
Abstract
Observational and clinical studies have associated increased cancer risks with primary or acquired immunodeficiencies, autoimmunity, and use of immunotherapies to treat chronic inflammation (e.g. autoimmunity) or support organ engraftment. Understanding of the relationship between immune status and cancer risk is generally grounded in two juxtaposing paradigms: that the immune system protects the host via surveillance of tumors and oncogenic viruses (e.g. immunosurveillance model) and that chronic inflammation can augment tumor growth and metastasis (inflammation model). Whereas these models support a role of immune status in many cancers, they are insufficient to explain the disproportionate increase in B-cell lymphoma risk observed across patient populations with either chronic immunosuppression or inflammation. Evaluation for the presence of Epstein-Barr virus (EBV) in lymphomas obtained from various populations demonstrates a variable role for the virus in lymphomagenesis across patient populations. An evaluation of the DNA alterations found in lymphomas and an understanding of B-cell ontogeny help to provide insight into the unique susceptibility of lymphocytes, primarily B-cells, to oncogenic transformation. EBV-independent B-cell oncogenic transformation is driven by chronic antigenic stimulation due to either inflammation (as seen in patients with autoimmune disease or a tissue allograft) or to unresolved infection (as seen in immunosuppressed patients), and the transformation arises as a result of DNA damage from genomic recombination and mutation during class switching and somatic hypermutation. This model explains the increased background rate of lymphoma in some patients with autoimmunity, and highlights the challenge of resolving the confounding that occurs between disease severity and use of targeted immunotherapies to treat chronic inflammation. The ability to distinguish between disease- and treatment-related risk of lymphoma and an appreciation of the etiology of B-cell transformation is central to an improved risk assessment by scientists, clinicians and regulators, including the approval, labeling, and chronic use of immunotherapies.
Epidemiology of immunomodulation and lymphoma
It is perhaps axiomatic to state that immunosuppression is associated with an increased risk of cancer. As reviewed below, this proposition is founded on a wealth of clinical and experimental evidence, including long-term surveillance of patients chronically treated with immunosuppressive therapies and humans with congenital or acquired immunodeficiencies. To understand the relationship between immune status and lymphoma risk, we review the epidemiology of cancer in patients with primary immunodeficiencies, Human Immunodeficiency Virus (HIV) infection/Acquired Immunodeficiency Syndrome (AIDS), organ transplantation, and autoimmunity.
Lymphoma in patients with primary immunodeficiencies
More than 200 distinct primary immunodeficiencies have been identified as arising from congenital defects in functional immunity (Marodi & Notarangelo, 2007) and are classified by an underlying functional or molecular mechanism (Notarangelo et al., Citation2009). The risk of a primary immunodeficiency is estimated to occur in 1 in 10 000 births (Salavoura et al., Citation2008), and most of these diseases are classified as rare, very rare, or extremely rare (Notarangelo et al., Citation2009). Cancer has been identified as the second leading cause of mortality in these patients (Mueller & Pizzo, 1995), and the presence of certain immunodeficiencies rank as among some of the highest reported risk factors for tumor development in humans (Filipovich et al., Citation1992). These cancers commonly develop in pediatric patients, and thus reflect a nearly 10 000-times higher risk of cancer, as compared to pediatric populations without these immune disorders (Ioachim, Citation1990; Mueller & Pizzo, 1995; Salavoura et al., Citation2008).
The type of malignancy in patients with primary immunodeficiencies is highly dependent on the nature of the defect, although lymphomas are among the most common cancer types arising in these patients (see Table 1). Among 500 cancer cases reported to the Immunodeficiency Cancer Registry, non-Hodgkin’s lymphomas (predominantly B-cell) accounted for one-half of all tumors across a mechanistically diverse range of immunodeficiencies, and 75% of tumors in patients with Severe Combined Immunodeficiency (SCID) and Wiskott-Aldrich syndrome (Filipovich et al., Citation1994). The development of cancer in patients with primary immunodeficiencies is commonly characterized by an unusually early age of onset and, in the case of lymphoma, an atypical distribution. For example, patients with either SCID or hypo-gammaglobulinemia are diagnosed with NHL at a median age of 0.5–1.6 and 1.2–4.0 years, respectively, whereas patients with IgA deficiency, hyper-IgM Syndrome, Wiskott-Aldrich Syndrome, and ataxia telangiectasia are all diagnosed at a median age of 10 years or less (Filipovich et al., Citation1994; Frizzera et al., Citation1980; Kersey et al., Citation1973).
Among patients with X-linked lymphoproliferative disease (Duncan’s Disease), 50% experience fatal infectious mononucleosis, 30% experience malignant lymphoma, and 30% experience dysgammaglobulinemia, all due to a unique sensitivity towards Epstein-Barr Virus (EBV) attributable to impaired activation-induced apoptosis of T- and B-cells (Nagy & Klein, 2010). The original case report of this disease documented the death of six boys in the Duncan family over a 13-year period from infectious mononucleosis, agammaglobulinemia, or malignant lymphoma; all of these boys had been infected with EBV (Purtilo et al., Citation1975, Citation1981).
Chronic inflammation towards foreign antigens or a breakdown in self-tolerance towards autoantigens in patients with various primary immunodeficiencies not only underlies certain cancers, but autoimmunity, which has been identified in patients with autoimmune lymphoproliferative syndrome (ALPS), autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), hyper-IgM syndrome, complement deficiencies, MHC I deficiency, Omenn syndrome, Wiscott-Aldrich syndrome, and other primary immunodeficiencies (Arkwright et al., Citation2002). In particular, autoimmune disease is observed universally amongst patients with the T-cell deficiency disorders IPEX (immunodysregulation, polyendocrinopathy, enteropathy X-linked syndrome), APECED, and Omenn syndrome as a result of a deficiency in peripheral tolerance towards autoantigens mediated by regulatory T-cells (Arason et al., Citation2010). Recognition of this relationship between primary immunodeficiencies and autoimmunity is an important consideration as a driver of lymphoma risk, as described later in this review.
Lymphoma and post-transplant lymphoproliferative disorders in patients following organ transplantation/tissue engraftment
Organ transplant recipients treated with immunosuppressive agents demonstrate a disproportionally high incidence of various cancers relative to the general population (Grulich et al., Citation2007; Kasiske et al., Citation2004; Penn, Citation1999, Citation2000), including non-Hodgkin’s lymphoma (8–20-fold increase risk) and Hodgkin’s lymphoma (4-fold increased risk). Immunosuppression to maintain organ or tissue engraftment is associated with post-transplant lymphoproliferative disorders (PTLD), which encompass a heterogeneous range of conditions from reactive, polyclonal hyperplasias to aggressive NHL. Most PTLD are of B-cell origin, frequently arise in extranodal sites (including the engrafted organ), display a range of clinical aggressiveness, and can often be reversed by reduction or termination of immunosuppression (Hopwood & Crawford, 2000; Thorley-Lawson & Gross, 2004).
PTLD arising early after organ engraftment/immunosuppression are typically EBV+ and frequently polyclonal or oligoclonal. In contrast, PTLD arising later (>1 year) are commonly EBV−, and often true monoclonal lymphoid malignancies. As discussed later in this review, these findings suggest that, whereas impaired anti-viral immunity contributes to early lymphoma development in organ transplant recipients, lymphomas arising later in these patients may also be driven by other factors, including chronic antigenic stimulation from the allograft.
The predominant categories of PTLD include plasmacytic hyperplasia, polymorphic PTLD, and monomorphic B-cell lymphoma (including diffuse large B-cell lymphoma and Burkitt/Burkitt-like lymphoma) (Capello et al., Citation2003). Risk factors for developing PTLD include the intensity and duration of the immunosuppression, degree of HLA mismatch (mismatch increases risk), EBV-status at the time of transplant (EBV-negative at transplant increases risk), and, in the case of hematopoietic stem cell transplant, whether the donor cells are depleted of T-cells (depletion increases risk) or B-cells (depletion decreases risk) (Gottschalk et al., Citation2005).
Lymphoma in patients with HIV/AIDS
AIDS is a pandemic disease caused by HIV infection, which results in progressive destruction of CD4+ T-cells. AIDS patients are highly susceptible to opportunistic infection by bacteria, viruses, fungi, and parasites, and an increased risk of cancer. Epidemiology studies have demonstrated an increased incidence/risk across cancer types as patients progress from HIV disease-free to HIV+ to AIDS (Engels et al., Citation2006, Citation2008). With some notable exceptions, the relative risk of cancer in patients with HIV/AIDS is broadly similar across cancer types to those found in post-transplant patients, as demonstrated by a meta-analysis of seven studies involving ∼440 000 HIV/AIDS patients (Grulich et al., Citation2007). Increased relative risks among AIDS patients compared to the US population have been identified for a number of cancers, including CNS lymphoma (3–100-fold), NHL (40–76-fold), and Hodgkin’s lymphoma (∼11-fold) (Boshoff & Weiss, 2002; Engels et al., Citation2008; Grulich et al., Citation2007; Howlader et al., Citation2011; Schabet, Citation1999).
Both chronic immune system activation and immune exhaustion are characteristic of HIV disease, and the depletion of CD4+ T-cells results in the loss of effector T-cells as well as (more variably) regulatory T-cells (TReg) (Arguello et al., Citation2012; Holmes et al., Citation2008; Sachdeva et al., Citation2010). Whereas the loss of effector T-cells may impair tumor and viral immunosurveillance (Vesely et al., Citation2011), the loss of TReg in HIV patients may increase autoimmunity (Louthrenoo, Citation2008; Walker et al., Citation2008) and underlie reported B-cell dysfunction including hyper-gamma-globulinemias and polyclonal B-cell hyperactivity (Moir & Fauci, 2008). Whether these B-cell disorders may contribute to the observed increase in lymphoma risk in HIV patients remains uncertain, although this is a plausible explanation for the substantial fraction of EBV-negative lymphomas observed in HIV patients, as described below.
Lymphoma in patients with autoimmunity
A number of studies have identified increased cancer risk, particularly lymphoma, in patients with rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), Sjögren syndrome, and other autoimmune conditions (see and ). For example, a recent large case-control study involving over 44 000 lymphoid malignancy cases and 122 500 controls generally identified up to a doubled risk (defined as a statistically increased odds ratio (OR)) of NHL (variously including diffuse large B-cell lymphoma, T-cell NHL, or follicular lymphoma) for patients with RA, Sjögren syndrome, SLE, sarcoidosis, or systemic sclerosis (see , Anderson et al., Citation2009). In contrast to other autoimmune diseases, the risk of lymphoma seems reduced in patients with multiple sclerosis in this study. Among NHL cancer sub-types, diffuse large B-cell lymphoma is commonly observed among patients with autoimmunity (see Table 2, Baecklund et al., 2006a; Keystone, Citation2011).
Table 1. Lymphoma risk in individuals with primary immunodeficiencies.
Table 2. Relative risk (as odds ratios with 95% confidence intervals) of non-Hodgkin’s lymphoma cancer (and sub-types) in individuals with autoimmunity compared to the general population.
Table 3. Relative risk (as standardized incidence ratios (95% confidence intervals)) of malignancies in patients with rheumatoid arthritis.
Estimating the relative risk of cancer in autoimmune patients is challenged by poor precision because of the low number of cases of cancer arising in the relatively rare population of patients with autoimmunity (see Kaiser, Citation2008). Recently, several meta-analyses have been conducted to circumvent these technical limitations in individual reports in an effort to ascertain risks. Kaiser (Citation2008) conducted a meta-analysis of studies reporting associations between RA and lymphoma risk and confirmed a 2–3-fold increased risk of lymphoma in patients with RA, with evidence of a statistical association in 20 of the 26 evaluated studies (Kaiser, Citation2008). These associations are consistent with a meta-analysis of 14 publications by Smitten et al. (Citation2008) that estimated a doubled risk of lymphoma in RA patients (standardized incidence ratio (SIR): 2.08, 95% confidence interval (CI): 1.80–2.39), with a higher risk of Hodgkin’s lymphoma (SIR: 3.29, 95% CI: 2.56–4.22) than non-Hodgkin’s lymphoma (SIR: 1.95, 95% CI: 1.70–2.24).
Lymphoma risk appears to increase with autoimmune disease severity (Llorca et al., Citation2007). Among patients with RA, patients classified as having high inflammatory scores have been reported to have 25.8- (95% CI: 3.1–213.0: Baecklund et al., Citation2004) and 71.3-fold (95% CI: 24.1–211.4: Baecklund et al., 2006b) greater risk of lymphoma compared to patients with low inflammatory score. Additionally, there may be a generally greater risk of lymphoma in patients with Sjögren syndrome compared to patients with RA (). For example, the risk of NHL was found to be highest among patients with primary Sjögren syndrome (8.7 [95% CI = 4.3–15.5, n = 11]) compared to secondary Sjögren syndrome (4.5 [95% CI = 1.5–10.6, n = 5]) and RA (2.2 [95% CI = 1.5–3.1, n = 34]), possibly reflecting differential tissue/organ involvement with these autoimmune diseases (Kauppi et al., Citation1997). These results are supported by findings from a meta-analysis of cancer outcomes among patients with systemic lupus erythematosus (SLE, six studies), primary Sjögren syndrome (five studies), and RA (nine studies) by Zintzaras et al. (2005) that found that patients with primary Sjögren syndrome had the highest relative NHL risk (18.8 as standardized incidence ratio [95% CI: 9.5–37.3]) compared to SLE (7.4 [95% CI: 3.3–17.0]) and RA (3.9 [95% CI: 2.5–5.9]).
Because immunosuppressive therapies, including methotrexate, azathioprine, and tumor necrosis factor (TNF)-α inhibitors are used in patients with autoimmunity, experimental and epidemiological studies must address competing effects on cancer of the underlying disease and the immunomodulatory therapy, which can independently contribute to cancer risk. However, patients with greater disease severity are more likely to receive increasingly potent therapies, thus, it may not be possible to disaggregate effects of disease from effects of immunotherapy in contributing to cancer risk, and most available studies do not address this confounding factor (Kaiser, Citation2008). For example, in a meta-analysis of nine randomized controlled trials, a relative risk of 3.4 (95% CI = 1.4–8.2) was reported for RA patients treated with high dose vs low dose anti-TNF antibody therapy, which may reflect a combination of either effect of therapy, underlying disease state, or both (Bongartz et al., Citation2006). However, an increased risk of lymphoma was not confirmed in other meta-analyses of RA patients treated with anti-TNF therapies compared to either placebo or other therapies (see and Keystone, Citation2011).
Table 4. EBV differentially contributes to lymphoma burden across patient populations.
Concern about the cancer risk associated with chronic use of TNFα inhibitors in patients with autoimmunity, which initially arose from a case series of lymphoproliferative disorders in rheumatic patients (Brown et al., Citation2002), resulted in an FDA Arthritis Drugs Advisory Committee meeting, which confirmed an increased rate of lymphoma in TNFα-treated patients over the background rate in the general population; however, they were unable to assess the incremental risk relative to untreated RA patients (US FDA, Citation2003). Controversy and debate on this issue has continued with subsequent studies, including a number of meta-analyses, that either support or refute an incremental increase in lymphoma risk in autoimmune patients treated with immuno-suppressive therapies over untreated patients (Baecklund et al., 2006b; Bongartz et al., Citation2006; Kaiser, Citation2008; Keystone, Citation2011; Lopez-Olivo et al., Citation2012; Mariette et al., Citation2002). Overall, these data suggest that, if anti-rheumatic therapies contribute to an increased lymphoma incidence, the magnitude of the increase appears to be within the margin of uncertainty associated with the background risk of lymphoma from autoimmunity.
Epstein-Barr Virus and lymphoma risk in patients with altered immune status
EBV (also called human herpesvirus 4, HHV-4) is a ubiquitous double stranded DNA γ-herpesvirus that is thought to have infected as much as 95% of the world population (CDC, Citation2011). EBV infection has strong associations with B-cell malignancies such as Burkitt’s lymphoma, Hodgkin’s lymphoma, nasopharyngeal carcinoma, and PTLD (Thorley-Lawson & Gross, 2004). EBV preferentially infects B-cells through binding the major EBV glycoprotein gp350 to the CD21 receptor on the surface of B-cells, typically resulting in asymptomatic disease, but occasionally causing infectious mononucleosis. EBV infection of naïve B-cells in vitro results in their transformation through the expression of six EBV nuclear antigens (EBNAs 1, 2, 3A, 3B, 3C, and –LP) and three latent membrane proteins (LMPs 1, 2A, and 2B); of these latent proteins, EBNA2 and LMP1 are absolutely required for B-cell transformation (Kuppers, Citation2003; Thorley-Lawson & Gross, 2004; Young & Rickinson, 2004).
Various patterns of viral gene usage have been identified for latent B-cell infection according to the B-cell differentiation and activation state (Thorley-Lawson, Citation2001; Thorley-Lawson & Gross, 2004). Newly-infected cells, including B-cells transformed in vitro, express all EBV nuclear antigens and latent membrane proteins (called the ‘growth program’ or latency III). These cells may subsequently undergo a germinal center reaction, with a more restricted EBV gene expression (latency II, expressing EBNA1, LMP1, LMP2A). During the germinal center reaction, clonal B-cell expansion increases the size of the B-cell pool infected with EBV, from which a sub-set will differentiate into resting memory B-cells, with minimal or no viral gene expression (perhaps LMP2A, latency 0); the low viral gene expression in memory B-cells creates a niche permissive for viral survival by effectively hiding the virus from host immunity (Kuppers, Citation2003; Thorley-Lawson & Gross, 2004). Different EBV+ malignancies are associated with unique expression of viral latency programs. Specifically, Burkitt’s lymphoma expresses EBNA1 (latency I), Hodgkin lymphoma generally demonstrates latency II gene expression, and PTLD and HIV-related lymphomas demonstrate latency III gene expression (Kuppers, Citation2003; Gottschalk et al., Citation2005).
Expression of latent EBV proteins LMP1 and LMP2A mimics normal B-cell signaling pathways that drive germinal center and memory B-cell development. LMP-1 mimics B-cell activation processes normally driven by ligand-mediated CD40-CD40L interactions, and activates growth and survival pathways that confer phenotypic transformation (Kilger et al., Citation1998; Soni et al., Citation2007). Although LMP2A is not involved in cell transformation, its expression allows B-cells to bypass normal differentiation checkpoints and to colonize peripheral lymphoid organs in the absence of signaling through the B-cell receptor (Caldwell et al., Citation1998).
Natural killer (NK) cells and T-cells are the main innate and adaptive immune cells that control EBV infection, respectively. During acute EBV infection (infectious mononucleosis), 1–10% of peripheral B-cells may be infected with EBV, and 1–40% of the total CD8+ T-cell population may have specificity towards EBV epitopes. During latent infection, EBV-infected B-cells are rarely found in the periphery (1–50/million B-cells), whereas up to 3% of memory CD8+ T-cells demonstrate specificity towards EBV antigens. Among long-term asymptomatic EBV carriers, particularly dominant anti-viral specificities have been identified for both CD8+ T-cells [towards immediate early proteins (i.e. BZLF1, BRLF1), lytic proteins (i.e. BMRF1, BMLF1), and latent nuclear antigens (i.e. EBNA3A, 3B, 3C)] and CD4+ T-cells [towards latent nuclear antigens (i.e. EBNA1, EBNA2, EBNAC3)]. A concomitant increase in both EBV replication and EBV+ cells in blood can be found with loss of CD4+ and/or CD8+ T-cells during immuno-suppression, demonstrating that lymphoproliferative diseases associated with latent EBV infection likely arise from impaired surveillance by EBV latent antigen-specific CD4+ and CD8+ T-cells (Hislop et al., Citation2007; Hopwood & Crawford, 2000).
Although various lymphomas are associated with EBV, EBV differentially contributes to disease burden across patient populations (). Among patients that develop lymphoma within the first year post-transplantation, virus can be detected in > 90% of cases (Macsween & Crawford, 2003), and EBV is universally associated with CNS lymphomas in patients with HIV (Boshoff & Weiss, 2002; Macsween & Crawford, 2003; Rabkin, Citation2001). However, EBV is associated with only ∼50% of lymphomas arising after 1 year post-transplantation (Young & Rickinson, 2004), and 28–66% of non-CNS lymphomas among HIV patients (Balandraud et al., Citation2005; Rabkin, Citation2001; Macsween & Crawford, 2003). Among patients with primary immunodeficiencies, EBV RNA was present in biopsy samples (detected using in situ hybridization) in some patients with Wiskott-Aldrich Syndrome (3 of 5) and SCID (1 of 3), but not in patients with other primary immunodeficiencies (n = 5; Filipovich et al., Citation1994), patients with antibody deficiencies (n = 10; Gompels et al., Citation2003), or patients with common variable immunodeficiency (CVID) presenting with mucosal-associated lymphomas (Cunningham-Rundles et al., Citation2002). As discussed above, patients with X-linked lymphoproliferative disorder appear to have a unique sensitivity to EBV based on a genetic defect in functional SAP protein, which normally regulates activation-induced lymphocyte apoptosis (Nagy & Klein, 2010).
A relatively low EBV+ frequency has been observed in lymphoma samples from RA patients, with rates ranging from those found in the general population (2–10%) to ∼25% (). Given that six of eight reported studies were published post-2000, this suggests that use of highly active disease-modifying drugs in RA patients does not substantially increase the EBV+ rate in their lymphoma, as is seen in post-transplant patients. However, to our knowledge no studies have evaluated the rate of EBV-infection among lymphoma samples derived from autoimmune patients stratified by their clinical history of drug use; such information could meaningfully inform whether use of anti-TNF therapies or other drugs sufficiently modify host immune status towards EBV and drive lymphoma risk.
Taken as a whole, although EBV may have a causal association with lymphomas in many immunosuppressed patient populations, other factors can contribute to disease development, and a viral etiology for these lymphomas cannot always be presumed.
B-cell susceptibility to transformation with immunomodulation
The association of cancer with both immunosuppression and inflammation illustrates the complex role the immune system plays in preventing or causing cancer. In particular, B-cells appear to have a unique susceptibility to transformation with immunomodulation. Chronic antigenic stimulation and inflammation have been proposed to underlie lymphomas and other lymphoproliferative disorders in response to a variety of stimuli, including unresolved infection, autoimmunity, and chronic inflammation associated with the host response to tissue/organ engraftment (see Aljurf et al., Citation2003; Suarez et al., Citation2006; Schuetz et al., Citation2010). These lymphoproliferative disorders are hypothesized to arise from transformation of chronically-activated lymphocytes that progressively transition from a non-malignant, reversible, polyclonal response to a malignant, irreversible, clonal disease. What drives the susceptibility of B-cells to transformation with chronic antigenic stimulation?
DNA hypermutation and recombination are uniquely necessary for normal B-cell development
Mature B-cells are uniquely susceptible to DNA damage arising from their reliance on DNA hypermutation and recombination during their differentiation. Upon antigenic stimulation, mature B-cells undergo extensive proliferation, somatic hypermutation (SHM), and class switch recombination (CSR) in germinal centers (specialized microenvironments in secondary lymphoid tissues; Shlomchik & Weisel, 2012). BCL-6 is the central transcriptional repressor controlling the germinal center reaction that expands the pool of antigen-specific B-cells, increases antibody affinity, and alters antibody effector activity; BCL-6 and Pax5 down-regulation are necessary for B-cell exit from the germinal center reaction and for differentiation of post-germinal center B-cell into long-lived plasma cells and memory cells (Basso & Dalla-Favera, 2010; Klein & Dalla-Favera, 2008). Somatic hypermutation increases antibody affinity through a process of random DNA point mutations in the variable region of the antibody gene (Peled et al., Citation2008). Class switch recombination alters the effector function of an antibody and involves DNA recombination in the Ig heavy chain (CH) to replace one constant region with another (Stavnezer et al., Citation2008).
Somatic hypermutation and CSR are both mediated by activation-induced cytidine deaminase (AID), which deaminates cytosine to produce uracil:guanosine mismatches in single stranded DNA. AID has some sequence specificity, and preferentially deaminates cytosine in W(A/G)R(A/T)C motifs (Frieder et al., Citation2006; Pham et al., Citation2003; Peled et al., Citation2008). Repair of AID-mediated cytosine deamination occurs through mismatch repair and short-patch (single base pair) or long-patch (≥2–8 base pairs) base excision repair processes. In base excision repair, the uracil DNA glycosylase UNG creates an abasic site (Imai et al., Citation2003; Rada et al., Citation2002; Schrader et al., Citation2005), and single-strand breaks are created by apurinic/apyrimidinic endonucleases (APE) 1 and 2 (Guikema et al., Citation2007); double-strand breaks can occur when two opposite strand lesions occur in proximity. CSR uses end-joining recombination of double strand breaks that occur in or near G-rich tandem repeated DNA sequences (i.e. switch regions) located 5’ of each CH gene (except for Cδ) to eliminate the intervening DNA and alter the effector activity of the antibody (Frieder et al., Citation2006; Stavnezer et al., Citation2008).
During SHM, C:T transition mutations can arise directly from replication errors following AID-mediated cytosine deamination. However, the observation that ∼60% of DNA mutations involve A:T bases suggests mismatch and base excision repair errors and use of low-fidelity, error-prone DNA polymerases (e.g. pol ζ, pol θ, and pol ι) incorporate single and multiple base pair mutations and diversify the complementarity-determining region of the variable chain (reviewed in Xu et al., Citation2005; Poltoratsky et al., Citation2007; Peled et al., Citation2008). Successive rounds of SHM, clonal expansion, and competition of germinal center B-cells for survival based on affinity (positive selection) drives the evolution of B-cells with increasing affinity for antigen during the germinal center reaction (Basso & Dalla-Favera, 2010; Chan & Brink, 2012; Shlomchik & Weisel, 2012). Germinal center B-cells survive and proliferate in the presence of DNA damage through BCL6-mediated transcriptional suppression of ATR, which is a critical sensor of DNA damage and mediator of DNA damage responses (Ranuncolo et al., Citation2007) and suppression of apoptotic processes that respond to DNA damage mediated by p53 (Phan & Dalla-Favera, Citation2004) and p21 (Phan et al., Citation2005). Although the co-ordinated processes taking place in the germinal center B-cell regulate both the random and specific DNA alterations necessary to yield high affinity antibody with specific effector activity, they also increase the likelihood of inappropriate mutation or recombination that underlie various B-cell lymphomas.
B-cell lymphomas occur at different steps of B-cell development and represent their malignant counterpart
With few exceptions, B-cell non-Hodgkin lymphomas (B-NHL) demonstrate immunoglobulin (Ig) variable chain (V) SHM, indicating that they derive from cells that have undergone a germinal center reaction (Stevenson et al., Citation1998). Post-germinal B-cell lymphomas commonly share distinctive phenotypes of the normal, healthy B-cells at different stages of their differentiation, presumably reflecting their malignant counterpart (Shaffer et al., 2002). The two hallmark genetic lesions found in B-NHL are aberrant SHM and balanced translocations/homologous recombinations that reflect errors during SHM and CSR (Kuppers & Dalla-Favera, 2001; Klein & Dalla-Favera, 2008). During SHM, AID preferentially targets IgV regions with a mutation rate of 10−5–10−3/basepair/generation, compared to a basal genomic mutation rate of ∼10−9 (Peled et al., Citation2008). However, AID also produces mutations in non-IgV loci including B-cell receptor genes (Gordon et al., Citation2003) and the B-cell cancer-associated genes BCL-6 (Pasqualucci et al., Citation1998; Shen et al., Citation1998), fas (Muschen et al., Citation2000), Pim- 1, Pax5, MYC, and Rhoh (Pasqualucci et al., Citation2001). Up to 25% of genes expressed in germinal center B-cells may be mutated by AID, although the mutation rate is 10–100-fold lower than that found in the IgV region (Liu et al., Citation2008).
In addition to the SHM errors described above, reciprocal chromosomal translocations are found in many B-cell lymphomas, characteristically involving the Ig CH switch regions or IgV regions involved in SHM. These translocations lead to inappropriate expression of genes at reciprocal breakpoints that regulate a variety of cellular functions including gene transcription, cell cycle, and apoptosis (Kuppers & Dalla-Favera, 2001; Kuppers, Citation2005). Among many identified translocations characteristic to lymphoma, common translocations have been associated with specific lymphomatous disease, including BCL6:IgH (90% of follicular lymphomas), BCL6 with various genes (35% of diffuse large B-cell lymphoma (DBLCL) and 48% in lymphocyte predominant Hodgkin’s lymphoma), BCL2:IgH (15–30% of DLBCL), MYC:IgH or IgL (15% of DLBCL and 100% of Burkitt’s lymphoma), and PAX5:IgH (see , Kuppers, Citation2005).
Figure 1. Lymphomas arise at different stages of B-cell differentiation, most commonly after antigenic stimulation during the germinal center reaction. Antigen-stimulated B-cells form germinal centers in lymph nodes under the regulation of BCL6 and PAX5. DNA mutation and recombination occur in an effort to develop and expand a pool of B-cell clones that produce high affinity antibody (via somatic hypermutation) with the appropriate effector activity (via class-switch recombination) towards antigen. Errors arising from these hyper-mutable events give rise to malignant transformation at discrete stages in this process, and the specific DNA alterations contribute to the diversity of lymphomatous phenotypes. See text for details. (Figure adapted from Klein & Dalla-Favera (Citation2008)).
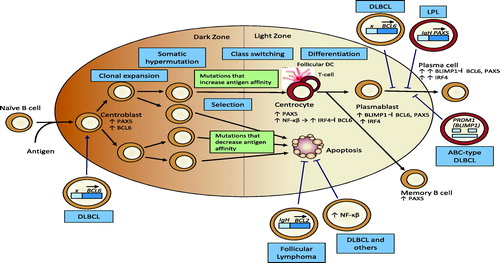
Thus, the same molecular mechanisms that underlie the intended genetic changes to the variable and constant regions of Ig genes during CSR and SHM contribute DNA mutations outside of IgV regions, translocation errors that result in dysregulation of genes controlling germinal center B-cell differentiation, or translocation errors that result in inappropriate expression of genes not normally expressed in differentiating germinal center B-cells (Klein & Dalla-Favera, 2008). Thus, lymphomas not only represent the malignant counterpart of B-cells at different stages of their development, but they can be sub-classified by common genetic lesions that typify a specific disease. Application of advanced genetic tools will continue to characterize the nature of the DNA alterations associated with given lymphoma classes, which will likely drive advances in both patient stratification and therapeutic preference. Our growing understanding of the cellular and molecular basis of lymphomagenesis also helps us to rationalize the existing epidemiology and clinical data that demonstrate increased lymphoma risk under broadly diverging conditions of chronic B-cell stimulation, whether by EBV, allograft, autoimmunity, or unresolved infection.
Discussion
This review confirms a strong relationship between immune status, whether immuno-suppression or immune activation, and cancer risk in human populations. Four general models have been proposed to describe the inter-relation between immune function and cancer risk. In the tumor immunosurveillance model, both innate and adaptive immune cells, primarily NK, cytotoxic T-lymphocytes (CTL), and NKT cells in concert with key cytokines and mediators (e.g. interferon [IFN]-γ, perforin, granzymes) protect the host from tumors (reviewed in Dunn et al., Citation2002; Wilczynski & Duechler, 2010; Vesely et al., Citation2011). Similarly, in the oncogenic virus model, the host response to viral infection is mediated by both innate (NK, NKT, and granulocyte) and adaptive (CTL) anti-viral responses, which contribute to viral clearance, the production of pro-inflammatory cytokines (interleukin [IL]-12, IL-18, IL-6, or TNFα), and the cellular production of IFNγ that both directly interferes with viral replication and promotes antiviral immunity (Biron & Brossay, 2001; Guidotti & Chisari, 2001; Malmgaard, Citation2004). In the chronic inflammation model, inflammation contributes to tumor development and growth through a number of mechanisms. Whereas tumor initiation is hypothesized to arise from DNA damage associated with reactive oxygen and nitrogen species, cytokines and inflammatory mediators establish a permissive environment for tumor growth and metastases, angiogenesis, increased cell survival, reduced cell death, and altered differentiation (Coussens & Werb, 2002; Colotta et al., Citation2009; de Visser et al., Citation2005; Federico et al., Citation2007; Mantovani et al., Citation2008). Finally, in the lymphomagenesis model, B-cells are driven into a germinal center response under conditions of either chronic immune system activation (i.e. autoimmunity) or immune suppression (i.e. from either EBV recrudescence or persistent/unresolved infection of other pathogens); this state of genomic hyper-mutability coupled to a high replication rate sets the stage for an increased risk for fixing critical errors in the genome that may result in lymphomagenic B-cell transformation.
The available clinical and observational data in human populations makes it difficult to isolate the effects of impaired tumor immunosurveillance, clearance of oncogenic viruses, and chronic inflammation as drivers of cancer, particularly lymphoma. However, it is likely that each of the identified immune-oncogenic mechanisms contribute differentially to tumor development in distinct populations.
When considering these mechanisms as causes for concern in therapeutic drug development, the data also suggest different methods may be necessary to control tumor development in different populations based on our evolving knowledge. For example, the available data suggest two plausible, possibly overlapping, etiologies for increased lymphoma risk in autoimmune patients treated with anti-TNF agents: (1) immunosuppression of tumor- and viral-surveillance mechanisms normally mediated by TNFα; or (2) background autoimmunity that increases the likelihood of spontaneous B-cell transformation. Whereas the first etiology suggests that anti-TNF therapy may increase the risk of lymphoma and dose reduction may be warranted, the second etiology suggests underlying disease should be better controlled, and an increased immunomodulation to control disease should be considered. Identifying the preferred strategy can only be resolved through careful design of epidemiology studies capturing clinical history (including measures of disease severity and medication use) or applying new tools to the problem, such as improved biomarkers of disease activity and immune status during the course of therapy, and knowledge of the viral state of individuals prior to therapeutic initiation and over the course of treatment. Such data may provide practitioners with greater knowledge to continually evaluate the risks and benefits of a therapeutic treatment regimen for a given individual.
Impressive insights are being obtained from the clinical use of immunotherapies in the treatment of cancer, demonstrating progress in our understanding of how we may eliminate or immunize patients to their cancer (e.g. Mellman et al., Citation2011). Similarly, impressive steps continue to be made to create increasingly selective immunomodulators for treatment of chronic inflammation or autoimmunity so as to reduce infection and cancer risk. Whether and how we can develop increasingly specific immunotherapies to selectively modulate the immune system in the treatment of chronic inflammation will depend, in part, on our ability to understand and predict potential cancer risks from these therapies. This requires that we increase our understanding of the immunologic basis of both autoimmunity and cancer, and to walk the fine line between them. Developing in vitro and in vivo tools to mechanistically assess the effects of a therapeutic regimen on key components of the immune response, to distinguish impacts on the identified paradigms relating the immune system and cancer, and to monitor patient outcome are key to this effort, and deserve further investigation.
Declaration of interest
R. A. Ponce, T. Gelzleichter, H. G. Haggerty, M. Holdren, and H. Lebrec are employed by, and own stock in, biotechnology/pharmaceutical companies developing immunomodulatory therapies.
Acknowledgments
This article reflects the views of the authors and should not be construed to represent FDA’s views or policies.
References
- Abasolo, L., Judez, E., Descalzo, M. A., et al. 2008. Cancer in rheumatoid arthritis: occurrence, mortality, and associated factors in a South European population. Semin. Arthritis Rheum. 37:388–397
- Aljurf, M. D., Owaidah, T. W., Ezzat, A., et al. 2003. Antigen- and/or immune-driven lymphoproliferative disorders. Ann. Oncol. 14:1595–1606
- Anderson, L. A., Gadalla, S., Morton, L. M., et al. 2009. Population-based study of autoimmune conditions and the risk of specific lymphoid malignancies. Int. J. Cancer 125:398–405
- Arason, G. J., Jorgensen, G. H., and Ludviksson, B. R. 2010. Primary immunodeficiency and autoimmunity: Lessons from human diseases. Scand. J. Immunol. 71:317–328
- Arguello, R. J., Balbaryski, J., Barboni, G., et al. 2012. Altered frequency and phenotype of CD4+ forkhead box protein 3+ T-cells and its association with autoantibody production in human immunodeficiency virus-infected paediatric patients. Clin. Exp. Immunol. 168:224–233
- Arkwright, P. D., Abinun, M., and Cant, A. J. 2002. Autoimmunity in human primary immuno-deficiency diseases. Blood 99:2694–2702
- Askling, J., Baecklund, E., Granath, F., et al. 2009. Anti-TNF therapy in rheumatoid arthritis and risk of malignant lymphomas: Relative risks and time trends in the Swedish Biologics Register. Ann. Rheum. Dis. 68:648–653
- Askling, J., Fored, C. M., Baecklund, E., et al. 2005. Hematopoietic malignancies in rheumatoid arthritis: Lymphoma risk and characteristics after exposure to TNF antagonists. Ann. Rheum. Dis. 64:1414–1420
- Baecklund, E., Askling, J., Rosenquist, R., et al. 2004. Rheumatoid arthritis and malignant lymphomas. Curr. Opin. Rheumatol. 16:254–261
- Baecklund, E., Backlin, C., Iliadou, A., et al. 2006a. Characteristics of diffuse large B-cell lymphomas in rheumatoid arthritis. Arthritis Rheum. 54:3774–3781
- Baecklund, E., Iliadou, A., Askling, J., et al. 2006b. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum. 54:692–701
- Baecklund, E., Sundstrom, C., Ekbom, A., et al. 2003. Lymphoma subtypes in patients with rheumatoid arthritis: Increased proportion of diffuse large B-cell lymphoma. Arthritis Rheum. 48:1543–1550
- Balandraud, N., Roudier, J., and Roudier, C. 2005. What are the links between Epstein-Barr virus, lymphoma, and TNF antagonism in rheumatoid arthritis? Semin. Arthritis Rheum. 34:31–33
- Basso, K., and Dalla-Favera, R. 2010. BCL6: Master regulator of the germinal center reaction and key oncogene in B-cell lymphomagenesis. Adv. Immunol. 105:193–210
- Bernatsky, S., Boivin, J. F., Joseph, L., et al. 2005. An international cohort study of cancer in systemic lupus erythematosus. Arthritis Rheum. 52:1481–1490
- Biron, C. A., and Brossay, L. 2001. NK cells and NKT cells in innate defense against viral infections. Curr. Opin. Immunol. 13:458–464
- Bongartz, T., Sutton, A. J., Sweeting, M. J., et al. 2006. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: Systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 295:2275–2285
- Boshoff, C., and Weiss, R. 2002. AIDS-related malignancies. Nat. Rev. Cancer 2:373–382
- Brown, S. L., Greene, M. H., Gershon, S. K., et al. 2002. TNF antagonist therapy and lymphoma development: 26 cases reported to the Food and Drug Administration. Arthritis Rheum. 46:3151–3158
- Caldwell, R. G., Wilson, J. B., Anderson, S. J., and Longnecker, R. 1998. Epstein-Barr virus LMP2A drives B-cell development and survival in the absence of normal B-cell receptor signals. Immunity 9:405–411
- Capello, D., Cerri, M., Muti, G., et al. 2003. Molecular histogenesis of post-transplantation lymphoproliferative disorders. Blood 102:3775–3785
- CDC. (Centers for Disease Control and prevention). 2011. Diseases and conditions. US Public Health Service, Atlanta, GA. Available online at: http://www.cdc.gov/DiseasesConditions/
- Chan, T. D., and Brink, R. 2012. Affinity-based selection and the germinal center response. Immunol. Rev. 247:11–23
- Chaturvedi, A. K., Mbulaiteye, S. M., and Engels, E. A. 2008. Underestimation of relative risks by standardized incidence ratios for AIDS-related cancers. Ann. Epidemiol. 18:230–234
- Cibere, J., Sibley, J., and Haga, M. 1997. Rheumatoid arthritis and the risk of malignancy. Arthritis Rheum. 40:1580–1586
- Cibere, J., Sibley, J., and Haga, M. 2001. Systemic lupus erythematosus and the risk of malignancy. Lupus 10:394–400
- Colotta, F., Allavena, P., Sica, A., et al. 2009. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 30:1073–1081
- Coussens, L. M., and Werb, Z. 2002. Inflammation and cancer. Nature 420:860–867
- Cunningham-Rundles, C., Cooper, D. L., Duffy, T. P., and Strauchen, J. 2002. Lymphomas of mucosal-associated lymphoid tissue in common variable immunodeficiency. Am. J. Hematol. 69:171–178
- Cunningham-Rundles, C., Siegal, F. P., Cunningham-Rundles, S., and Lieberman, P. 1987. Incidence of cancer in 98 patients with common varied immunodeficiency. J. Clin. Immunol. 7:294–299
- Dawson, T. M., Starkebaum, G., Wood, B. L., et al. 2001. Epstein-Barr virus, methotrexate, and lymphoma in patients with rheumatoid arthritis and primary Sjogren's syndrome: Case series. J. Rheumatol. 28:47–53
- de Visser, K. E., Korets, L. V., and Coussens, L. M. 2005. De novo carcinogenesis promoted by chronic inflammation is B-lymphocyte-dependent. Cancer Cell 7:411–423
- Dunn, G. P., Bruce, A. T., Ikeda, H., et al. 2002. Cancer immuno-editing: From immunosurveillance to tumor escape. Nat. Immunol. 3:991–998
- Ekstrom, K., Hjalgrim, H., Brandt, L., et al. 2003. Risk of malignant lymphomas in patients with rheumatoid arthritis and in their first-degree relatives. Arthritis Rheum. 48:963–970
- Engels, E. A., Biggar, R. J., Hall, H. I., et al. 2008. Cancer risk in people infected with human immunodeficiency virus in the United States. Int. J. Cancer 123:187–194
- Engels, E. A., Cerhan, J. R., Linet, M. S., et al. 2005. Immune-related conditions and immune-modulating medications as risk factors for non-Hodgkin's lymphoma: A case-control study. Am. J. Epidemiol. 162:1153–1161
- Engels, E. A., Pfeiffer, R. M., Goedert, J. J., et al. 2006. Trends in cancer risk among people with AIDS in the United States 1980–2002. AIDS 20:1645–1654
- Federico, A., Morgillo, F., Tuccillo, C., et al. 2007. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 121:2381–2386
- Filipovich, A. H., Mathur, A., Kamat, D., and Shapiro, R. S. 1992. Primary immunodeficiencies: Genetic risk factors for lymphoma. Cancer Res. 52:5465s–5467s
- Filipovich, A. H., Mathur, A., Kamat, D., et al. 1994. Lymphoproliferative disorders and other tumors complicating immunodeficiencies. Immunodeficiency 5:91–112
- Franklin, J., Lunt, M., Bunn, D., et al. 2006. Incidence of lymphoma in a large primary care derived cohort of cases of inflammatory polyarthritis. Ann. Rheum. Dis. 65:617–622
- Frieder, D., Larijani, M., Tang, E., et al. 2006. Antibody diversification: Mutational mechanisms and oncogenesis. Immunol. Res. 35:75–88
- Frizzera, G., Rosai, J., Dehner, L. P., et al. 1980. Lymphoreticular disorders in primary immunodeficiencies: New findings based on an up-to-date histologic classification of 35 cases. Cancer 46:692–699
- Geborek, P., Bladstrom, A., Turesson, C., et al. 2005. TNF blockers do not increase overall tumor risk in patients with rheumatoid arthritis, but may be associated with increased risk of lymphomas. Ann. Rheum. Dis. 64:699–703
- Gompels, M. M., Hodges, E., Lock, R. J., et al. 2003. Lymphoproliferative disease in antibody deficiency: A multi-centre study. Clin. Exp. Immunol. 134:314–320
- Gordon, M. S., Kanegai, C. M., Doerr, J. R., and Wall, R. 2003. Somatic hypermutation of the B-cell receptor genes B29 (Igβ, CD79b) and mb1 (Igα, CD79a). Proc. Natl. Acad. Sci. USA 100:4126–4131
- Gottschalk, S., Rooney, C. M., and Heslop, H. E. 2005. Post-transplant lymphoproliferative disorders. Annu. Rev. Med. 56:29–44
- Gridley, G., McLaughlin, J. K., Ekbom, A., et al. 1993. Incidence of cancer among patients with rheumatoid arthritis. J. Natl. Cancer Inst. 85:307–311
- Grulich, A. E., van Leeuwen, M. T., Falster, M. O., and Vajdic, C. M. 2007. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: A meta-analysis. Lancet 370:59–67
- Guidotti, L. G., and Chisari, F. V. 2001. Non-cytolytic control of viral infections by the innate and adaptive immune response. Annu. Rev. Immunol. 19:65–91
- Guikema, J. E., Linehan, E. K., Tsuchimoto, D., et al. 2007. APE1- and APE2-dependent DNA breaks in immunoglobulin class switch recombination. J. Exp. Med. 204:3017–2306
- Hakulinen, T., Isomaki, H., and Knekt, P. 1985. Rheumatoid arthritis and cancer studies based on linking nationwide registries in Finland. Am J Med 78:29–32
- Hislop, A. D., Taylor, G. S., Sauce, D., and Rickinson, A. B. 2007. Cellular responses to viral infection in humans: Lessons from Epstein-Barr virus. Annu. Rev. Immunol. 25:587–617
- Holmes, D., Jiang, Q., Zhang, L., and Su, L. 2008. Foxp3 and Treg cells in HIV-1 infection and immunopathogenesis. Immunol. Res. 41:248–266
- Hopwood, P., and Crawford, D. H. 2000. The role of EBV in post-transplant malignancies: A review. J. Clin. Pathol. 53:248–254
- Hoshida, Y., Xu, J. X., Fujita, S., et al. 2007. Lymphoproliferative disorders in rheumatoid arthritis: Clinicopathological analysis of 76 cases in relation to methotrexate medication. J. Rheumatol. 34:322–331
- Howlader, N., Noone, A., Krapcho, M., et al. 2011. SEER Cancer Statistics Review, 1975–2008. Bethesda, MD: N. C. Institute. Available online at: http://seer.cancer.gov/csr/1975_2008/ </csr/1975_2008/>. based on November 2010 SEER data submission, posted to the SEER web site, April 15, 2011
- Imai, K., Slupphaug, G., Lee, W. I., et al. 2003. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat. Immunol. 4:1023–1028
- Ioachim, H. L. 1990. The opportunistic tumors of immune deficiency. Adv. Cancer Res. 54:301–317
- Kaiser, R. 2008. Incidence of lymphoma in patients with rheumatoid arthritis: A systematic review of the literature. Clin. Lymphoma Myeloma 8:87–93
- Kamel, O. W., Holly, E. A., van de Rijn, M., et al. 1999. A population based, case control study of non-Hodgkin's lymphoma in patients with rheumatoid arthritis. J. Rheumatol. 26:1676–1680
- Kasiske, B. L., Snyder, J. J., Gilbertson, D. T., and Wang, C. 2004. Cancer after kidney transplantation in the United States. Am. J. Transplant. 4:905–913
- Kauppi, M., Pukkala, E., and Isomaki, H. 1997. Elevated incidence of hematologic malignancies in patients with Sjogren's syndrome compared with patients with rheumatoid arthritis (Finland). Cancer Causes Control 8:201–204
- Kersey, J. H., Spector, B. D., and Good, R. A. 1973. Primary immunodeficiency diseases and cancer: The immunodeficiency-cancer registry. Int. J. Cancer 12:333–347
- Keystone, E. C. 2011. Does anti-TNFα therapy affect risk of serious infection and cancer in patients with rheumatoid arthritis?: A review of long-term data. J. Rheumatol. 38:1552–1562
- Kilger, E., Kieser, A., Baumann, M., and Hammerschmidt, W. 1998. Epstein-Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J. 17:1700–1709
- Kinlen, L. J., Webster, A. D., Bird, A. G., et al. 1985. Prospective study of cancer in patients with hypogammaglobulinaemia. Lancet 1:263–266
- Klein, U., and Dalla-Favera, R. 2008. Germinal centres: Role in B-cell physiology and malignancy. Nat. Rev. Immunol. 8:22–33
- Kuppers, R. 2003. B cells under influence: Transformation of B-cells by Epstein-Barr virus. Nat. Rev. Immunol. 3:801–812
- Kuppers, R. 2005. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 5:251–262
- Kuppers, R., and Dalla-Favera, R. 2001. Mechanisms of chromosomal translocations in B-cell lymphomas. Oncogene 20:5580–5594
- Liu, M., Duke, J. L., Richter, D. J., et al. 2008. Two levels of protection for the B-cell genome during somatic hyper-mutation. Nature 451:841–845
- Llorca, J., Lopez-Diaz, M. J., Gonzalez-Juanatey, C., et al. 2007. Persistent chronic inflammation contributes to the development of cancer in patients with rheumatoid arthritis from a defined population of northwestern Spain. Semin. Arthritis Rheum. 37:31–38
- Lopez-Olivo, M. A., Tayar, J. H., Martinez-Lopez, J. A., et al. 2012. Risk of malignancies in patients with rheumatoid arthritis treated with biologic therapy: A meta-analysis. JAMA 308:898–908
- Louthrenoo, W. 2008. Rheumatic manifestations of human immunodeficiency virus infection. Curr. Opin. Rheumatol. 20:92–99
- Macsween, K. F., and Crawford, D. H. 2003. Epstein-Barr virus-recent advances. Lancet Infect. Dis. 3:131–140
- Malmgaard, L. 2004. Induction and regulation of IFNs during viral infections. J. Interferon Cytokine Res. 24:439–454
- Mantovani, A., Allavena, P., Sica, A., and Balkwill, F. 2008. Cancer-related inflammation. Nature 454:436–444
- Mariette, X., Cazals-Hatem, D., Warszawki, J., et al. 2002. Lymphomas in rheumatoid arthritis patients treated with methotrexate: A 3-year prospective study in France. Blood 99:3909–3915
- Marodi, L., and Notarangelo, L. D. 2007. Immunological and genetic bases of new primary immunodeficiencies. Nat. Rev. Immunol. 7:85–861
- Mellemkjaer, L., Andersen, V., Linet, M. S., et al. 1997. Non-Hodgkin's lymphoma and other cancers among a cohort of patients with systemic lupus erythematosus. Arthritis Rheum. 40:761–768
- Mellemkjaer, L., Hammarstrom, L., Andersen, V., et al. 2002. Cancer risk among patients with IgA deficiency or common variable immunodeficiency and their relatives: A combined Danish and Swedish study. Clin. Exp. Immunol. 130:495–500
- Mellemkjaer, L., Linet, M. S., Gridley, G., et al. 1996. Rheumatoid arthritis and cancer risk. Eur. J. Cancer 32A:1753–1757
- Mellman, I., Coukos, G., and Dranoff, G. 2011. Cancer immunotherapy comes of age. Nature 480:480–489
- Moir, S., and Fauci, A. S. 2008. Pathogenic mechanisms of B-lymphocyte dysfunction in HIV disease. J. Allergy Clin. Immunol. 122:12–19
- Morrell, D., Cromartie, E., and Swift, M. 1986. Mortality and cancer incidence in 263 patients with ataxia-telangiectasia. J. Natl. Cancer Inst. 77:89–92
- Mueller, B. U., and Pizzo, P. A. 1995. Cancer in children with primary or secondary immuno-deficiencies. J. Pediatr. 126:1–10
- Muschen, M., Re, D., Jungnickel, B., et al. 2000. Somatic mutation of the CD95 gene in human B-cells as a side-effect of the germinal center reaction. J. Exp. Med. 192:1833–1840
- Nagy, N., and Klein, E. 2010. Deficiency of the proapoptotic SAP function in X-linked lymphoproliferative disease aggravates Epstein-Barr virus (EBV)-induced mononucleosis and promotes lymphoma development. Immunol. Lett. 130:13–18
- Nived, O., Bengtsson, A., Jonsen, A., et al. 2001. Malignancies during follow-up in an epidemiologically defined systemic lupus erythematosus inception cohort in southern Sweden. Lupus 10:500–504
- Notarangelo, L. D., Fischer, A., Geha, R. S., et al. 2009. Primary immunodeficiencies: 2009 update. J. Allergy Clin. Immunol. 124:1161–1178
- Olsen, J. H., Hahnemann, J. M., Borresen-Dale, A. L., et al. 2001. Cancer in patients with ataxia-telangiectasia and in their relatives in the nordic countries. J. Natl. Cancer Inst. 93:121–127
- Parikh-Patel, A., White, R. H., Allen, M., and Cress, R. 2008. Cancer risk in a cohort of patients with systemic lupus erythematosus (SLE) in California. Cancer Causes Control 19:887–894
- Pasqualucci, L., Migliazza, A., Fracchiolla, N., et al. 1998. BCL-6 mutations in normal germinal center B-cells: Evidence of somatic hypermutation acting outside Ig loci. Proc. Natl. Acad. Sci. USA 95:11816–11821
- Pasqualucci, L., Neumeister, P., Goossens, T., et al. 2001. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 412:341–346
- Peled, J. U., Kuang, F. L., Iglesias-Ussel, M. D., et al. 2008. The biochemistry of somatic hypermutation. Annu. Rev. Immunol. 26:481–511
- Penn, I. 1999. Post-transplant malignancies. Transplant. Proc. 31:1260–1262
- Penn, I. 2000. Post-transplant malignancy: Role of immunosuppression. Drug Safety 23:101–113
- Perry, G. S. 3rd, Spector, B. D., Schuman, L. M., et al. 1980. The Wiskott-Aldrich syndrome in the United States and Canada (1892–1979). J. Pediatr. 97:72–78
- Pettersson, T., Pukkala, E., Teppo, L., and Friman, C. 1992. Increased risk of cancer in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 51:437–439
- Pham, P., Bransteitter, R., Petruska, J., and Goodman, M. F. 2003. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature 424:103–107
- Phan, R. T., and Dalla-Favera, R. 2004. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B-cells. Nature 432:635–639
- Phan, R. T., Saito, M., Basso, K., et al. 2005. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B-cells. Nat. Immunol. 6:1054–1060
- Poltoratsky, V., Prasad, R., Horton, J. K., and Wilson, S. H. 2007. Down-regulation of DNA polymerase beta accompanies somatic hypermutation in human BL2 cell lines. DNA Repair (Amst.) 6:244–253
- Purtilo, D. T., Cassel, C. K., Yang, J. P., and Harper, R. 1975. X-linked recessive progressive combined variable immunodeficiency (Duncan's disease). Lancet 1:935–940
- Purtilo, D. T., Sakamoto, K., Saemundsen, A. K., et al. 1981. Documentation of Epstein-Barr virus infection in immunodeficient patients with life-threatening lymphoproliferative diseases by clinical, virological, and immunopathological studies. Cancer Res. 41:4226–4236
- Rabkin, C. S. 2001. AIDS and cancer in the era of highly active anti-retroviral therapy (HAART). Eur. J.Cancer 37:1316–1319
- Rada, C., Williams, G. T., Nilsen, H., et al. 2002. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 12:1748–1755
- Ramsey-Goldman, R., Mattai, S. A., Schilling, E., et al. 1998. Increased risk of malignancy in patients with systemic lupus erythematosus. J. Invest. Med. 46:217–222
- Ranuncolo, S. M., Polo, J. M., Dierov, J., et al. 2007. Bcl-6 mediates the germinal center B-cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nat. Immunol. 8:705–714
- Sachdeva, M., Fischl, M. A., Pahwa, R., et al. 2010. Immune exhaustion occurs concomitantly with immune activation and decrease in regulatory T-cells in viremic chronically HIV-1-infected patients. J. Acquir. Immune Defic. Syndr. 54:447–454
- Salavoura, K., Kolialexi, A., Tsangaris, G., and Mavrou, A. 2008. Development of cancer in patients with primary immunodeficiencies. Anticancer Res. 28:1263–1269
- Schabet, M. 1999. Epidemiology of primary CNS lymphoma. J. Neurooncol. 43:199–201
- Schrader, C. E., Linehan, E. K., Mochegova, S. N., et al. 2005. Inducible DNA breaks in Ig S regions are dependent on AID and UNG. J. Exp. Med. 202:561–568
- Schuetz, C., Niehues, T., Friedrich, W., and Schwarz, K. 2010. Autoimmunity, autoinflammation and lymphoma in combined immunodeficiency (CID). Autoimmun. Rev. 9:477–482
- Setoguchi, S., Solomon, D. H., Weinblatt, M. E., et al. 2006. TNFα antagonist use and cancer in patients with rheumatoid arthritis. Arthritis Rheum. 54:2757–2764
- Shaffer, A. L., Rosenwald, A., and Staudt, L. M. 2002. Lymphoid malignancies: The dark side of B-cell differentiation. Nat. Rev. Immunol. 2:920–932
- Shen, H. M., Peters, A., Baron, B., et al. 1998. Mutation of BCL-6 gene in normal B-cells by the process of somatic hypermutation of Ig genes. Science 280:1750–1752
- Shlomchik, M. J., and Weisel, F. 2012. Germinal center selection and the development of memory B and plasma cells. Immunol. Rev. 247:52–63
- Smedby, K. E., Hjalgrim, H., Askling, J., et al. 2006. Auto-immune and chronic inflammatory disorders and risk of non-Hodgkin lymphoma by subtype. J. Natl. Cancer Inst. 98:51–60
- Smitten, A. L., Simon, T. A., Hochberg, M. C., and Suissa, S. 2008. A meta-analysis of the incidence of malignancy in adult patients with rheumatoid arthritis. Arthritis Res. Ther. 10:R45
- Soni, V., Cahir-McFarland, E., and Kieff, E. 2007. LMP1 TRAFficking activates growth and survival pathways. Adv. Exp. Med. Biol. 597:173–187
- Staal, S. P., Ambinder, R., Beschorner, W. E., et al. 1989. A survey of Epstein-Barr virus DNA in lymphoid tissue. Frequent detection in Hodgkin's disease. Am. J. Clin. Pathol. 91:1–5
- Stavnezer, J., Guikema, J. E., and Schrader, C. E. 2008. Mechanism and regulation of class switch recombination. Annu. Rev. Immunol. 26:261–292
- Stevenson, F., Sahota, S., Zhu, D., et al. 1998. Insight into the origin and clonal history of B-cell tumors as revealed by analysis of immunoglobulin variable region genes. Immunol. Rev. 162:247–259
- Suarez, F., Lortholary, O., Hermine, O., and Lecuit, M. 2006. Infection-associated lymphomas derived from marginal zone B-cells: A model of antigen-driven lymphoproliferation. Blood 107:3034–3044
- Swerdlow, A. J. 2003. Epidemiology of Hodgkin's disease and non-Hodgkin's lymphoma. Eur. J. Nucl. Med. Mol. Imaging 30:S3–12
- Thomas, E., Brewster, D. H., Black, R. J., and Macfarlane, G. J. 2000. Risk of malignancy among patients with rheumatic conditions. Int. J. Cancer 88:497–502
- Thorley-Lawson, D. A. 2001. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 1:75–82
- Thorley-Lawson, D. A., and Gross, A. 2004. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. New Engl. J. Med. 350:1328–1337
- U.S. Food and Drug Administration. 2003. Arthritis Drugs Advisory Committee: Safety update on TNFα antagonists. Washington, DC: Arthritis Drugs Advisory Committee. Available online at: http://www.fda.gov/ohrms/dockets/ac/03/briefing/3930b1.htm
- Vajdic, C. M., Mao, L., van Leeuwen, M. T., et al. 2010. Are antibody deficiency disorders associated with a narrower range of cancers than other forms of immunodeficiency? Blood 116:1228–1234
- Vesely, M. D., Kershaw, M. H., Schreiber, R. D., and Smyth, M. J. 2011. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 29:235–271
- Walker, U. A., Tyndall, A., and Daikeler, T. 2008. Rheumatic conditions in human immunodeficiency virus infection. Rheumatology (Oxford) 47:952–959
- Wilczynski, J. R., and Duechler, M. 2010. How do tumors actively escape from host immunosurveillance? Arch. Immunol. Ther. Exp. (Warsz.) 58:435–448
- Wolfe, F., and Michaud, K. 2004. Lymphoma in rheumatoid arthritis: The effect of methotrexate and anti-TNF therapy in 18 572 patients. Arthritis Rheum. 50:1740–1751
- Wolfe, F., and Michaud, K. 2007. The effect of methotrexate and anti-TNF therapy on the risk of lymphoma in rheumatoid arthritis in 19 562 patients during 89 710 person-years of observation. Arthritis Rheum. 56:1433–1439
- Xu, Z., Fulop, Z., Zhong, Y., et al. 2005. DNA lesions and repair in immunoglobulin class switch recombination and somatic hypermutation. Ann. NY Acad. Sci. 1050:146–162
- Young, L. S., and Murray, P. G. 2003. Epstein-Barr virus and oncogenesis: From latent genes to tumours. Oncogene 22:5108–5121
- Young, L. S., and Rickinson, A. B. 2004. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4:757–768
- Zintzaras, E., Voulgarelis, M., and Moutsopoulos, H. M. 2005. The risk of lymphoma development in autoimmune diseases: A meta-analysis. Arch. Intern. Med. 165:2337–2344