
Abstract
Bisphenol A (BPA), a monomer of polycarbonate plastics and epoxide resin, acts as an endocrine-active compound and has been shown to enhance the inflammatory response to allergen challenge. Previous reports in rodents have demonstrated that perinatal BPA exposure alters airway inflammation following sensitization and challenge to ovalbumin in juvenile and adult offspring. Additionally, a high concentration of BPA has been shown to enhance mediator release in mast cell lines. This study aimed to determine if short-term BPA exposure, at levels relevant to human exposure, enhances mast cell release of histamine and cysteinyl leukotrienes (CysLTs). Primary murine bone marrow-derived mast cells (BMMC) produced from the femurs of female C57BL/6 mice were stimulated with BPA or estradiol (E2) in vitro. It was observed that both BPA and E2 increased BMMC histamine release over a range of nanomolar concentrations (1–1000 nM). The estrogen receptor (ER) antagonist ICI 182,780 partially blocked the ability of E2, but not BPA, to elevate histamine release. BPA also increased CysLT release, which was not abrogated by ER inhibition. It was also observed that the ability of BPA to enhance histamine and CysLT release was inhibited by blocking the extracellular signal-regulated kinase (ERK) pathway with U0126 or by chelating extracellular calcium (Ca2+) using EGTA. In summary, these experiments are the first to demonstrate that acute BPA exposure enhances mast cell histamine and CysLT release in vitro—an effect that is not dependent on an ER-mediated mechanism. Instead, BPA-induced mast cell histamine and CysLT release may be mediated, in part, by the ERK pathway and extracellular Ca2+ concentrations. These data suggest that exposure to BPA at levels relevant to human exposure may provoke an acute inflammatory response in atopic individuals via enhanced mast cell activation.
Introduction
The prevalence of asthma, a chronic inflammatory disease of the airways characterized by wheezing, shortness of breath, chest tightness, and coughing (Murdoch & Lloyd, Citation2010), has steadily increased since the 1970s (Anandan et al., Citation2010; Lai et al., Citation2009; To et al., Citation2012). Current estimates suggest 300 million individuals have been diagnosed with asthma globally, and that asthma accounts for nearly 300,000 deaths yearly (Holgate et al., Citation2007; WHO, Citation2007). Asthma pathogenesis arises from a complex interplay of genetic pre-disposition and environmental exposure (Holgate et al., Citation2007). The observation that asthma prevalence tends to be highest in industrialized areas suggests that one or more products of industrialization may be contributing to asthma pathogenesis (Anandan et al., Citation2010; Asher, Citation2010; Masoli et al., Citation2004; Crinnion, Citation2012; To et al., Citation2012).
Observational epidemiologic studies have suggested that endogenous and exogenous estrogen may contribute to the pathogenesis and severity of asthma (Bonds & Midoro-Horiuti, Citation2013); asthma prevalence among adults is higher in women than in men (Leynaert et al., Citation2012; Vink et al., Citation2010), and peaks in estrogen and progesterone corresponding to the ovulation cycle have been associated with worsened asthma symptoms (Thornton et al., Citation2012; Vrieze et al., Citation2003). Additionally, a higher prevalence of adult-onset asthma has been reported in patients undergoing hormone replacement therapy (Barr et al., Citation2004; Dratva, Citation2010). Lab-based approaches also indicate that estradiol, progesterone, and synthetic xenoestrogens can enhance mast cell degranulation (Jensen et al., Citation2010; Kennedy et al., Citation2012; Lee et al., Citation2011; Narita et al., Citation2007; Zaitsu et al., Citation2007), and, since mast cells play a central role in atopic asthma, it has been hypothesized that widespread exposure to synthetic xenoestrogens (Latini et al., Citation2010; Phillips an&d Foster, Citation2008; Yang et al., Citation2006) may be contributing to the increased prevalence of asthma by enhancing mast cell activation.
Mast cells play a critical role in the development of atopic asthma and other allergic diseases by initiating an acute inflammatory response (Amin, Citation2012). Activated mast cells release pre-formed mediators stored in cytoplasmic secretory granules, including histamine, leukotriene (LT)C4, and prostaglandin (PG)D2, each capable of inducing bronchoconstriction, mucus secretion, and edema, as well as pro-inflammatory cytokines (i.e., interleukin [IL]-4, IL-5, IL-13, and tumor necrosis factor [TNF]-α), that contribute to IgE production and eosinophil recruitment (Boyce, Citation2003; Bradding et al., Citation2006).
Recently, the endocrine-active compound bisphenol A (BPA), a monomer of poly-carbonate plastics and epoxide resin, has been associated with worsened asthma symptoms in humans (Donohue et al., Citation2013; Spanier et al., Citation2012; Vaidya & Kulkarni, Citation2012) in addition to numerous other adverse health effects (Golub et al., Citation2010; Rubin, Citation2011). Additionally, the development of asthma in juvenile mice (Midoro-Horiuti et al., Citation2010; Nakajima et al., Citation2012), and altered airway inflammatory responses in adult mice (Bauer et al., Citation2012) following perinatal BPA exposure have also been reported. Human exposure to BPA is widespread (Vandenberg et al., Citation2007), with National Health and Nutrition Examination Survey (NHANES) data reporting detectable levels of total BPA in the urine of 95% of participants (Calafat et al., Citation2005). BPA is capable of disrupting normal endocrine signaling by weakly binding to steroid receptors including estrogen receptors (ERs) and thyroid hormone receptor (Zoeller et al., Citation2005). BPA has also been demonstrated to bind to the membrane-bound G-protein coupled ER 1 (GPER, also GRP30) (Bouskine et al., Citation2009; Sheng & Zhu, Citation2011), as well as the nuclear receptor estrogen-related receptor (ERR)-γ (Matsushima et al., Citation2007). BPA levels in human samples including urine, serum, blood, and saliva can vary depending on sample type, study population, and detection method, although reports repeatedly indicated that levels of BPA in humans are in the nanomolar range (Vandenberg et al., Citation2007, Citation2012). For example, typical total BPA levels in adult serum and urine range from 1–90 nM and 5–10 nM, respectively (Vandenberg et al., Citation2007). Previous reports conducted using a high level of BPA (50 μM) have shown that BPA can enhance mast cell activation (Lee & Lim, Citation2010; Lee et al., Citation2012; Park & Lim, Citation2010; Shim & Lim, Citation2009).
The study reported here tested the hypothesis that BPA at levels relevant to human exposure enhances bone marrow-derived mast cell (BMMC) release of histamine and cysteinyl LTs (CysLTs). Furthermore, the requirements of ER-alpha (ERα), the extracellular signal-regulated kinase (ERK) pathway, and extracellular calcium ions (Ca2+) in BPA-induced mediator increases were investigated. It was observed that BPA exposure increased histamine and CysLT release from mast cells, and that these responses could not be attenuated with an ER antagonist. Yet, BPA-induced histamine and CysLT increases were found to require ERK signaling and extracellular Ca2+. These observations suggest a role for BPA in exacerbating inflammatory responses through enhancing mast cell degranulation.
Materials and methods
Animals
Wild-type female C57BL/6 mice (8-weeks-of-age) were purchased from Charles River (Wilmington, MA) and housed in a University of Michigan Animal facility. Rooms were maintained at 70 °C and a 25–30% relative humidity, and with a 12-hour light–dark cycle. All mice had ad libitum access to standard chow and filtered water throughout the study. Animals were treated according to National Institutes of Health guidelines for the use of experimental animals with approval of the University of Michigan Committee for the Use and Care of Animals.
Generation and culture of BMMC
Following euthanasia by CO2 inhalation, femurs obtained from the mice were lavaged with RPMI (Life Technologies, Invitrogen, Carlsbad, CA). Primary BMMC were generated by culturing bone marrow cells in RPMI containing 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin (Invitrogen) supplemented with 10 ng/mL murine IL-3 (Sigma, St. Louis, MO) and 10 ng/ml murine stem cell factor (Sigma) at 37 °C in 5% CO2. Throughout incubation, culture media and culture flasks were changed once weekly. After 4 weeks in culture, cells were spun onto glass slides using a cytocentrifuge (STAT SPIN, Norwood, MA), and the mast cell phenotype was confirmed when 95% of the cells were positive for c-kit. For the immunocytostaining, c-kit was probed using anti-c-kit antibody produced in rabbit (Cell Signaling, Beverly, MA), secondarily probed using a goat anti-rabbit avidin biotin complex kit (Vector, Burlingame, CA), and ultimately visualized using a diaminobenzidine kit (Vector), each according to manufacturer instructions.
Stimulation of BMMC for pro-inflammatory mediator release
Differentiated BMMC were collected by centrifugation, re-suspended in RPMI containing 1% penicillin/streptomycin, enumerated using a hemocytometer, and plated in flat-bottom 96-well plates at a concentration of 2 × 105 cells/well. Plated BMMC were treated with vehicle control (0.01% ethanol), BPA (0.1, 1, 10, 100, or 1000 nM; National Toxicology Program standard), or 17β-estradiol (E2; 0.1, 1, 10, 100, or 1000 nM) (Sigma) for 30 min at 37 °C with 5% CO2 to induce BMMC release of histamine and CysLTs. The concentrations of BPA tested are considered not to be cytotoxic to mast cells according to a report by Lee & Lim (Citation2010). As a positive control, cells were also treated with 1 μM calcium ionophore A23187 (Sigma) to induce mass release of pro-inflammatory mediators. In subsequent experiments, plated BMMC were pre-treated with the ER antagonist ICI 182,780 (ICI; 0.1, 1, or 10 μM) (Sigma), the ERK1/2 inhibitor U0126 (10 μM; Sigma), or the Ca2+ chelator ethylene glycol tetraacetic acid (EGTA; 3 mM) (Sigma) for 1 h at 37 °C before treatment with 10 nM BPA or E2 for 30 min. After the allotted time, cell culture media were collected and stored at −80 °C until analysis.
Histamine determination
Analysis of histamine was conducted according to the protocol previously described by Zhao et al. (Citation2001). Briefly, 30 μl from collected supernatants were distributed on 384-well plates (Corning Life Sciences, Tewksbury, MA). To each sample, 6 μl of 1 M NaOH and 1.5 μl of 10 mg/ml o-phthaldialdehyde (Sigma) prepared in methanol was added to induce histamine derivatization. After incubation at room temperature for 4 min, 5 μl of 3 M HCl was added to each well to halt the derivatization. Fluorescence in each well was then read at 530 nm (360 nm excitation, 450 nm emission) using a SpectraMax Gemini M2e fluorometer (Molecular Devices, Sunnyvale, CA). Values were expressed as percentage histamine release of the vehicle control (indicated as ‘% histamine release’ in figures).
CysLT determination
Levels of CysLTs produced by BMMC following stimulation were determined using commercial enzyme immunoassay (EIA) kits (Cayman Chemical, Ann Arbor, MI), according to manufacturer instructions.
Statistical analysis
Data are expressed as mean ± SEM with an n of 3–6 separate experiments, each using a minimum of three replicates per treatment condition. Analyses were conducted in Prism Graph Software using one-way analysis of variance with the Bonferroni test ( and ) or a Student’s t-test ( and ) for separation of the means. In all cases, a p value < 0.05 was considered statistically significant.
Figure 1. (a) Percentage increase in histamine release from BMMC following 30-min treatment with E2 (open bars) (n = 6) or BPA (solid bars) (n = 6). (b) CysLT release following 30-min treatment with BPA (n = 4). Bars represent mean ± SEM. *p < 0.05 and **p ≤ 0.001 compared to untreated cells.
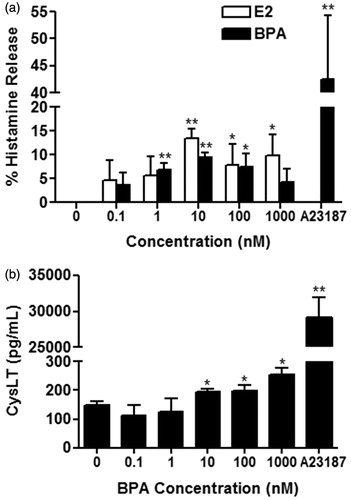
Figure 2. (a) Percentage increase in histamine release from BMMC pre-treated with ICI for 1 h before treatment with 10 nM E2 (open bars) (n = 3) or 10 nM BPA (solid bars) (n = 3) for 30 min. (b) CysLT release from BMMC pre-treated with (shaded bar) or without (solid bar) 1 μM ICI before treatment with 10 nM BPA (n = 4). Bars represent mean ± SEM. *p < 0.05 compared to no ICI treatment. †p < 0.05 compared to control.
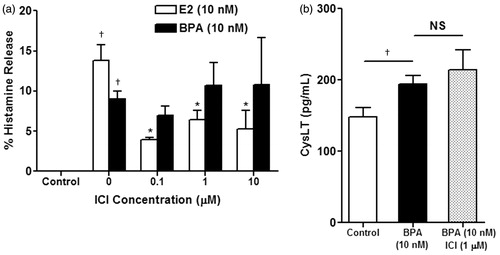
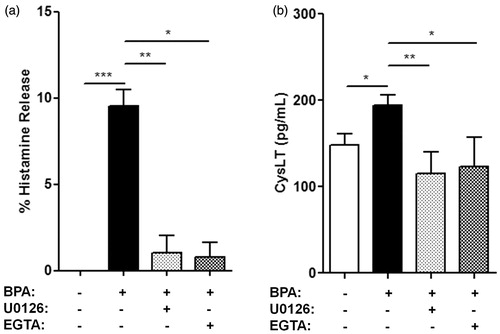
Figure 3. Percentage increase in (a) histamine release and (b) CysLT release from BMMC pre-treated with 10 μM U0126 (shaded bar) or 3 mM EGTA (checkered bar) for 1 h before treatment with 10 nM BPA for 30 min. Bars represent mean ± SEM. n = 3 for all experiments. *p < 0.05, **p < 0.005, and ***p < 0.001.
Results
BPA enhances BMMC histamine and CysLT release
To determine if short-term BPA exposure affects release of pro-inflammatory mediators from mast cells, BMMC release of histamine and CysLTs was determined after treatment with varying concentrations (0.1–1000 nM) of BPA for 30 min (). Cells exposed to 1 nM (p = 0.001), 10 nM (p < 0.0001), and 100 nM (p = 0.030) BPA displayed significantly increased histamine release compared to vehicle control, with cells in the 10 nM exposure group displaying the highest mean increase in histamine release (i.e., 9.0%) among the BPA-exposed cells (). Increased histamine release was not significant following exposure to BPA at 0.1 nM (p = 0.076) or 1000 nM (p = 0.061). As a comparison, cells exposed to E2 also displayed increased histamine release in 10 nM (p = 0.0004) and 100 nM (p = 0.046) exposure groups, as well as at 1000 nM (p = 0.024), compared to vehicle control. Treatment with 0.1 nM (p = 0.145) and 1 nM (p = 0.191) E2 did not result in significantly increased histamine release. Cells in the 10 nM E2 exposure group displayed the highest mean increase in histamine release (i.e., 13.5%) among E2-treated cells. There was no difference in histamine release between BPA- and E2-exposed cells treated with matching concentrations. CysLT release from treated BMMC was increased at 10 nM (p = 0.013), 100 nM (p = 0.029), and 1000 nM (p = 0.002) BPA compared to vehicle control, with no statistical differences between concentrations ().
ER antagonism on BPA-induced histamine and CysLT release from BMMC
BMMC have previously been demonstrated to express only the ERα sub-unit, and not to express the ER-beta (ERβ) sub-unit (Jensen et al., Citation2010; Zaitsu et al., Citation2007). Thus, the role of ERα in BPA-induced increased release of histamine and CysLTs from BMMC was investigated using the ER antagonist ICI (). Pre-treatment with 0.1 μM (p = 0.178), 1 μM (p = 0.627), or 10 μM (p = 0.674) ICI before treatment with 10 nM BPA for 30 min did not alter histamine release compared to BPA treatment alone (). However, pre-treatment with ICI at 0.1 μM (p = 0.034), 1 μM (p = 0.048), or 10 μM (p = 0.049) before treatment with 10 nM E2 decreased E2-induced histamine increases from BMMC compared with E2 alone, but did not completely abolish the response (). Pre-treatment with 1 μM ICI before treatment with 10 nM BPA did not alter CysLT release (p = 0.463) from BMMC compared to BPA treatment alone ().
ERK inhibition on BPA-induced histamine and CysLT release from BMMC
To examine the role of the ERK pathway in BPA-induced increases in histamine and CysLT release, cells were pre-treated with the inhibitor U0126 (). Pre-treatment with U0126 followed by treatment with 10 nM BPA for 30 min nearly completely inhibited the increase in histamine release from BMMC noted with 10 nM BPA alone (p = 0.003) (). A similar response was observed regarding CysLT release. Pre-treatment with U0126 followed by treatment with 10 nM BPA resulted in decreased CysLT release compared to 10 nM BPA alone (p = 0.004) ().
Extracellular Ca2+ chelation on BPA-induced histamine and CysLT release from BMMC
A requirement of extracellular Ca2+ for BPA-induced increases in mast cell histamine and CysLT release was studied by pre-treating the cells and media with EGTA before treatment with 10 nM BPA (). Pre-treatment with the Ca2+ chelator followed by BPA treatment nearly completely inhibited increased histamine release due to 10 nM BPA alone (p = 0.025) (). Similarly, EGTA pre-treatment followed by 10 nM BPA treatment inhibited CysLT release compared to BPA alone (p = 0.037), bringing EGTA-pre-treated CysLT levels in line with those of vehicle control CysLT levels (p = 0.427) ().
Discussion
This study is the first to demonstrate that concentrations of BPA relevant to human exposure are capable of stimulating BMMC to release the pro-inflammatory mediators histamine and CysLTs. Furthermore, up-regulated levels of histamine induced by BPA treatment closely matched the increased levels of histamine induced by E2, both of which produced non-monotonic dose response curves for increased histamine release. Although increases in histamine and CysLT release following BPA treatment were modest, these responses are important because they suggest that low levels of this environmental compound can promote allergic responses in the absence of IgE-mediated mast cell activation.
Since BPA is a known xenoestrogen, we hypothesized that BPA (and E2) may be acting through ERα to cause changes in mediator release. This idea was tested using the ER inhibitor ICI. Our results do not support a role for ERα in BPA-induced changes in histamine or CysLT release, although data suggest that E2-induced histamine increase is partially dependent on ERα. Additionally, we sought to examine the role of the ERK pathway in the BPA-induced responses, since this pathway is integral for mast cell production of CysLTs and other lipid mediators (Kambayashi & Koretzky, Citation2007) and is also a common pathway used in ER-initiated signaling (Levin, Citation2011). A requirement for the ERK pathway in the BPA-induced mast cell stimulation was tested by pre-treating cells with the inhibitor U0126; this agent actively inhibits ERK kinase 1/2 (MEK1/2) and, thus, passively inhibits ERK1/2 and gene transcription down-stream. Pre-treatment with the inhibitor resulted in diminished BMMC histamine and CysLT release compared to that induced by 10 nM BPA alone. This outcome provides support for an important role of the ERK pathway in BPA-mediated increases in histamine and CysLT release.
Lastly, this study examined the role of extracellular Ca2+ on the ability of BPA to alter mediator release, since cytoplasmic influx of Ca2+ is necessary for mast cells to induce exocytosis of granular contents (Kambayashi & Koretzky, Citation2007). Pre-treatment with the extracellular Ca2+ chelator EGTA reduced histamine and CysLT release from BMMC exposed to 10 nM BPA, suggesting a necessity for extracellular Ca2+ in the BPA-mediated response. Collectively, this study suggests that levels of BPA relevant to human exposure activate BMMC to increase histamine and CysLT release, and do so in an ERK-dependent and Ca2+-dependent manner.
While other studies have demonstrated that BPA can enhance histamine release from HMC-1 (a human mast cell line) and RBL-2H3 (a rat mast cell line) cells, our report is the first, to our knowledge, to demonstrate increased histamine (and CysLT) release following BPA exposure in BMMC. Observations from the current study are in agreement with other reports indicating that physiologic levels of E2 (Jensen et al., Citation2010; Zaitsu et al., Citation2007) and progesterone (Jensen et al., Citation2010) can enhance the release of β-hexosaminidase (β-hex; a degranulatory enzyme) and LTC4 from mast cells. Additionally, treatment of mast cells with other synthetic xenoestrogens including endosulfan, dieldrin, dichloro-diphenyl-dichloroethylene (DDE), nonyl-phenol, Aroclor 1242, Aroclor 1254, di-(2-ethylhexyl) phthalate, and 4-tert-octylphenol (Kennedy et al., Citation2012; Lee et al., Citation2011; Narita et al., Citation2007), at concentrations comparable to those used in the current study, increases β-hex release.
Previous studies examining the effect of BPA exposure on mast cell mediator release have done so using a BPA concentration above the range of human exposure (Lee & Lim, Citation2010; Lee et al., Citation2012; Park & Lim, Citation2010; Shim & Lim, Citation2009). Shim & Lim (Citation2009) demonstrated that treatment of HMC-1 cells with 50 μM BPA resulted in increased histamine, IL-4, and interferon (IFN)-γ release and up-regulated phosphorylation of ERK1/2, while Lee & Lim (Citation2010) reported increased release of IL-6 and TNFα under the same conditions. Increased release of histamine, IL-4, and IFNγ following treatment with the same BPA concentration was also reported in RBL-2H3 cells (Park and Lim, Citation2010). Lee et al. (Citation2012) reported increased release of IL-6 and TNFα and increased phosphorylation of ERK1/2 in RBL-2H3 cells following treatment with 50 μM BPA, and also reported increased in vivo levels of β-hex and histamine in BALB/c mice dosed with 5 mg/kg/day for 4 weeks. Thus, in addition to histamine and CysLTs, BPA is capable of enhancing the release of multiple other pro-inflammatory mediators from mast cells, all of which will contribute to more robust allergic responses.
The requirement of ERα for E2-induced mast cell release of mediators has been clearly demonstrated by Zaitsu et al. (Citation2007). Pre-treatment with the ER antagonist tamoxifen followed by E2 treatment in both RBL-2H3 cells and HMC-1 cells resulted in diminished β-hex and LTC4 release compared to E2 alone (Zaitsu et al., Citation2007). In addition, BMMC from ERα knockout (KO) mice did not exhibit up-regulated β-hex or LTC4 release displayed by BMMC from wild-type (WT) mice when treated with various concentrations of E2 (Zaitsu et al., Citation2007). However, the requirement of ERα for enhanced mediator release induced by synthetic xenoestrogens, including BPA, remains unclear. Of the synthetic xenoestrogens examined by Narita et al. (Citation2007), one displayed no difference in β-hex release between BMMC of WT mice and ERα KO mice (Arocolr 1242), while another displayed a difference at only one particular dose (Arocolr 1252, 1 nM). Interestingly, β-hex release from mast cells following treatment with DDE, dieldrin, and nonylphenol resulted in a U-shaped dose response curve, indicating that ERα was required at low and high concentrations of the xenoestrogens, but not at mid-range doses (Narita et al., Citation2007). One synthetic xenoestrogen even displayed significantly greater β-hex release in the ERα KO BMMC compared to WT BMMC at one particular dose (endosulfan, 1 nM) (Narita et al., Citation2007).
Considering the variability in requirement of ERα for synthetic xenoestrogens to stimulate mast cells (Narita et al., Citation2007), it was not surprising that we report no effect on ERα antagonism with regard to histamine and CysLT release following treatment with 10 nM BPA. We suspect that BPA-mediated mast cell mediator release may be explained by interactions with non-classical ERs, especially GPER. At doses relevant to human exposure, BPA has been demonstrated to interact with GPER (Bouskine et al., Citation2009; Sheng & Zhu, Citation2011). Furthermore, Dong et al. (Citation2011) reported that BPA induced activation of ERK in ERα/β-negative breast cancer cell lines, and that siRNA blocking GPER inhibited BPA-induced ERK activation.
The requirement of ERK activation and Ca2+ influx for mast cell activation and degranulation is well understood. Allergen-mediated mast cell activation occurs when IgE molecules bound to high-affinity Fcε receptors (FcεRI) on a mast cell surface cross-link with a single ligand. Downstream signaling rapidly induces ERK1/2 phosphorylation, thereby helping to mediate mast cell activation by initiating synthesis of lipid mediators (Kambayashi & Koretzky, Citation2007). Similarly, IgE cross-linking and subsequent signaling result in a critically important influx of Ca2+ that mediates cellular degranulation (Kambayashi & Koretzky, Citation2007). Our findings that mast cell mediator release enhanced by BPA is blocked by ERK inhibition and Ca2+ influx inhibition suggest that BPA relies on these critical mechanisms to stimulate mast cells.
Conclusions
The finding that BPA, at levels relevant to human exposure, can enhance mast cell release of histamine and CysLTs suggests that exposure to this endocrine-disrupting chemical can exacerbate symptoms associated with allergen-mediated inflammation. Due to the current environmental situation where humans are regularly exposed to multiple synthetic estrogens (Latini et al., Citation2010; Phillips & Foster, Citation2008; Yang et al., Citation2006), it will be important for future studies to examine the effect of simultaneous exposures to multiple xenoestrogens on mast cell activation and function in vitro and on allergic inflammatory diseases, such as asthma, in vivo.
Declaration of interest
This work was supported by the NIH grants HL077417 (PM) and ES017524 (DCD), the Flight Attendant Medical Research Institute award CIA-103071 (PM), and the University of Michigan National Institutes of Environmental Health Sciences (NIEHS) Core Center (P30 ES017885). Support for EO was provided by Institutional Training Grant T32 ES007062.
References
- Amin, K. 2012. The role of mast cells in allergic inflammation. Resp. Med. 106:9–14
- Anandan, C., Nurmatov, U., van Schayck, O. C., and Sheikh, A. 2010. Is the prevalence of asthma declining? Systematic review of epidemiological studies. Allergy 65:152–167
- Asher, M. I. 2010. Recent perspectives on global epidemiology of asthma in childhood. Allergol. Immunopathol. 38:83–87
- Barr, R. G., Wentowski, C. C., Grodstein, F., et al. 2004. Prospective study of postmenopausal hormone use and newly diagnosed asthma and chronic obstructive pulmonary disease. Arch. Intern. Med. 164:379–386
- Bauer, S. M., Roy, A., Emo, J., et al. 2012. The effects of maternal exposure to bisphenol A on allergic lung inflammation into adulthood. Toxicol. Sci. 130:82–93
- Bonds, R. S., and Midoro-Horiuti, T. 2013. Estrogen effects in allergy and asthma. Curr. Opin. Allergy Clin. Immunol. 13:92–99
- Bouskine, A., Nebout, M., Brucker-Davis, F., et al. 2009. Low doses of bisphenol A promote human seminoma cell proliferation by activating PKA and PKG via a membrane G-protein–coupled estrogen receptor. Environ. Health Perspect. 117:1053–1058
- Boyce, J. A. 2003. The role of mast cells in asthma. Prostagland. Leukotr. Essen. Fatty Acids 69:195–205
- Bradding, P., Walls, A. F., and Holgate, S. T. 2006. Role of the mast cell in the pathophysiology of asthma. J. Allergy Clin. Immunol. 117:1277–1284
- Calafat, A. M., Kuklenyik, Z., Reidy, J. A., et al. 2005. Urinary concentrations of bisphenol A and 4-nonylphenol in a human reference population. Environ. Health Perspect. 113:391–395
- Crinnion, W. J. 2012. Do environmental toxicants contribute to allergy and asthma? Alt. Med. Rev. 17:6–18
- Dong, S., Terasaka, S., and Kiyama, R. 2011. Bisphenol A induces a rapid activation of ERK1/2 through GPR30 in human breast cancer cells. Environ. Pollut. 159:212–218
- Donohue, K. M., Miller, R. L., Perzanowski, M. S., et al. 2013. Prenatal and post-natal bisphenol A exposure and asthma development among inner-city children. J. Allergy Clin. Immunol. 131:736–742
- Dratva, J. 2010. Use of estrogen only hormone replacement therapy associated with increased risk of asthma onset in post-menopausal women. Evid. Based Med. 15:190–191
- Golub, M. S., Wu, K. L., Kaufman, F. L., et al. 2010. Bisphenol A: Developmental toxicity from early prenatal exposure. Birth Defects Res. B Dev. Reprod. Toxicol. 89:441–466
- Holgate, S. T., Davies, D. E., Powel, R. M., et al. 2007. Local genetic and environmental factors in asthma disease pathogenesis: Chronicity and persistence mechanisms. Eur. Respir. J. 29:793–803
- Jensen, F., Woudwyk, M., Teles, A., et al. 2010. Estradiol and progesterone regulate the migration of mast cells from periphery to the uterus and induce their maturation and degranulation. PLoS One 5:e14409
- Kambayashi, T., and Koretzky, G. A. 2007. Proximal signalling events in FcεRI-mediated mast cell activation. J. Allergy Clin. Immunol. 119:544–552
- Kennedy, R. H., Pelletier, J. H., Tupper, E. J., et al. 2012. Estrogen mimetic 4-tert-octylphenol enhances IgE-mediated degranulation of Rbl-2H3 mast cells. J. Toxicol. Environ. Health 75:1451–1455
- Lai, C. K., Beasley, R., Crane, J., et al. 2009. Global variation in the prevalence and severity of asthma symptoms: Phase three of the International Study of Asthma and Allergies in Childhood (ISAAC). Thorax 64:467–483
- Latini, G., Knipp, G., Mantovani, A., et al. 2010. Endocrine disruptors and human health. Mini Rev. Med. Chem. 10:846–855
- Lee, J., and Lim, K. T. 2010. Plant-originated glycoprotein (36 kDa) suppresses IL-4 and -10 in bisphenol A-stimulated primary cultured mouse lymphocytes. Drug Chem. Toxicol. 33:421–429
- Lee, J., Oh, P. S., and Lim, K. T. 2011. Allergy-related cytokines (LI-4 and TNFα) are induced by di-(2-ethylhexyl) phthalate and attenuated by plant-originated glycoprotein (75 kDa) in HMC-1 cells. Env. Toxicol. 26:364–372
- Lee, J., Lee, S. J., and Lim, K. T. 2012. CTB glycoprotein (75kDa) inhibits IgE releasing, TNFα and IL-6 expressed by bisphenol A in vivo and in vitro. Food Chem. Toxicol. 50:2109–2117
- Levin, E. R. 2011. Minireview: Extranuclear steroid receptors: Roles in modulation of cell functions. Mol. Endocrinol. 25:377–384
- Leynaert, B., Sunyer, J., Carcia-Esteban, R., et al. 2012. Gender differences in prevealnce, diagnosis and incidence of allergic and non-allergic asthma: A population-based cohort. Thorax 67:625–631
- Masoli, M., Fabian, D., Holt, S., and Beasley, R. 2004. The global burden of asthma: Executive Summary of the GINA Dissemination Committee report. Allergy 59:469–478
- Matsushima, A., Kakuta, Y., Teramoto, T., et al. 2007. Structural evidence for endocrine disruptor bisphenol A binding to human nuclear receptor ERRγ. J. Biochem. 142:517–524
- Midoro-Horiuti, T., Tiwari, R., Watson, C. S., and Goldblum, R. M. 2010. Maternal bisphenol A exposure promotes the development of experimental asthma in mouse pups. Environ. Health Perspect. 118:273–277
- Murdoch, J. R., and Lloyd, C. M. 2010. Chronic inflammation and asthma. Mutat. Res. 690:24–39
- Nakajima, Y., Goldblum, R. M., and Midoro-Horiuti, T. 2012. Fetal exposure to bisphenol A as a risk factor for the development of childhood asthma: An animal model study. Environ. Health. 11:1–7
- Narita, S., Goldblum, R. M., Watson, C. S., et al. 2007. Environmental estrogens induce mast cell degranulation and enhance IgE-mediated release of allergic mediators. Environ. Health Perspect. 115:48–52
- Park, C. H., and Lim, K. T. 2010. Phytoglycoprotein (75 kDa) suppresses release of histamine and expression of IL-4 and IFNγ in BPA-treated RBL-2H3 cells. Immunol. Invest. 39:141–185
- Phillips, K. P., and Foster, W. G. 2008. Endocrine toxicants with emphasis on human health risks. J. Toxicol. Environ. Health B. Crit. Rev. 11:149–151
- Rubin, B. S. 2011. Bisphenol A: An endocrine disruptor with widespread exposure and multiple effects. J. Steroid Biochem. Mol. Biol. 127:27–34
- Sheng, Z. G., and Zhu, B. Z. 2011. Low concentrations of bisphenol A induce mouse spermatogonial cell proliferation by G protein-coupled receptor 30 and estrogen receptor-α. Environ. Health Perspect. 119:1775–1780
- Shim, J. U., and Lim, K. T. 2009. Inhibitory effect of glycoprotein isolated from Cudrania tricuspidata bureau on expression of inflammation-related cytokine in bisphenol A-treated HMC-1 cells. Inflammation 32:211–217
- Spanier, A. J., Kahn, R. S., Kunselman, A. R., et al. 2012. Prenatal exposure to bisphenol A and child wheeze from birth to 3 years of age. Environ. Health Perspect. 120:916–920
- Thornton, J., Lewis, J., Lebrun, C. M., and Licskai, C. J. 2012. Clinical characteristics of women with menstrual-linked asthma. Respir. Med. 106:1236–1243
- To, T., Stanojevic, S., Moores, G., et al. 2012. Global asthma prevalence in adults: Findings from the cross-sectional world health survey. BMC Public Health 12:204–211
- Vaidya, S. V., and Kulkarni, H. 2012. Association of urinary bisphenol A concentration with allergic asthma: Results from the National Health and Nutrition Examination Survey 2005–2006. J. Asthma. 49:800–806
- Vandenberg, L. N., Chahoud, I., Heindel, J. J., et al. 2012. Urinary, circulating, and tissue biomonitoring studies indicate widespread exposure to bisphenol A. Cien. Saude Colet. 17:407–434
- Vandenberg, L. N., Hauser, R., Marcus, M., et al. 2007. Human exposure to bisphenol A (BPA). Reprod. Toxicol. 24:139–177
- Vink, N. M., Postma, D. S., Schouten, J. P., et al. 2010. Gender differences in asthma development and remission during transition through puberty: The TRacking Adolescents’ Individual Lives Survey (TRAILS) study. J. Allergy Clin. Immunol. 126:498–504
- Vrieze, A., Postma, D. S., and Kerstjens, H. A. 2003. Perimenstrual asthma: A syndrome without known cause or cure. J. Allergy Clin. Immunol. 112:271–282
- World Health Organization (WHO). 2007. Global surveillance, prevention, and control of chronic respiratory diseases: A comprehensive approach. WHO: Geneva
- Yang, M., Park, M. S., and Lee, H. S. 2006. Endocrine disrupting chemicals: Human exposure and health risks. J. Environ. Sci. Health C. Environ. Carcinog. Ecotoxicol. Rev. 24:183–224
- Zaitsu, M., Narita, S., Lambert, K. C., et al. 2007. Estradiol activates mast cells via a non-genomic estrogen receptor-alpha and calcium influx. Mol. Immunol. 44:1977–1985
- Zhao, Z. Z., Sugerman, P. B., Walsh, L. J., and Savage. N. W. 2001. A fluorometric microassay for histamine release from human gingival mast cells. J. Periodontal Res. 36:233–236
- Zoeller, R. T., Bansal, R., and Parris, C. 2005. Bisphenol A, an environmental contaminant that acts as a thyroid hormone receptor antagonist in vitro, increases serum thyroxine, and alters RC3/neurogranin expression in the developing rat brain. Endocrinology 146:607–612