Abstract
Virtually all drugs that contain a primary aromatic amine are associated with a high incidence of idiosyncratic drug reactions (IDRs), suggesting that this functional group has biological effects that may be used as biomarkers to predict IDR risk. Most IDRs exhibit evidence of immune involvement and the ability of aromatic amines to form reactive metabolites and redox cycle may be responsible for initiation of an immune response through induction of cell stress, as postulated by the Danger Hypothesis. If true, danger signals could be biomarkers of IDR risk. A previous attempt to test the Danger Hypothesis found that sulfamethoxazole (SMX), the only aromatic amine tested, was also the only drug not associated with an increase of cell stress genes in mice. To ensure that these observations were not species-specific, and to determine biomarkers of IDR risk common to aromatic amines, rats were treated with SMX and two other aromatic amine drugs, dapsone (DDS) and aminoglutethimide (AMG), and hepatic gene expression was determined using microarrays. As in mice, SMX induced minimal gene changes in the rat, and none indicated cell stress, whereas DDS and AMG induced several changes including up-regulation of enzymes such as aldo-keto reductase, glutathione-S-transferase, and aldehyde dehydrogenase, which may represent danger signals. Early insulin-induced hepatic gene (Eiih) was up-regulated by all three drugs. Some mRNA changes were observed in the Keap-1-Nrf2-ARE pathway; however, the pattern was significantly different for each drug. Overall, the most salient finding was that the changes in the liver were minimal, even though aromatic amines cause a high incidence of IDRs. The liver generates a large number of reactive species; however, the ability of aromatic amines to be bioactivated by cells of the immune system may be why they cause a high incidence of IDRs.
| Abbreviations | ||
| IDR, | = | Idiosyncratic drug reaction; |
| SMX, | = | Sulfamethoxazole; |
| DDS, | = | Dapsone; |
| AMG, | = | Aminoglutethimide; |
| CYP450, | = | Cytochrome P450; |
| FDR, | = | False discovery rate; |
| Eiih, | = | early insulin-induced hepatic gene; |
| β2M, | = | β2-microglobulin; |
| Dusp1, | = | Dual specificity phosphatase 1; |
| Sgk, | = | Serum/glucocorticoid-regulated kinase 1; |
| Txnrd, | = | Thioredoxin reductase; |
| GSH-S-Tr, | = | Glutethione-S-Transferase |
Introduction
Drugs that contain the primary aromatic amine functional group are almost always associated with a high incidence of idiosyncratic drug reactions (IDRs), and this is considered a structural alert for drug development (Uetrecht, Citation2002). The inability to predict which drug candidates will cause such reactions, and the fact that they are usually not detected until very late in development or after the drug has been marketed, markedly increases the risks associated with the development of new/novel drugs.
The exact mechanisms of IDRs remain unclear; however, many lines of evidence suggest that most IDRs are mediated by the adaptive immune system (Uetrecht, Citation2009). The Danger Hypothesis has been proposed as a mechanism for the initiation of immune-mediated IDRs (Li and Uetrecht, Citation2010). This hypothesis posits that the tissues are responsible for initiating an immune response through the release of danger signals in response to cell stress or damage (Matzinger, Citation1994). This hypothesis compliments other hypotheses such as the hapten hypothesis because it may result in activation of antigen-presenting cells leading to costimulatory signals, referred to as Signal 2, that are required for activation of T-helper cells (). In the context of IDRs, a drug, or more likely its reactive metabolite, could induce cell stress or damage, and this might contribute to the ability of a drug to cause IDRs.
Figure 1. The Danger Hypothesis applied to IDRs. The drug or its reactive metabolite can form antigenic adducts with endogenous molecules that are presented to T-cells on major histocompatibility complexes of antigen-presenting cells (Signal 1). Additionally, the drug or reactive metabolite may induce cell stress or damage through processes such as redox cycling and oxidative damage. This may lead to the release of danger signals to up-regulate co-stimulatory molecules such as B7 and CD40 on antigen-presenting cells to activate T-cells (Signal 2). Signals 1 and 2 are required concurrently to initiate an adaptive immune response and certain drugs have the ability to induce both signals.
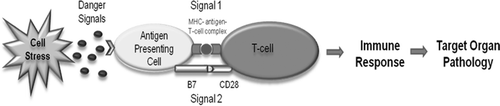
To date, many potential danger signals have been identified such as heat shock proteins, high-mobility group protein B1, ATP, cytokines, and nuclear DNA (Gallucci and Matzinger, Citation2001). However, it is difficult to rigorously test whether the induction of danger signals is an important biomarker of IDR risk. Danger signals vary widely in their characteristics; the only consistent feature being that they are always endogenous molecules, either actively secreted or released due to destruction of cell membrane integrity in the event of cell stress or damage. If the release of danger signals could be used as a biomarker to predict the risk that a drug candidate would cause IDRs, it would have a profound effect on the process of drug development.
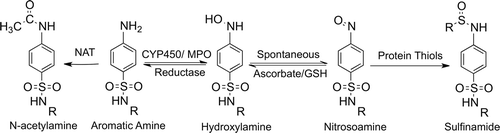
The IDRs associated with aromatic amine drugs are thought to be due to the ease with which they form reactive metabolites (). Aromatic amines can be oxidized by cytochrome P450s (CYP450s) and also myeloperoxidase to hydroxylamine metabolites, which are not very reactive, but can auto-oxidize to electrophilic nitroso metabolites that can covalently bind endogenous proteins. The nitroso metabolite can be easily reduced back to the parent amine with antioxidants, and this redox cycling could lead to oxidative stress. Thus, aromatic amine drugs have the potential to form antigenic substances and to cause cell stress and damage, which could produce danger signals and initiate immune-mediated IDRs.
Figure 2. Metabolic scheme for the formation of reactive metabolites of aromatic amine drugs. Oxidation of aromatic amines to hydroxylamines can occur through enzymes such as CYP450s or myeloperoxidase (MPO); this is prevented by N-acetylation. Hydroxylamines are not very reactive but can auto-oxidize to electrophilic nitroso metabolites. The nitrosoamine can react with thiol-containing nucleophiles such as glutathione or endogenous proteins to form sulfinamide adducts. Additionally, the nitrosoamine can be reduced back to the parent drug, leading to redox cycling and oxidative stress.
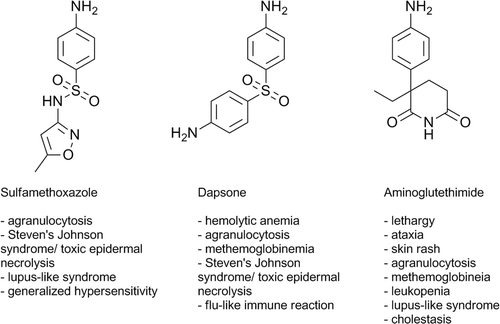
Sulfamethoxazole (SMX) is an aromatic amine antimicrobial that is widely used in combination with trimethoprim for many indications including prophylaxis against and to treat Pnuemocystis carinii pneumonia in HIV-infected individuals. SMX can cause a generalized hypersensitivity reaction and is also associated with among the highest incidences of Stevens-Johnson syndrome and toxic epidermal necrolysis, which can be very serious and even fatal (Chan et al., Citation1990; Mockenhaupt, Citation2011).
Previously we tried to test the Danger Hypothesis with a study of changes in gene expression induced by tienilic acid, which causes idiosyncratic liver injury. Its reactive metabolite binds principally to CYP450 that is unlikely to cause much cell stress. However, it binds to several other proteins and it induced several changes in gene expression in the liver that likely represented danger signals (Pacitto et al., Citation2007). In contrast, treatment of mice with SMX, which was meant as a positive control because it can covalently bind to a wider variety of proteins and also induce oxidative stress, did not lead to up-regulation of genes that were likely to represent danger signals, and the few changes that were induced in the liver were mostly down-regulation of gene expression (Pacitto et al., Citation2007). It is possible that down-regulation of mRNAs represents a danger signal. However, metabolism of SMX in mice is limited (Farrell et al., Citation2003), and the metabolism of SMX in rats more closely resembles its metabolism in humans (Gill et al., Citation1997). Thus, a global screen of hepatic gene expression was performed to determine the effects of SMX in the rat to investigate whether it can induce cell stress leading to the release of danger signals. In addition, two other aromatic amine drugs associated with a high incidence of IDRs, dapsone (DDS) and aminoglutethimide (AMG; ), were tested to determine whether similar expression profiles exist between aromatic amine drugs that could potentially indicate a similar mechanism of action and be useful as biomarkers for drugs with the aromatic amine functional group. As the major site of drug metabolism and target for IDRs, the liver (or hepatocytes) are extensively used for reactive metabolite and toxicity studies during drug development. Therefore, we chose the liver in which to study changes in gene expression induced by aromatic amines; possible downstream effects of these gene changes were also tested.
Figure 3. Structures of the aromatic amine drugs used in this study and some associated IDRs.
Materials and methods
Reagents
Sulfamethoxazole (SMX) and dapsone (DDS) were purchased from Sigma (Oakville, Ontario, Canada). AMG was purchased from Toronto Research Chemical (North York, Ontario, Canada). Nevirapine was obtained from Boehringer Ingelheim (Ridgefield, CT). Clozapine was provided by Novartis (Dorval, Quebec, Canada). RNeasy Mini kits were obtained from Qiagen (Missis-sauga, Ontario, Canada). Gene chips were purchased from Affymetrix (Santa Clara, CA). Light Cycler FastStart DNA Master SYBR Green I was purchased from Roche Applied Science (Laval, Quebec, Canada). Primers were obtained from Integrated DNA Technologies (Coralville, IA).
Animals
Male Brown Norway rats (≈ 8-weeks-old, 200–250 g) were purchased from Charles River (Montreal, Quebec, Canada). Rats were housed under standard conditions (doubly housed in plastic cages, standard rat chow, automatic watering, 12:12 h light:dark cycle, and 22°C). Food and water were provided ad libitum. Rats were acclimatized for 1 week before the start of experiments. In other studies performed with penicillamine and nevirapine in our laboratory, the Brown Norway rat was more susceptible to IDRs induced by these drugs (i.e., autoimmunity and skin rash, respectively) than other strains; for this reason, this strain was selected for use in the current study. The study protocol was approved by and performed in accordance with the University of Toronto Faculties of Medicine and Pharmacy Animal Care Committee.
Treatments
The treatment protocol was similar to that of Pacitto et al. (Citation2007), with some modifications. Each treatment had four rats per group unless otherwise stated. The drugs were administered by oral gavage at doses of 150 mg/kg (SMX), 20 mg/kg (DDS), and 80 mg/kg (AMG). These dosages were chosen from previous rat studies (Schwartz and Rieder, Citation1970; Nicholls et al. Citation1984; Gill et al., Citation1997; Helton et al., Citation2000) to mimic peak drug concentrations in the blood of patients taking these drugs for therapy (≈37 µg/ml for 1000 mg SMX (Goodman et al., Citation2006), 1.7 µg/ml for 100 mg DDS (Goodman et al., Citation2006), and 3 µg/ml for 250 mg AMG (Lonning et al., Citation1985)).
Drugs were suspended in 0.5% methylcellulose due to the poor solubility of DDS and AMG. For the microarray study, rats were euthanized by CO2 asphyxiation after 12 h drug treatment. Treatments for follow-up studies were performed as described previously using several different timepoints, with the exception that for timepoints > 24 h, rats were given an additional dose of drug every 24 h. At necropsy, the liver was extracted using aseptic techniques, immersed in RNAlater RNA Stabilization Reagent (Qiagen), and stored according to manufacturer′s instructions at −80°C until RNA extraction. Liver tissue was also immersed in phosphate buffered saline (PBS, pH 7.4) with protease inhibitor (Sigma) for activity assays. PBS was used instead of lysis buffer to minimize possible interferences by the solvent with assay matrices.
RNA extraction
Liver tissue (≈20–30 mg) stored in RNAlater RNA Stabilization Reagent was homogenized with a rotor stator homogenizer (IKA Ultra-Turrax T25 S1, Janke & Kunel, Staufen, Germany). Total RNA was extracted using an RNeasy Mini Kit (Qiagen) as per manufacturer′s protocol. Extracted RNA was eluted in 40 µl of RNase-free water and stored at −80°C. The quality and purity of the extracted RNA was initially measured using UV spectrophotometry based on the A260/A280 ratio before a further quality check through capillary electrophoresis using the Agilent Bioanalyzer (Affymetrix).
Microarray analysis
Affymetrix RatGene 1.0 ST chips were used for detection of changes in gene expression. Microarray processing of the liver samples was performed at the microarray facility in The Centre for Applied Genomics (Hospital for Sick Children, Toronto, Ontario, Canada) based on Affymetrix protocols. For microarray data, Expression Console software was used for quality control analysis before fold-changes were determined using Partek software. The Robust Multi-array Analysis method was used for background correction, which included Quantile Normalization and Median Polish summarization. Data was log-base 2 transformed and fold-change values were obtained for treatments as compared to the control group. One-way ANOVA analysis was performed to determine the statistical significance based on differences between the treatment and control mean intensities for each gene. Attempts were made to obtain more accurate p-values by performing multiple test corrections using the false discovery rate (FDR).
Real-time (RT)-PCR of early insulin-induced hepatic gene (Eiih)
Extracted RNA samples were converted to cDNA using the Omniscript RT Kit (Qiagen), with oligo(dT15) primers (Roche) and RNase inhibitor (Roche), as per the manufacturer′s protocol. Quality was checked using the spectrophotometry method as previously described. For each sample, 2 µg of RNA was converted to cDNA in a total reaction volume of 20 µl. Using Light Cycler FastStart DNA Master SYBR Green 1 (Roche), PCR was performed using a Light Cycler instrument (Roche) as per the following conditions: pre-incubation at 95°C for 10 min and amplification for 45 cycles (denaturation at 95°C for 15 s, annealing at 60°C for 5 s, and elongation at 72°C for 10 s). Primers (Integrated DNA Technologies) for Eiih were as follows: forward primer 5′-AGCTCTCCAGCTCTGGATTCTT-3′, reverse primer 5′-CACACCCAGAACAGAGTCTTAC-3′. β2-Microglobulin (β2M) was used as the housekeeping gene and the primers (Integrated DNA Technologies) were as follows: forward primer 5′-TCAGTTCCACCCACCTCAGATAGA-3′, reverse primer 5′-TGTGAGCCAGGATGTAGAAAGAC-3′. Data was analyzed using the RelQuant software and normalized to a calibrator control.
An additional two drugs, nevirapine and clozapine, were both tested in the rat for Eiih expression using RT-PCR. The treatment protocol, tissue extraction, and PCR analysis were similar to previously stated. Nevirapine and clozapine were administered at 150 mg/kg and 50 mg/kg, respectively, through oral gavage, and samples were taken 6 and 12 h after treatment.
Activity assays
Thioredoxin reductase (Txnrd) and glutathione-S-transferase (GSH-S-Tr) activity in the liver was measured using a Txnrd assay kit from Cayman Chemical (Ann Arbour, MI) and a GSH-S-Tr activity colorimetric assay kit from Abcam (Cambridge, MA), respectively, as per manufacturer′s instructions. Assayed samples contained a total protein concentration of 4 mg/ml in PBS with proteinase inhibitor. The levels of sensitivity of the Txnrd and GSH-S-Tr kits/assays were 0.015 and 0.025 µmol/min/ml, respectively.
Statistical analysis
Data obtained from PCR and activity assays were analyzed by ANOVA using GraphPad Prism 5. Bonferroni post-tests were used to determine statistical significance between treatment and control groups.
Results
Microarray quality
The extracted rat liver RNA was of good quality as demonstrated via UV spectrophotometry and the Agilent Bioanalyzer methods. The A260/A280 ratios of the samples were in the range of 1.96–2.29, indicating good RNA quality. Only one sample in the SMX treatment had a RNA Integrity Number < 7, but was still sufficient for microarray processing. Expression Console software revealed no outlier arrays and the Spearman Rank Correlation (r2) was > 0.982, indicating a good correlation between signal values of different gene chips. Thus, all arrays were used for microarray data analysis.
SMX-induced hepatic gene expression
SMX induced very few gene changes >2-fold expression in the rat 12 h after treatment (see Appendix A). When FDR was applied for multiple test corrections, none of the changes were significant (p < 0.05); of the gene changes that occurred, none seemed to explicitly indicate cell stress or initiation of an immune response. Interestingly, genes involved with the acute cell response were down-regulated, including dual specificity phosphatase 1 (Dusp1) and serum/glucocorticoid-regulated kinase (Sgk).
DDS-induced hepatic gene expression
In contrast to SMX, a greater number of genes were up-regulated > 2-fold with DDS treatment in the rat (see Appendix B). However, only changes in six genes were statistically significant (p < 0.05), with the FDR applied: aldo-keto reductase 7A3 (Akr7a3, 5.38-fold), glutathione-S-transferase Yc2 subunit (Yc2, 3.18-fold), aldehyde oxidase 1 (Aox1, 1.63-fold), ferritin light polypeptide (Ftl1, 1.44-fold), UDP glucosyltransferase 1A3 (Ugt1a3, 1.39-fold), and glutathione reductase (Gsr, 1.38-fold). Up-regulation of genes such as Akr7a3, Yc2, Ugt1a3, and Gsr could indicate cell stress due to their function in drug metabolism and cytoprotection against oxidative stress. Again, Dusp1 and Sgk were among the most down-regulated genes, similar to what was observed with SMX.
AMG-induced hepatic gene expression
AMG induced the most gene changes of the aromatic amine drugs tested (see Appendix C). Among these changes, 552 genes were significantly different using the FDR. Of these genes, quite a few were involved in antioxidant and drug metabolizing pathways, including CYP450 oxidoreductase (Por), aldehyde dehydrogenase 1A1 (Aldh1a1), UDP glucuronosyltransferase 2B1 (Udpgtr2), epoxide hydrolase 1(Ephx1), aldo-keto reductase 7A3 (Akr7a3), glutathione-S-transferase Yc2 sub-unit (Yc2), thioredoxin reductase 1 (Txnrd1), glutathione reductase (Gsr), and UDP glycosyltransferase 1A3 (Ugt1a3), which could indicate that cell stress was induced. Additionally, several heat shock proteins were up-regulated such as Hspca, Hsph1, Hspa1b, and Hspb8 that could be potential danger signals to initiate an immune response. The pattern of decreased gene expression was also similar to the other aromatic amine drugs, as Dusp1 and Sgk were among the most down-regulated.
Aromatic amine gene expression profile
Although the gene changes were different for each aromatic amine drug, in general, aromatic amines induced greater down-regulated than up-regulated gene changes (). Among these, hepatic protein EIIH (Eiih; aka early insulin-induced hepatic gene) was the most highly up-regulated gene in all three drugs tested. Increased Eiih gene expression was confirmed through RT-PCR for both DDS and AMG treatment, but not SMX (). Further time-course investigation using RT-PCR found that Eiih was expressed only acutely, with the highest expression at 8 and 12 h after AMG treatment (). Other drugs known to cause IDRs such as nevirapine and clozapine also increased expression of Eiih at early time points in treated rats ().
Table 1. Similar genes differentially regulated at least 1.4-fold change in the same direction in all aromatic amine drugs tested 12 h after treatment.
Figure 4. Eiih expression induced by aromatic amine drugs 12 h after treatment. Among the treatment groups (solid bars), AMG induced a significant increase in Eiih expression as compared to control (open bars). DDS also induced an increase in Eiih; however, no change was observed for SMX. Rats were given 150 mg SMX/kg, 20 mg DDS/kg, or 80 mg AMG/kg, and liver samples were taken for RT-PCR analysis. Eiih expression was normalized to β2-microglobulin (β2M) as a housekeeping gene and a calibrator control. RT-PCR was run in triplicate (n = 4; *** p < 0.001).
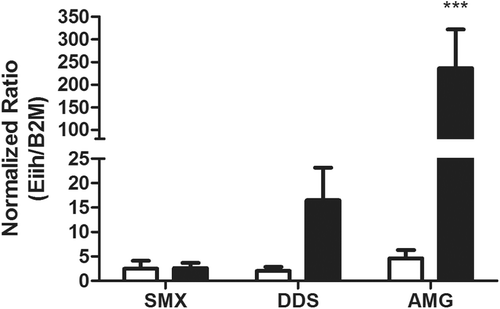
Figure 5. Time-course of hepatic Eiih expression induced by AMG. Eiih was expressed the greatest 8 and 12 h after AMG treatment (solid bars) as compared to control (open bars). Rats were treated with 80 mg AMG/kg, and liver samples were taken for RT-PCR analysis. Eiih expression was normalized to β2-microglobulin (β2M) as a housekeeping gene and a calibrator control. RT-PCR was run in triplicate (n = 2).
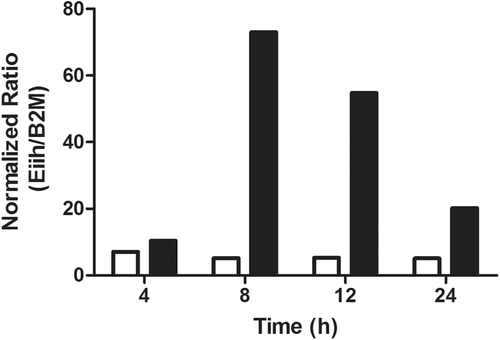
Figure 6. Hepatic gene expression of Eiih induced by several other drugs associated with IDRs. Clozapine (CLZ), an atypical anti-psychotic drug, induced an increase in Eiih 12 h after treatment of rats, and nevirapine (NVP), an anti-retroviral drug, increased Eiih expression at both 6 and 12 h after treatment of rats. Data expressed as fold-change between treated and controls (n = 4; * p < 0.05, ** p < 0.01).
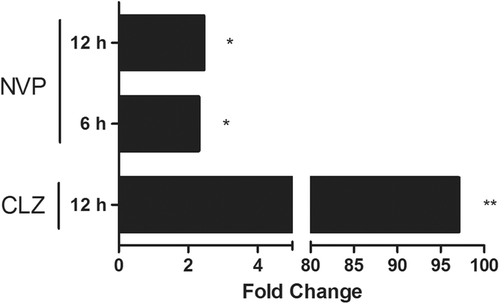
A greater number of similarities were also observed between DDS and AMG in terms of genes regulated by the Keap1-Nrf2-ARE pathway (). Further testing of downstream effects of these gene changes found that enzyme activity did not always correlate with changes in mRNA expression. Greater Txnrd activity was found after SMX treatment when Txnrd1 gene expression was low compared to that induced by DDS or AMG, whereas gene expression of Txnrd1 was higher with the other two drugs, when Txnrd activity was low (). Nevertheless, a time-course study with AMG found that Txnrd activity was only increased at 48 h after treatment (). GSH-S-Tr activity was also tested, but no significant changes were observed except for a possible increase in activity 48 h after AMG treatment ().
Table 2. Keap1-Nrf2-ARE-regulated genes that were changed ≥1.4 fold by at least one of the aromatic amine drugs (SMX, DDS, or AMG) tested 12 h after treatment.
Figure 7. Hepatic Txnrd activity after treatment with aromatic amine drugs. Thioredoxin reductase activity was increased at both 12 and 24 h after SMX treatment (a) but not with the other two aromatic amine drugs (n = 4; *** p < 0.001). However, a time-course study (b) found that AMG appeared to increase thioredoxin reductase activity 48 h after treatment (n = 2). Rats were treated with 150 mg SMX/kg, 20 mg DDS/kg, or 80 mg AMG/kg before liver samples were taken for activity assay. For timepoints > 24 h, rats were given an additional dose of drug every 24 h.
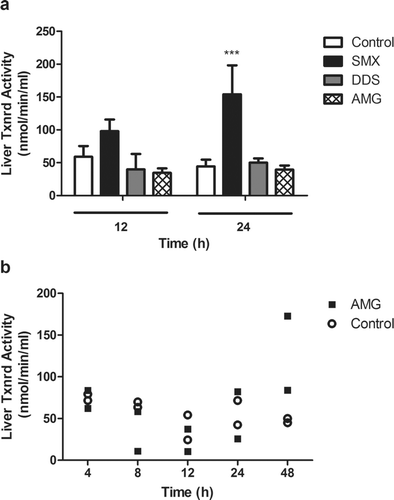
Figure 8. Time-course of hepatic GSH-S-Tr activity in AMG-treated rats. No significant change was observed in GST activity after treatment with 80 mg AMG/kg (solid squares) in the liver as compared to controls (open circles). For timepoints > 24 h, rats were given an additional dose of drug every 24 h (n = 2).
Discussion
If biomarkers that predict the risk that a drug candidate will cause an unacceptable risk of IDRs could be found it would have a profound effect on the process of drug development. The liver is a logical place to look because it is the site of most reactive metabolite formation, and it is the target of IDRs that are most likely to lead to drug withdrawal. Therefore, when drug candidates are screened for reactive metabolite formation, this is done almost exclusively in the liver or with hepatocytes. Accordingly, much of the screening for toxicities of drug candidates is focused on the liver.
Drugs that are primary aromatic amines represent a good starting point because this functional group is notorious for being associated with IDRs. Microarray technology represents the easiest way to examine a wide variety of possible biomarkers. The initial goal of these experiments was to verify whether the changes in hepatic gene expression induced by SMX were similar to our previous study performed in mice and whether there were changes that could be danger signals, biomarkers, or both. We chose here to focus on early time-points because subtle signs of cell stress, through changes in mRNA expression, would likely happen rapidly upon drug treatment and thus be good predictors. In comparison, effects on host immune responses would not be expected to occur—or to be detected—as early. Our findings indicate that, as in mice, SMX induced minimal hepatic gene changes in the rat. Further, of these changes, none met the strict criteria set here for denoting statistical significance.
It was surprising that there was a significant difference between the gene expression profile of SMX and DDS because they are structurally very similar with similar pharmacology, although DDS causes hemolytic anemia and methemoglobinemia and SMX does not (Uetrecht, Citation1989); this presumably reflects differences in the amount of redox cycling and distribution to red cells (Naisbitt et al., Citation1999). AMG is a first generation aromatase inhibitor used to treat estrogen-responsive breast and prostate cancer. AMG is structurally different from the other two drugs; in particular, there is no electron-withdrawing group on the aromatic ring to decrease the electron density of the aromatic amine, and this should change its redox potential. AMG was included to determine the extent of the involvement of the aromatic amine group in the induction of IDRs to determine whether the mere presence of the moiety is enough to induce similar changes. Interestingly, AMG induced the most significant number and magnitude of gene changes. This is not surprising because AMG is associated with a variety of adverse reactions in ≈ 50% of patients that take this drug, although only 1% of patients experience idiosyncratic hematologic toxicities including thrombocytopenia, leucopenia, pancytopenia, and agranulocytosis (Young et al. Citation1984). To some degree the similarities between DDS and AMG correlate with the fact that they are more prone to induce blood dyscrasias and bone marrow toxicity than SMX (Young et al., Citation1984; Coleman, Citation2001). Covalent adducts of SMX hydroxylamine have been found to localize on the surface of normal human epidermal keratinocytes in vitro, whereas covalent adducts of DDS hydroxylamine were found to localize intracellularly (Roychowdhury et al., Citation2005). These distinct covalent binding patterns may play a role in the observed differences and types of IDRs induced because intracellular adducts may have a greater ability to induce a strong immune response.
DDS and AMG induced some genes that could be considered danger signals. Our previous microarray studies on drugs including tienilic acid, carbamazepine, phenytoin, and D-penicillamine have all shown induction of cell stress through the up-regulation of genes involved with the Keap1-Nrf2-ARE pathway (Seguin et al., Citation2005; Pacitto et al., Citation2007; Lu et al., Citation2008), and this may potentially be a sign of danger to the immune system. The Keap1-Nrf2-ARE pathway is responsible for regulating the transcription of a variety of genes involved with detoxification and cytoprotection. The up-regulation of these genes suggests that cell stress occurred and there is an increased need to modulate potential damage that could eventually lead to an immune response.
In the present study, the same general pattern of increased expression of Keap1-Nrf2-ARE-regulated genes was observed, although mainly for DDS and AMG. Nevertheless, although the changes induced by SMX were not statistically significant, the same Keap1-Nrf2-ARE-regulated genes as DDS and AMG appeared to be slightly elevated. Txnrd is an enzyme analogous to glutathione reductase that functions to provide reducing equivalents to thioredoxin to act as an antioxidant, and it is crucial in maintaining cell redox status (Mustacich and Powis, Citation2000). Surprisingly, it was SMX that induced the greatest increase in Txnrd activity. Furthermore, there was a lag period between gene induction and Txnrd activity during AMG treatment. This implies that gene expression does not necessarily correlate with functional changes and that gene expression was likely induced before protein expression, which may explain the inconsistent relationship between gene and protein activity. This also raises an important issue as to whether gene changes are meaningful on their own. The activities of certain proteins are governed by post-translational modifications or by cytoplasmic ‘regulators′. In the canonical pathway, Nrf2 is kept inactivated by Keap1 in the cytoplasm, and only when Keap1 undergoes proteolytic cleavage is Nrf2 released to migrate into the nucleus where it then binds to the antioxidant response element and initiates transcription (Nguyen et al., Citation2009). Thus, changes in gene expression of Nrf2 may not be useful because activation is regulated post-translationally.
The acute cell response genes Sgk and Dusp1 were consistently the most down-regulated with all aromatic amine drugs tested. Sgk, a serine/threonine protein kinase involved in regulating cellular metabolism, proliferation, and differentiation, can be induced transiently by gluco-corticoids and serum (Webster et al., Citation1993). Dusp1, a nuclear phosphatase involved in regulating cellular signalling pathways, has been reported as inducible in early response to LPS, heat shock, and oxidative stress (Patterson et al., Citation2009). Down-regulation of these genes opposes what would be expected upon drug treatment; however, decreased Sgk expression upon tienilic acid treatment was attributed to the short half-life of Sgk mRNA (Webster et al., Citation1993; Pacitto et al., Citation2007). It is also plausible that a lack of signal could alert the cell to stress and initiate an immune response, although the exact mechanism is unclear.
Possibly the most interesting change was the increase in Eiih mRNA that was observed with all three drugs, although we failed to confirm increases in Eiih expression with RT-PCR for SMX. Additional testing of two other drugs associated with IDRs: clozapine and nevirapine, found a rapid increase in Eiih expression. These other drugs are not primary aromatic amines, and therefore it is possible that this is a more general biomarker of IDR risk. However, without testing a variety of drugs including negative controls that are not associated with a significant risk of IDRs it would be premature to conclude that this represents a useful biomarker of IDR risk. Unfortunately, little is known about the function of this gene. To date, there has only been one published study on Eiih that found it was induced early and acutely upon insulin treatment in rats with expression mainly localized to the liver, intestine, and islets of Langerhan, and there is some indication for its involvement in carbohydrate metabolism (Coffy et al., Citation2005). Its predicted protein arrangement suggests that it has the potential to be secreted or membrane bound, which implies that Eiih could possibly be a danger signal if secreted or act as a receptor-signalling complex on cellular membranes.
Although there were changes in gene expression induced by these primary aromatic amines, especially AMG, which could represent a danger signal, the most salient finding of this study is that primary aromatic amines do not appear to cause many changes in gene expression in the liver. Given that the primary aromatic amine is such a notorious structural alert and is readily oxidized to reactive metabolites in the liver and can also redox cycle, this is surprising. SMX and AMG frequently cause liver injury: the incidence of elevated γ-glutamyltransferase is greater than 60% in AMG-treated patients (Nagel et al., Citation1982). However, SMX-induced liver injury is usually part of a more generalized hypersensitivity IDR, and the more severe AMG-induced liver injury is more commonly cholestatic rather than hepatocellular. The most likely explanation for these results is that, although this functional group is readily oxidized to reactive metabolites in the liver, the liver is well equipped to deal with reactive metabolites. For example, in the case of an animal model of nevirapine-induced skin rash, there was more covalent binding in the liver than in the skin, but there were many more significant changes in gene expression in the skin (> 400, unpublished observation, manuscript in preparation). It is likely that what makes the aromatic amine functional group a structural alert is that it can also be oxidized by myeloperoxidase, which is present in antigen presenting cells. The formation of reactive metabolites by antigen presenting cells has been shown to lead to their activation (Elsheikh et al., Citation2010), and this is likely to be a strong stimulus for the induction of an immune response. Specifically, dendritic cells treated with SMX and nitroso metabolite were found to increase expression of the co-stimulatory molecule CD40 (Sanderson et al., Citation2007). Similarly, keratinocytes were found to up-regulate their expression of heat shock protein 70 (which could be a potential danger signal) upon treatment with the hydroxylamine metabolite of SMX (Khan et al., Citation2007).
Even in the case of immune-mediated liver injury, it appears that activation of the immune system outside of the liver is necessary in order to cause an immune response in the liver that results in injury (Bowen et al., Citation2005). It is notable that all three of these drugs are associated with a relatively high incidence of idiosyncratic agranulocytosis; this presumably involves oxidation of the drug to reactive metabolites by myeloperoxidase, the major oxidizing enzyme in neutrophils. This clinical picture is most consistent with activation of the immune system outside of the liver with the immune response sometimes extending to the liver. Therefore, the present focus on the liver by the pharmaceutical industry may miss signals that predict IDR risk, especially for drugs that also cause IDRs outside of the liver. In particular, bioactivation of easily-oxidized drugs, such as those containing aromatic amines, by myeloperoxidase in neutrophils and antigen-presenting cells, may play an important role in the induction of many types of IDRs.
Appendices
Appendix A. SMX-induced hepatic gene changes at 12 h (fold-change between treated and controls).
Appendix B. DDS-induced hepatic gene changes at 12 h (fold-change between treated and controls).
Appendix C. AMG-induced hepatic gene changes at 12 h (fold-change between treated and controls).
Acknowledgments
We thank Xiaochu Zhang for providing the nevirapine liver samples. This research was funded by the Canadian Institutes of Health Research. J.P.U. is the Canada Research Chair in Adverse Drug Reactions.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Anunciado-Koza, R. P., Zhang, J., Ukropec, J., Bajpeyi, S., Koza, R. A., Rogers, R. C., Cefalu, W. T., Mynatt, R. L., Kozak, L. P. 2011. Inactivation of the mitochondrial carrier SLC25A25 (ATP-Mg2+/Pi Transporter) reduces physical endurance and metabolic efficiency in mice. J. Biol. Chem. 286:11659–11671.
- Babic, A. M., Kireeva, M. L., Kolesnikova, T. V., Lau, L. F. 1998. CYR61 a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 95:6355–6360.
- Bowen, D. G., Mccaughan, G. W., Bertolino, P. 2005. Intrahepatic immunity: A tale of two sites? Trends Immunol. 26:512–517.
- Chan, H. L., Stern, R. S., Arndt, K. A., Langlois, J., Jick, S. S., Jick, H., Walker, A. M. 1990. The incidence of erythema multiforme, Stevens-Jonson syndrome, and toxic epidermal necrolysis. Arch. Dermatol. 126:43–47.
- Chugh, A., Ray, A., Gupta, J. B. 2003. Squalene epoxidase as hypocholesterolemic drug target revisited. Prog. Lipid Res. 42:37–50.
- Coffy, S., Decaux, J., Girard, J., Keyzer, Y., Asfari, M. 2005. Identificaiton of a novel rat hepatic gene induced early by insulin, independently of glucose. Biochem. J. 385:165–171.
- Coleman, M. D. 2001. Dapsone-mediated agranulocytosis: Risks, possible mechanisms and prevention. Toxicology 162:53–60.
- Elsheikh, A., Lavergne, S. N., Castrejon, J. L., Farrell, J., Wang, H., Sathish, J., Pichler, W. J., Park, B. K., Naisbitt, D. J. 2010. Drug antigenicity, immunogenicity, and co-stimulatory signaling: Evidence for formation of a functional antigen through immune cell metabolism. J. Immunol. 185:6448–6460.
- Farrell, J., Naisbitt, D. J., Drummond, N. S., Depta, J. P., Vilar, R. J., Pirmohomed, M., Park, B. K. 2003. Characterization of sulfamethoxazole and sulfamethoxazole metabolite-specific T-cell responses in animals and humans. J. Pharmacol. Exp. Ther. 306:229–237.
- Gallucci, S., Matzinger, P. 2001. Danger signals SOS to the immune system. Curr. Opin. Immunol. 13:113–119.
- Gill, H. J., Hough, S. J., Naisbitt, D. J., Maggs, J. L., Kitteringham, N. R., Pirmohamed, M., Park, B. K. 1997. The relationship between the disposition and immunogenicity of sulfamethoazole in the rat. J. Pharmacol. Exp. Ther. 282:795–801.
- Goodman, L. S., Gilman, A., Brunton, L. L., Lazo, J. D., Parker, K. L. (Eds.). 2006. Goodman and Gilman′s the Pharmacological Basis of Therapeutics. New York: McGraw-Hill.
- Grusch, M., Drucker, C., Petervorosmarty, B., Erlach, N., Lackner, A., Losert, A., Macheiner, D., Schneider, W., Hermann, M., Groome, N. 2006. De-regulation of the activin/folli-statin system in hepatocarcinogenesis. J. Hepatol. 45:673–680.
- Helton, D. R., Osborne, D. W., Pierson, S. K., Buonarati, M. H., Bethem, R. A. 2000. Pharmacokinetic profiles in rats after intravenous, oral, or dermal administration of dapsone. Drug Metab. Dispos. 28:925–929.
- Khan, F. D., Vyas, P. M., Gaspari, A. A., Svensson, C. K. 2007. Effect of arylhydroxylamine metbaolites of sulfamethoxazole and dapsone on stress signal expression in human keratinocytes. J. Pharmacol. Exp. Ther. 323:771–777.
- Lada, A. T., Davis, M., Kent, C., Chapman, J., Tomoda, H., Omura, S., Rudel, L. L. 2003. Identification of ACAT1- and ACAT2-specific inhibitors using a novel, cell-based fluorescence assay: Individual ACAT uniqueness. J. Lipid Res. 45:378–386.
- Leppa, S., Bohmann, D. 1999. Diverse function of JNK singaling and c-Jun in stress response and apoptosis. Oncogene 18:6158–6162.
- Li, J., Uetrecht, J. 2010. The Danger Hypothesis applied to idiosyncratic drug reactions. In: Adverse Drug Reactions (Uetrecht, J., Ed.).Berlin: Springer-Verlag, pp. 473–509.
- Lonning, P. E., Schanche, J. S., Kvinnsland, S., Ueland, P. M. 1985. Single-dose and steady-state pharmacokinetics of aminoglutethimide. Clin. Pharmacokinet. 10:353–364.
- Lu, W., Li, X., Uetrecht, J. P. 2008. Changes in gene expression induced by carbamazepine and phenytoin: Testing the Danger Hypothesis. J. Immunotoxicol. 5:107–113.
- Machado, F. S., Johndrow, J. E., Esper, L., Dias, A., Bafica, A., Serhan, C. N., Aliberti, J. 2006. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nature Med. 12:330–334.
- Matzinger, P. 1994. Tolerance, danger, and the extended family. Ann. Rev. Immunol. 12:991–1045.
- Mockenhaupt, M. 2011. The current understanding of Stevens-Johnson syndrome and toxic epidermal necrolysis. Expert Rev. Clin. Immunol. 7:803–815.
- Mustacich, D., Powis, G. 2000. Thioredoxin reductase. Biochem. J. 346:1–8.
- Nagel, G. A., Wander, H. E., Blossey, H. C. 1982. Phase II study of aminoglutethimide and medroxyprogesterone acetate in the treatment of patients with advanced breast cancer. Cancer Res. (Suppl.) 42:3442s–3444s.
- Naisbitt, D. J., Hough, S. J., Gill, H. J., Pirmohamed, M., Kitteringham, N. R., Park, B. K. 1999. Cellular disposition of sulphamethoxazole and its metabolites: Implications for hyper-sensitivity. Br. J. Pharmacol. 126:1393–1407.
- Nguyen, T., Nioi, P., Pickett, C. B. 2009. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 284:13291–13295.
- Nicholls, P. J., Dalrymple, P. D., Eweiss, N., Douglas, J. S. 1984. Aminoglutethimide: Absorption, physiological disposition and pharmacokinetics. In: Aminoglutethimide as an Aromatase Inhibitor in the Treatment of Cancer: An International Symposium Held During the 13th International Congress of Chemotherapy. Vienna, Austria (Nagel, G. A.Santen, R. J., Eds.). Berne, Switzerland:Hans Huber, pp. 58–67.
- Pacitto, S. R., Uetrecht, J. P., Boutros, P. C., Popovic, M. 2007. Changes in gene expression induced by tienilic acid and sulfamethoxazole: Testing the Danger Hypothesis. J. Immunotoxicol. 4:253–266.
- Patterson, K. I., Brummer, T., O′brien, P. M., Daly, R. J. 2009. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 418:475–489.
- Roychowdhury, S., Vyas, P. M., Reilly, T. P., Gaspari, A. A., Svensson, C. K. 2005. Characterization of the formation and localization of sulfamethoxazole and dapsone-associated drug-protein adducts in human epidermal keratinocytes. J. Pharmacol. Exp. Ther. 314:43–52.
- Sanderson, J. P., Naisbitt, D. J., Farrell, J., Ashby, C. A., Tucker, M. J., Rieder, M. J., Pirmohamed, M., Clarke, S. E., Park, B. K. 2007. Sulfamethoxazole and its metabolite nitroso sulfamethoxazole stimulate dendritic cell costimulatory signaling. J. Immunol. 178:5533–5542.
- Schwartz, D. E., Rieder, J. 1970. Pharmacokinetics of sulfamethoxazole and trimethoprim in man and their distribution in the rat. Chemotherapy 15:337–355.
- Seguin, B., Boutros, P. C., Li, X., Okey, A. B., Uetrecht, J. P. 2005. Gene expression profiling in a model of D-penicillamine-induced autoimmunity in the Brown Norway rat: Predictive value of early signs of danger. Chem. Res. Toxicol. 18:1193–1202.
- Uetrecht, J. 1989. Dapsone and sulfapyridine. In: Clinics in Dermatology (Shear, N. H., Ed.).Philadelphia, PA: Lippincott, pp. 111–120.
- Uetrecht, J. 2002. N-Oxidation of drugs associated with idiosyncratic drug reactions. Drug Metab. Rev. 34:651–665.
- Uetrecht, J. 2009. Immune-mediated adverse drug reactions. Chem. Res. Toxciol. 22:24–34.
- Webster, M. K., Goya, L., Firestone, G. L. 1993. Immediate-early transcriptional regulation and rapid mRNA turnover of a putative serine/threonine protein kinase. J. Biol. Chem. 268:11482–11485.
- Welch, C., Santra, M. K., El-Assaad, W., Zhu, X., Huber, W. E., Keys, R. A., Teodoro, J. G., Green, M. R. 2009. Identification of a protein, G0S2, that lacks Bcl-2 homology domains and interacts with and antagonizes Bcl-2. Cancer Res. 69:6782–6789.
- Xu, H., Bai, L., Collins, J. F., Ghishan, F. K. 1999. Molecular cloning, functional characterization, tissue distribution, and chromosomal localization of a human, small intestinal sodium-phosphate (Na+-Pi) transporter (SLC34A2). Genomics 62:281–284.
- Young, J. A., Newcomer, L. N., Keller, A. M. 1984. Aminoglutethimide-induced bone marrow injury: Report of a case and review of the literature. Cancer 54:1731–1733.
- Zandbergen, F., Mandard, S., Escher, P., Tan, N. S., Nguan, S., Patsouris, D., Jatkoe, T., Rojas-Caro, S., Madore, S., Wahli, W., Tafuri, S., Müller, M., Kersten, S. 2005. The G0/G1 switch gene 2 is a novel PPAR target gene. Biochem. J. 392:313–324.