. M. and Biswal, S. 2004. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 114:1248–1259.
- Rangasamy, T., Guo, J., Mitzner, W. A., Roman, J., Singh, A., Fryer, A. D., Yamamoto, M., Kensler, T. W., Tuder, R. M., Georas, S. N. and Biswal, S. 2005. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 202:47–59.
- Ritz, S. A., Wan, J. and Diaz-Sanchez, D. 2007. Sulforaphane-stimulated phase II enzyme induction inhibits cytokine production by airway epithelial cells stimulated with diesel extract. Am J Physiol Lung Cell Mol Physiol. 292:L33–L39.
- Sacerdoti, D., Colombrita, C., Ghattas, M. H., Ismaeil, E. F., Scapagnini, G., Bolognesi, M., Li Volti, G. and Abraham, N. G. 2005. Heme oxygenase-1 transduction in endothelial cells causes downregulation of monocyte chemoattractant protein-1 and of genes involved in inflammation and growth. Cell Mol Biol (Noisy-le-grand). 51:363–370.
- Shokawa, T., Yoshizumi, M., Yamamoto, H., Omura, S., Toyofuku, M., Shimizu, Y., Imazu, M. and Kohno, N. 2006. Induction of heme oxygenase-1 inhibits monocyte chemoattractant protein-1 mRNA expression in U937 cells. J Pharmacol Sci. 100:162–166.
- Stockley, R. A. 2009. Progression of chronic obstructive pulmonary disease: Impact of inflammation, comorbidities and therapeutic intervention. Curr Med Res Opin. 25:1235–1245.
- Sussan, T. E., Rangasamy, T., Blake, D. J., Malhotra, D., El-Haddad, H., Bedja, D., Yates, M. S., Kombairaju, P., Yamamoto, M., Liby, K. T., Sporn, M. B., Gabrielson, K. L., Champion, H. C., Tuder, R. M., Kensler, T. W. and Biswal, S. 2009. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc Natl Acad Sci USA. 106:250–255.
- Suzuki, M., Betsuyaku, T., Ito, Y., Nagai, K., Nasuhara, Y., Kaga, K., Kondo, S. and Nishimura, M. 2008. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 39:673–682.
- Thimmulappa, R. K., Lee, H., Rangasamy, T., Reddy, S. P., Yamamoto, M., Kensler, T. W. and Biswal, S. 2006a. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 116:984–995.
- Thimmulappa, R. K., Mai, K. H., Srisuma, S., Kensler, T. W., Yamamoto, M. and Biswal, S. 2002. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 62:5196–5203.
- Thimmulappa, R. K., Scollick, C., Traore, K., Yates, M., Trush, M. A., Liby, K. T., Sporn, M. B., Yamamoto, M., Kensler, T. W. and Biswal, S. 2006b. Nrf2-dependent protection from LPS-induced inflammatory response and mortality by CDDO-Imidazolide. Biochem Biophys Res Commun. 351:883–889.
- Traves, S. L., Culpitt, S. V., Russell, R. E., Barnes, P. J. and Donnelly, L. E. 2002. Increased levels of the chemokines GROα and MCP-1 in sputum samples from patients with COPD. Thorax. 57:590–595.
- Ueda, A., Ishigatsubo, Y., Okubo, T. and Yoshimura, T. 1997. Transcriptional regulation of the human monocyte chemoattractant protein-1 gene. Cooperation of two NF-κB sites and NF-κB/Rel subunit specificity. J Biol Chem. 272:31092–31099.
- Venugopal, R. and Jaiswal, A. K. 1996. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci USA. 93:14960–14965.
- Viegi, G., Pistelli, F., Sherrill, D. L., Maio, S., Baldacci, S. and Carrozzi, L. 2007. Definition, epidemiology, and natural history of COPD. Eur Respir J. 30:993–1013.
- Wakabayashi, N., Slocum, S. L., Skoko, J. J., Shin, S. and Kensler, T. W. 2010. When NRF2 talks, who’s listening? Antioxid Redox Signal. 13:1649–1663.
- Yamamoto, C., Yoneda, T., Yoshikawa, M., Fu, A., Tokuyama, T., Tsukaguchi, K. and Narita, N. 1997. Airway inflammation in COPD assessed by sputum levels of interleukin-8. Chest. 112:505–510.
- Zakkar, M., Van der Heiden, K., Luong, l. e. A., Chaudhury, H., Cuhlmann, S., Hamdulay, S. S., Krams, R., Edirisinghe, I., Rahman, I., Carlsen, H., Haskard, D. O., Mason, J. C. and Evans, P. C. 2009. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler Thromb Vasc Biol. 29:1851–1857.
- Zhou, L., Tan, A., Iasvovskaia, S., Li, J., Lin, A. and Hershenson, M. B. 2003. Ras and mitogen-activated protein kinase kinase kinase-1 coregulate activator protein-1- and nuclear factor-κB-mediated gene expression in airway epithelial cells. Am J Respir Cell Mol Biol. 28:762–769.