Abstract
Maternal environmental exposures during pregnancy are known to affect disease onset in adult offspring. For example, maternal asthma exacerbations during pregnancy can worsen adult asthma in the offspring. Cigarette smoking during pregnancy is associated with future onset of cardiovascular disease, obesity and diabetes. However, little is known about the effect of maternal environmental exposures on offspring susceptibility to liver disease. This pilot study examined the long-term effect of maternal allergen challenge and/or cigarette smoking during pregnancy on hepatic inflammation and fibrosis in adult mouse offspring. Ovalbumin (OVA) or phosphate-buffered saline (PBS)-sensitized/challenged CD-1 dams were exposed to mainstream cigarette smoke (MCS) or filtered air from gestational day 4 until parturition. Eight weeks postnatally, offspring were sacrificed for comparison of hepatic histology and mRNA expression. Adult male offspring of OVA-sensitized/challenged dams exposed to MCS (OSM) displayed significantly increased liver fibrosis (9.2% collagen content vs. <4% for all other treatment groups). These mice also had 1.8-fold greater collagen 1A1 mRNA levels. From the results here, we concluded that maternal allergen challenge in combination with cigarette smoke exposure during pregnancy may be an important risk factor for liver disease in adult male offspring.
Introduction
Thirty percent of adults in the United States have non-alcoholic fatty liver disease largely due to the epidemic of obesity in the United States. Most are asymptomatic; however, one out of five eventually will develop extensive hepatic inflammation and fibrosis. The risk factors for disease progression are not well-defined. The developmental origin of health and disease hypothesis holds that events occurring during intrauterine life have profound consequences on the future health of the offspring (Rinaudo and Lamb, Citation2008). Intrauterine stress and obesity have been linked to predisposition to hypertension, cardiovascular diseases and diabetes (Williams and Poulton, Citation1999; Montgomery and Ekbom, Citation2002; Lawlor et al., Citation2004; Oken et al., Citation2005), which are all associated with non-alcoholic fatty liver disease (Diehl, Citation2004).
Epidemiological studies support a link between intrauterine stress and adult liver disease. In a study of 2100 women, Fraser et al. found an association between low birth weight, a common consequence of intrauterine stress, and increases in serum levels of the liver inflammation biomarkers alanine transaminase (ALT), γ-glutamyl transpeptidase (GGT) and alkaline phosphatase (ALP) (Fraser et al., Citation2008). Various cytokines, including interleukin (IL)-6, play key roles in triggering hepatic inflammation and fibrosis (Friedman, Citation2008). In addition, an increased incidence of liver cirrhosis-related mortality in adult Danish men who had a low birth weight has been described (Andersen and Osler, Citation2004). Maternal smoking decreases uterine blood flow to the placenta and may reduce birth weight, yet these offspring are actually at greater risk of obesity in adulthood (Pausová et al., Citation2003; Zdravkovic et al., Citation2005). Maternal allergy (e.g., asthma) has also been associated with reduced birth weight in humans (Cousins, Citation1999; Clifton et al., Citation2001, Citation2010; Bracken et al., Citation2003, Murphy et al., Citation2005; Mayhew et al., Citation2008; Scott et al., Citation2009) and in animal models (Fedulov and Kobzik, Citation2008; Lim and Kobzik, Citation2009), but it is unknown whether it affects adult onset of obesity.
In the studies reported here, we sought to determine whether fetal stress due to maternal allergy and/or in utero exposure to cigarette smoke (CS) alters adult offspring liver fat content, inflammation or cytokine mRNA expression using an animal model.
Methods and materials
Animals
Outbred CD-1 male and female mice (4-week-old) were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were housed in pairs (females) or individually (males) in polycarbonate cages with corncob bedding in a facility maintained at 23°C with a 30–50% relative humidity and 12-h interval light/dark cycle. Food (Purina lab chow) and tap water were provided ad libitum (except for during each daily 4-h smoke exposure). Mice were allowed to acclimate for 1 week prior to use. All animal procedures were conducted under an animal protocol approved by New York University Institutional Animal Care and Use Committee (IACUC).
Exposure protocol
After acclimatization, female mice were either sensitized by intraperitoneal (IP) injection with ovalbumin (OVA) or administered phosphate-buffered saline (PBS) and served as controls. The sensitized mice received an IP injection (5 μL) of PBS containing 10 mg OVA + 2 mg alum; the same injection was repeated 7 days later. One and two weeks following the second IP injection, isofluorane-sedated mice were intranasally (IN) challenged with 50 μL of OVA (2 µg/mL in PBS). To confirm OVA sensitization, blood samples were collected (by facial vein or tail bleeding) within 24 h of the final IN challenge and levels of total and OVA-specific IgE measured using an ELISA assay (Bethyl Laboratories, Montgomery, TX); blood was also recovered from the dams on gestational day (GD) 15 (via tail bleeding) to determine the persistence of IgE levels throughout pregnancy.
Sensitized and non-OVA-sensitized females were then paired with a single naïve male (two females/one male) for 4 days. The first day of pairing was considered GD0. At GD4, females were exposed to either mainstream cigarette smoke (MCS) or filtered air (control). Pregnant mice were exposed to MCS by (whole body) inhalation for 4 h/day (5 days/week) until parturition (i.e., ~GD21). During pregnancy and lactation, gestational parameters were monitored, including dam weight gain, incidence of pregnancy, litter size and daily litter weight gain; weight gain of the pups was determined by weighing the entire group and dividing by the total number of pups in each individual litter. After weaning, pups were separated into eight groups based upon exposure status of the mother: (1) PBS–air male (PAM); (2) PBS–air female (PAF); (3) PBS–smoke male (PSM); (4) PBS–smoke female (PSF); (5) OVA–air male (OAM); (6) OVA–air female (OAF); (7) OVA–smoke male (OSM); and (8) OVA–smoke female (OSF). Each group consisted of pups selected randomly from six to seven different dams for each exposure group. Mice were fed normal chow and water ad libitum until Week 8 postpartum. Serum samples were collected at sacrifice for analysis of transforming growth factor-β (TGFβ) levels by commercial ELISA (R&D Systems, Minneapolis, MN).
CS exposure
CS was generated using an automated cigarette-smoking machine controlled by an adjustable peristaltic pump programmed to produce an amount of carbon monoxide (CO) and total suspended particulates (TSP) approximately equivalent to a human smoking <1 pack of cigarettes/day, that is, >25 ppm and ~15 mg TSP/m3, respectively (Ng et al., Citation2006). CO levels were monitored throughout exposure using a 48C CO Analyzer (Thermo Environmental Instruments Inc., Franklin, MA). TSP were collected from the chamber every hour onto Pallflex Emfab filters (Pall Corporation, East Hills, NY). Filters were weighed before and after sampling, and mean TSP levels were determined gravimetrically. To ensure exposure to tobacco smoke, cotinine (a longer-lived nicotine metabolite) was measured in blood samples from both CS- and air-exposed dams immediately after exposure on GD15, using a Cotinine Micro-Plate ELISA kit (Orasure Technologies, Inc., Bethlehem, PA).
Histological staining
Formalin-fixed liver specimens from 8-week-old male and female offspring were paraffin-embedded. Three animals per treatment group were analyzed. Five millimeter sections were prepared and stained either with hematoxylin and eosin (H&E) or with 0.1% Sirius red F3B (Sigma, St. Louis, MO) in saturated picric acid (Sigma). The H&E sections were scored for inflammation and fibrosis by a liver histopathologist (who was blinded as to treatment group) using the Ludwig and Batts scoring method (Lefkowitch, Citation2007). Fibrosis area (expressed as percent [%] of total liver area) was assessed via the analysis of 40 low-power fields of Sirius red-stained liver sections per animal. Each field was visualized using a 10× magnification and then analyzed with a computerized Bioquant morphometry system (Bioquant, Nashville, TN). The subtraction of the vascular luminal area from the total field area yielded the net field area (Horani et al., Citation2007). To evaluate the relative fibrosis area, the measured Sirius red-stained collagen area was divided by the net field area, and then multiplied by 100.
Quantitative real-time PCR
Liver samples were stored in RLT buffer (Qiagen, Valencia, CA), and total RNA was extracted with RNeasy Mini Kit (Qiagen). Expression levels of mRNA fibrosis-related genes were assayed by qRT-PCR. Synthesis of cDNA was performed using 1 µg of total RNA per sample with random primers and reagents contained in qScript cDNA SuperMix (Quanta, Gaithersburg, MD). The cDNA was diluted 20 times in nuclease-free H2O, and 5 µL of each sample was loaded into 384-well plates for qRT-PCR. Each reaction included 5 µL 2X-FastStart SYBR Green Master Mix (Roche Applied Science, Indianapolis, IN) and 20 nM of primers. GAPDH was used as internal control.
The primers are as follows:
GAPDH (f) 5′-CAATGACCTTCATTGACC-3′
GAPDH (r) 5′-GATCTCGCTCCTGGAAGATG-3′
Collagen1 (f) 5′-GTCCCTGAAGTCAGCTGCATA-3′
Collagen1 (r) 5′-GTCCCTGAAGTCAGCTGCATA-3′
PDGFRβ (f) 5′-CTTTGTGCCAGATCCCACCA-3′
PDGFRβ (r) 5′-TCACTCGGCACGGAATTGTC-3′
ASMA (f) 5′-TCCTCCCTGGAGAAGAGCTAC-3′
ASMA (r) 5′-TATGGTGGTTTCGTGGATGC-3′
Plates were run in the Light Cycler 480 System (Roche). The reaction mixtures were subjected to 98°C for 5 min, then 45 cycles at 95°C for 10 sec, 55°C for 10 sec and 72°C for 12 sec. Relative quantification was calculated with the comparative threshold cycle (CT). The CT indicates the fractional cycle number at which the amount of amplified target genes reach a fixed threshold within the linear phase of gene amplification and is inversely related to the abundance of mRNA transcripts in the initial sample.
Statistical analysis
Since litter size can affect pup size, litter size was first checked for outliers using the Grubbs’ test. For comparisons of average litter size and mass/pup/litter between the treatment groups, one-way ANOVA was used. The chi-square test was used for comparison of male with female pup ratio between the groups. For group-to-group comparisons, two-way ANOVA was used with Bonferroni post-tests. A Student’s t-test was used when comparing RT-PCR results of mice grouped by sex, CS exposure or OVA sensitization. Correlations were calculated using Pearson’s correlation coefficient for parametric comparisons. For all statistical analyses, p < 0.05 was considered statistically significant.
Results
Examination of maternal serum demonstrated significant cotinine levels and induction of a specific immune response following maternal tobacco smoke exposure and OVA exposure, respectively. The mean cotinine value for dams exposed to CS was 23.1 ng/mL on GD15 and undetectable for non-exposed dams. OVA-specific IgE levels 24 h after the last IN OVA instillation were 14.5 (±15.8) for controls and 77.6 (±57.8) for OVA-exposed dams. At postnatal day 7, there was no significant difference in the average litter weight per pup per dam, the ratio of male-to-female offspring or the litter size between treatment groups ().
Table 1. Gestational parameters.
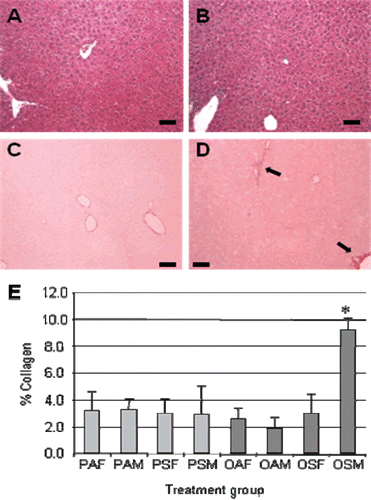
Liver sections of adult offspring stained with H&E were scored in a blinded fashion to assess hepatic steatosis, inflammatory grade and fibrosis stage. The OSM group (male offspring exposed in utero to CS and whose mothers were challenged with OVA during pregnancy) demonstrated Stage 2 hepatic fibrosis, whereas minimal fibrosis (Stage 0) was observed in all other exposure groups. Hepatic steatosis and inflammation were absent in each offspring group, including the OSM group ( and ). Sirius red staining (to demonstrate liver hepatic collagen content) confirmed increased fibrosis in the OSM treatment group. The fibrosis was primarily perivenular (or Zone 3) in nature as seen in fatty liver disease (–). Hepatic collagen was significantly increased by 4.7-fold in the OSM group compared with male offspring of OVA-sensitized dams exposed to filtered air (i.e., OAM group), and 3.0-fold elevated compared with male offspring of mice exposed to CS and PBS only (PSM group) () (both p < 0.050). The combination of maternal OVA sensitization and prenatal CS exposure apparently had a synergistic effect on hepatic collagen levels in male offspring.
Figure 1. Histological analyses of offspring livers. Representative images of (A and B) hematoxylin and eosin (H&E)-stained and (C and D) Sirius red-stained liver sections of (A and C) PAF and (B and D) OSM mice. In OSM mice, the increased hepatic fibrosis was primarily perivenular (D, solid arrows). (E) Bioquant colorimetric analysis of Sirius red staining indicated that combined maternal exposure to ovalbumin (OVA) and mainstream cigarette smoke (MCS) increased hepatic fibrosis in male offspring (OSM) compared with each other group. *p < 0.001 for OSM versus OAM, OSM versus PAM, and OSM versus PSM. Black bar represents 100 µm. Three mice were evaluated for each group.
Quantitative RT-PCR was performed to determine if the increased collagen deposition observed in OSM mice was associated with alterations in collagen mRNA levels or hepatic stellate cell activation. Hepatic stellate cells, upon activation into potent myofibroblasts, secrete collagen type I, α-1 (Col1A1). Overall, offspring of OVA-sensitized dams had 1.4-fold higher levels of Col1A1 mRNA than PBS-treated dams (p = 0.001); CS exposure did not have a similar effect. Bonferroni post-test analysis revealed a significant increase (1.8-fold, p < 0.01) in Col1A1 mRNA levels between OSM compared with PSM mice (), but the difference between OSM and OAM mice was not statistically significant. Col1A1 mRNA levels did not correlate with the mean % collagen staining for each group. The pattern of changes between treatment groups was similar for male and female mice.
Figure 2. Comparison of hepatic fibrosis-related mRNA expression. Mean mRNA expression levels for (A) collagen type I, α-1 (Col1A1) and (B) platelet-derived growth factor receptor-β (PDGFRβ) were compared by two-way ANOVA testing. Combined maternal exposure to ovalbumin (OVA) and mainstream cigarette smoke (MCS) significantly increased hepatic collagen 1A1 mRNA levels in male offspring (OSM) compared with male offspring of dams exposed only to MCS (PSM). There was a trend toward increased Col1A1 levels in all offspring of OVA-exposed dams compared with in PBS-exposed counterparts. PDGFRβ mRNA levels were significantly decreased in the OSM group compared with their PSM counterparts. Similar significant differences were seen for OAM versus PAM groups and OSF versus PSF groups. The mRNA expression pattern was similar between the male and female groups for both Col1A1 and PDGFRβ. *Value is statistically significantly different at p < 0.05.
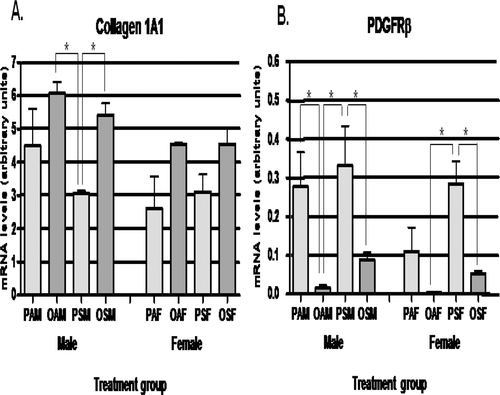
α-Smooth muscle actin (ASMA) mRNA levels, a general marker of hepatic stellate cell activation, were not significantly increased in the OSM mice (data not shown). Hepatic mRNA levels of platelet-derived growth factor receptor-β (PDGFRβ), which increases proliferation of hepatic stellate cells (Friedman, Citation2008), were significantly reduced by OVA exposure (). The PDGFRβ mRNA levels were reduced 18.3-fold in the OAM mice versus PAM mice (p < 0.01) and reduced 3.7-fold in the OSM mice versus PSM mice (p < 0.05). There was a trend toward increased maternal serum levels of total TGFβ, which stimulates collagen transcription in hepatic stellate cells (Friedman, Citation2008), in the OSM group compared with the PSM group (2.9-fold, p = 0.08). Significant differences in hepatic mRNA levels of IL-6, which also promote stellate cell activation (Friedman, Citation2008), were not observed (data not shown).
Discussion
Maternal sensitization and challenge with an allergen (i.e., OVA) acted synergistically with in utero CS exposure to induce increased hepatic fibrosis in adult male offspring. In addition to this histological effect, significant hepatic mRNA alterations were also evident in adult offspring mice. Cigarette smoking by adults is already recognized as a potential risk factor for increased hepatic fibrosis in several chronic liver diseases (Klatsky and Armstrong, Citation1992; Costenbader and Karlson, Citation2006, Dev et al., Citation2006; Zein et al., Citation2006; Mallat and Lotersztajn, Citation2009). Allergies/asthma alone in adults, however, is not known to be a risk factor for increased hepatic fibrosis.
Fetal stress has been described as an important risk factor for a number of chronic diseases in offspring including Type 2 diabetes (Montgomery and Ekbom, Citation2002; Oken et al., Citation2005) and hypertension (Williams and Poulton, Citation1999; Lawlor et al., Citation2004; Oken et al., Citation2005). Our results suggest that certain fetal stressors may also be a novel cause of liver fibrosis in adult male offspring. The degree of fetal stress in our study was relatively mild given that birth weight was not significantly affected. Based on offspring hepatic mRNA expression levels, increased collagen production was important in the observed increase in hepatic fibrosis in male offspring whose mothers had an allergic phenotype and were exposed in utero to CS. In general, men are more prone than women to liver fibrosis progression (Poynard et al., Citation1997, Citation2010). Increased mean hepatic collagen mRNA levels were found more generally in the adult offspring of OVA-sensitized dams. The mRNA data here indicate that OVA sensitization had a greater long-term effect on the measured hepatic mRNA levels than MCS exposure. Maternal allergy and/or asthma unexpectedly broadly affected adult hepatic mRNA expression in offspring. Several potential mechanisms may be involved in this effect.
Maternal asthma and smoking during pregnancy have previously been shown to have synergistic, sex-specific effects on the fetus (Scott et al., Citation2009; Clifton et al., Citation2010). In the latter study, placental insulin-like growth factor 1 (IGF1) release was increased for male fetuses of mothers with asthma and decreased for female fetuses of mothers who had asthma and smoked while pregnant. As the liver is the first fetal organ through which blood from the placenta flows, it may be particularly sensitive to placental IGF1 release. IGF1 is known to stimulate proliferation in vitro of hepatic stellate cells, the cell type primarily responsible for collagen deposition in the liver (Svegliati-Baroni et al., Citation1999; Pinzani and Marra, Citation2001; Liu et al., Citation2009). An increase in the number of stellate cells in the liver may contribute to increased liver fibrosis. Although there was no histological indication of increased hepatic stellate cell activation in the adult mice, histological analyses may not be sensitive enough to detect a small increase in the number of stellate cells. Alternatively the effect may have waned over time.
As mentioned above, female gender seems to be protective against the progression of hepatic fibrosis. The mechanism underlying this protective effect is uncertain, but studies suggest estrogens may suppress the rate of hepatic fibrosis progression (Yasuda et al., Citation1999; Di Martino et al., Citation2004). In a study of 472 women, Di Martino and colleagues found that hormone replacement therapy in postmenopausal women infected with hepatitis C reduced the rate of fibrosis progression compared with among untreated postmenopausal women. Yasuda et al. (Citation1999) utilized a rat model of hepatic fibrosis to examine the importance of estrogen levels on fibrosis progression. Both male and female rats were exposed to dimethylnitrosamine to induce hepatic fibrosis. As expected the fibrotic response was greater in the male mice. Estradiol treatment of the male mice reduced hepatic collagen deposition by reducing mRNA levels of Type 1 collagen. Ovariectomy had a profibrotic effect that could be suppressed by estradiol replacement. In isolated rat hepatic stellate cells, estradiol exposure in culture reduced stellate cell activation and production of Type 1 collagen. Based on these studies, the lack of fibrosis in the OSF group (female offspring exposed in utero to CS and whose mothers were challenged with OVA during pregnancy) in our study may have been due to direct effects of estrogens on hepatic stellate cell activation.
Fibrosis in the OSM group was observed in the absence of significant hepatic inflammation. Typically, hepatic inflammation is a precursor to the development of hepatic fibrosis in non-alcoholic fatty liver disease (Syn et al., Citation2009). In certain types of human liver disease (e.g., hereditary hemochromatosis), fibrosis progression appears to be independent of inflammation (Fleming et al., Citation2004). Hepatic inflammation could have been induced during pregnancy and resolved prior to examination of the adult offspring. Fibrosis may still be present in some adult offspring since it usually takes longer to resolve than inflammation. The increased Col1A1 mRNA levels in the offspring of OVA-exposed dams may no longer be clinically significant. Unfortunately offspring hepatic tissue from earlier time points was not available.
The decreased PDGFRβ mRNA levels in the adult offspring of OVA-exposed dams may reflect resolving inflammation and suggest that altered cytokine signaling persists into adulthood. In liver cirrhosis, PDGFβ levels and PDGFRβ mRNA levels are usually increased (Pinzani et al., Citation1996), though under some conditions, PDGFR mRNA levels are down-regulated upon an over-expression of PDGFβ (Gao et al., Citation2001). The significant differences in the analyzed mRNA levels fail to explain why fibrosis was only increased in the OSM mice since the changes were not specific to the OSM mice. Analysis of additional time points would be useful as mentioned above. Due to limited tissue availability microarray analysis to more broadly examine mRNA expression levels was not possible.
CS exposure during pregnancy has been shown by Izzotti et al. (Citation2003) to alter fetal hepatic mRNA expression levels and cause fetal hepatic genomic damage. In their study, transplacental CS was shown to up-regulate the expression of 116 mRNAs involved in metabolism, response to oxidative stress, DNA and protein repair and signal transduction. Additionally, the oral administration of N-acetylcysteine during pregnancy inhibited up-regulation of these mRNAs, suggesting that oxidative stress during pregnancy was involved in CS-induced hepatic alterations. Our study suggests that hepatic mRNA changes may persist into adulthood. Induction of epigenetic alterations during pregnancy could account for persistent mRNA changes in the male offspring. Environmental exposures have been shown to cause epigenetic changes affecting subsequent generations (Suter and Aagaard-Tillery, Citation2009), and fibrosis-related genes are known to be sensitive to epigenetic alterations (Niki et al., Citation1999; Mann et al., Citation2007; Ramani et al., Citation2010).
Twenty to thirty million residents of the United States have chronic liver disease and 20% of them will develop progressive liver fibrosis and cirrhosis. Maternal allergy and prenatal exposure to CS acted synergistically in this study to increase hepatic fibrosis in adult male offspring. Thus, maternal exposures during pregnancy may be risk factors for hepatic fibrosis in males later in life. Further studies in mice fed a high fat diet may indicate whether this affects progression of non-alcoholic fatty liver disease, the most common liver disease in the United States residents. Identification of risk factors will enable anti-fibrotic therapy to be tailored toward those most in need.
Acknowledgements
We would like to thank Carol Hoffman for her technical assistance. This work was supported by an American Liver Foundation Liver Scholar Award (JA), an American Gastroenterological Association Pilot Study Award (JAO), grant from the Institute for Science and Health (JTZ) and in part by the NYU NIEHS Center grant.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
References
- Andersen, A. M. and Osler, M. 2004. Birth dimensions, parental mortality, and mortality in early adult age: A cohort study of Danish men born in 1953. Int. J. Epidemiol. 33:92–99.
- Bracken, M. B.,Triche, E. W., Belanger, K., Saftlas, A., Beckett, W. S. and Leaderer, B. P. 2003. Asthma symptoms, severity, and drug therapy: A prospective study of effects on 2205 pregnancies. Obstet. Gynecol. 102:739–752.
- Clifton, V. L., Giles, W. B., Smith, R., Bisits, A. T., Hempenstall, P. A., Kessell, C. G. and Gibson, P. G. 2001. Alterations of placental vascular function in asthmatic pregnancies. Am. J. Respir. Crit. Care Med. 164:546–553.
- Clifton, V. L., Hodyl, N. A., Murphy, V. E., Giles, W. B., Baxter, R. C. and Smith, R. 2010. Effect of maternal asthma, inhaled glucocorticoids and cigarette use during pregnancy on the newborn insulin-like growth factor axis. Growth Horm. IGF Res. 20:39–48.
- Costenbader, K. H. and Karlson, E. W. 2006. Cigarette smoking and autoimmune disease: What can we learn from epidemiology? Lupus 15:737–745.
- Cousins, L. 1999. Fetal oxygenation, assessment of fetal well-being, and obstetric management of the pregnant patient with asthma. J. Allergy Clin. Immunol. 103:S343–S349.
- Dev, A., Patel, K., Conrad, A., Blatt, L. M. and McHutchison, J. G. 2006. Relationship of smoking and fibrosis in patients with chronic hepatitis C. Clin. Gastroenterol. Hepatol. 4:797–801.
- Di Martino, V., Lebray, P., Myers, R. P., Pannier, E., Paradis, V., Charlotte, F., Moussalli, J., Thabut, D., Buffet, C. and Poynard, T. 2004. Progression of liver fibrosis in women infected with hepatitis C: Long-term benefit of estrogen exposure. Hepatology 40:1426–1433.
- Diehl, A. M. 2004. Fatty liver, hypertension, and the metabolic syndrome. Gut 53:923–924.
- Fedulov, A. V. and Kobzik, L. 2008. Immunotoxicologic analysis of maternal transmission of asthma risk. J. Immunotoxicol. 5:445–452.
- Fleming, R. E., Britton, R. S., Waheed, A., Sly, W. S. and Bacon, B. R. 2004. Pathogenesis of hereditary hemochromatosis. Clin. Liver Dis. 8:755–773, vii.
- Fraser, A., Ebrahim, S., Smith, G. D. and Lawlor, D. A. 2008. The associations between birthweight and adult markers of liver damage and function. Paediatr. Perinat. Epidemiol. 22:12–21.
- Friedman, S. L. 2008. Mechanisms of hepatic fibrogenesis. Gastroenterology 134:1655–1669.
- Gao, C., Miyazaki, M., Kondo, T., Tsuji, T., Sakaguchi, M. and Namba, M. 2001. Over-expression of platelet-derived growth factor B and down-regulation of PDGF-receptor α in human immortalized fibroblasts. Int. J. Oncol. 18:871–875.
- Horani, A., Muhanna, N., Pappo, O., Melhem, A., Alvarez, C. E., Doron, S., Wehbi, W., Dimitrios, K., Friedman, S. L. and Safadi, R. 2007. Beneficial effect of glatiramer acetate (Copaxone) on immune modulation of experimental hepatic fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 292:G628–G638.
- Izzotti, A., Balansky, R. M., Cartiglia, C., Camoirano, A., Longobardi, M. and De Flora, S. 2003. Genomic and transcriptional alterations in mouse fetus liver after transplacental exposure to cigarette smoke. FASEB J. 17:1127–1129.
- Klatsky, A. L. and Armstrong, M. A. 1992. Alcohol, smoking, coffee, and cirrhosis. Am. J. Epidemiol. 136:1248–1257.
- Lawlor, D. A., Najman, J. M., Sterne, J., Williams, G. M., Ebrahim, S. and Davey Smith, G. 2004. Associations of parental, birth, and early life characteristics with systolic blood pressure at 5 years of age: Findings from the Mater-University study of pregnancy and its outcomes. Circulation 110:2417–2423.
- Lefkowitch, J. H. 2007. Liver biopsy assessment in chronic hepatitis. Arch. Med. Res. 38:634–643.
- Lim, R. H. and Kobzik, L. 2009. Maternal transmission of asthma risk. Am. J. Reprod. Immunol. 61:1–10.
- Liu, L. X., Huang, S., Zhang, Q. Q., Liu, Y., Zhang, D. M., Guo, X. H. and Han, D. W. 2009. Insulin-like growth factor binding protein-7 induces activation and transdifferentiation of hepatic stellate cells in vitro. World J. Gastroenterol. 15:3246–3253.
- Mallat, A. and Lotersztajn, S. 2009. Cigarette smoke exposure: A novel cofactor of NAFLD progression? J. Hepatol. 51:430–432.
- Mann, J., Oakley, F., Akiboye, F., Elsharkawy, A., Thorne, A. W. and Mann, D. A. 2007. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: Implications for wound healing and fibrogenesis. Cell Death Differ. 14:275–285.
- Mayhew, T. M., Jenkins, H., Todd, B. and Clifton, V. L. 2008. Maternal asthma and placental morphometry: Effects of severity, treatment and fetal sex. Placenta 29:366–373.
- Montgomery, S. M. and Ekbom, A. 2002. Smoking during pregnancy and diabetes mellitus in a British longitudinal birth cohort. BMJ 324:26–27.
- Murphy, V. E., Gibson, P., Talbot, P. I. and Clifton, V. L. 2005. Severe asthma exacerbations during pregnancy. Obstet. Gynecol. 106:1046–1054.
- Ng SP, Steinetz BG, Lasano SG, and Zelikoff JT. Hormonal changes accompanying cigarette smoke-induced preterm births in a mouse model. Exp Biol Med (Maywood). 2006 Sep;231(8):1403–9.
- Niki, T., Rombouts, K., De Bleser, P., De Smet, K., Rogiers, V., Schuppan, D., Yoshida, M., Gabbiani, G. and Geerts, A. 1999. A histone deacetylase inhibitor, trichostatin A, suppresses myofibroblastic differentiation of rat hepatic stellate cells in primary culture. Hepatology 29:858–867.
- Oken, E., Huh, S. Y., Taveras, E. M., Rich-Edwards, J. W. and Gillman, M. W. 2005. Associations of maternal prenatal smoking with child adiposity and blood pressure. Obes. Res. 13:2021–2028.
- Pausová, Z., Paus, T., Sedová, L. and Bérubé, J. 2003. Prenatal exposure to nicotine modifies kidney weight and blood pressure in genetically susceptible rats: A case of gene-environment interaction. Kidney Int. 64:829–835.
- Pinzani, M. and Marra, F. 2001. Cytokine receptors and signaling in hepatic stellate cells. Semin. Liver Dis. 21:397–416.
- Pinzani, M., Milani, S., Herbst, H., DeFranco, R., Grappone, C., Gentilini, A., Caligiuri, A., Pellegrini, G., Ngo, D. V., Romanelli, R. G. and Gentilini, P. 1996. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am. J. Pathol. 148:785–800.
- Poynard, T., Bedossa, P. and Opolon, P. 1997. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 349:825–832.
- Poynard, T., Lebray, P., Ingiliz, P., Varaut, A., Varsat, B., Ngo, Y., Norha, P., Munteanu, M., Drane, F., Messous, D., Bismut, F. I., Carrau, J. P., Massard, J., Ratziu, V. and Giordanella, J. P. 2010. Prevalence of liver fibrosis and risk factors in a general population using non-invasive biomarkers (FibroTest). BMC Gastroenterol. 10:40.
- Ramani, K., Yang, H., Kuhlenkamp, J., Tomasi, L., Tsukamoto, H., Mato, J. M. and Lu, S. C. 2010. Changes in the expression of methionine adenosyltransferase genes and S-adenosylmethionine homeostasis during hepatic stellate cell activation. Hepatology 51:986–995.
- Rinaudo, P. F. and Lamb, J. 2008. Fetal origins of perinatal morbidity and/or adult disease. Semin. Reprod. Med. 26:436–445.
- Scott, N. M., Hodyl, N. A., Murphy, V. E., Osei-Kumah, A., Wyper, H., Hodgson, D. M., Smith, R. and Clifton, V. L. 2009. Placental cytokine expression covaries with maternal asthma severity and fetal sex. J. Immunol. 182:1411–1420.
- Suter, M. A. and Aagaard-Tillery, K. M. 2009. Environmental influences on epigenetic profiles. Semin. Reprod. Med. 27:380–390.
- Svegliati-Baroni, G., Ridolfi, F., Di Sario, A., Casini, A., Marucci, L., Gaggiotti, G., Orlandoni, P., Macarri, G., Perego, L., Benedetti, A. and Folli, F. 1999. Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: Differential effects on signal transduction pathways. Hepatology 29:1743–1751.
- Syn, W. K., Choi, S. S. and Diehl, A. M. 2009. Apoptosis and cytokines in non-alcoholic steatohepatitis. Clin. Liver Dis. 13:565–580.
- Williams, S. and Poulton, R. 1999. Twins and maternal smoking: Ordeals for the fetal origins hypothesis? A cohort study. BMJ 318:897–900.
- Yasuda, M., Shimizu, I., Shiba, M. and Ito, S. 1999. Suppressive effects of estradiol on dimethylnitrosamine-induced fibrosis of the liver in rats. Hepatology 29:719–727.
- Zdravkovic, T., Genbacev, O., McMaster, M. T. and Fisher, S. J. 2005. The adverse effects of maternal smoking on the human placenta: A review. Placenta 26 Suppl A:S81–S86.
- Zein, C. O., Beatty, K., Post, A. B., Logan, L., Debanne, S. and McCullough, A. J. 2006. Smoking and increased severity of hepatic fibrosis in primary biliary cirrhosis: A cross validated retrospective assessment. Hepatology 44:1564–1571.