
Abstract
Isoniazid (INH) is associated with one of the highest incidences of idiosyncratic drug-induced liver failure of any commonly prescribed drug. The mechanism of this liver injury remains uncertain, and a valid animal model would greatly facilitate mechanistic studies. Most studies of INH-induced liver toxicity have been acute studies performed in rats with high doses of the drug, and this is very different from the idiosyncratic liver injury that occurs in humans. It has previously been demonstrated that covalent binding of INH in the liver of mice is greater than in rats and more like that in humans. Therefore, mice should be a better species in which to develop an animal model of INH-induced liver injury. Treatment of Cbl-b−/− and PD1−/− mice, which have impaired immune tolerance, resulted in greater injury than their C57BL/6 background, but not liver failure. This suggested that the injury was mediated by the adaptive immune system; however, Rag−/− mice, which do not have competent T- and B-cells, sustained more liver injury than C57BL/6 wild-type mice. This suggested that the adaptive immune system also played a protective role. INH treatment also led to a decrease in the inflammatory cytokines IL-1α and IL-12, which suggests that the drug may have immunosuppressive properties. In short, a mouse model was developed of INH-induced liver injury in which the immune system appears to play a both protective and pathogenic role, but this study was unable to develop a model of INH-induced liver failure.
Introduction
Isoniazid (INH) remains a first line drug for the treatment of tuberculosis, even though it is probably associated with the highest risk of idiosyncratic liver failure of any commonly prescribed drug (Centers for Disease Control and Prevention, 2010; Maddrey & Boitnott, Citation1973). The mechanism of INH-induced hepatotoxicity remains uncertain. Mechanistic studies are difficult to perform because it is important to obtain samples before the injury is severe so that the events leading up to the injury can be studied, but it is impossible to predict which patient will develop serious toxicity. In addition, it has not been possible to reproduce liver injury in animals with the same characteristics as the idiosyncratic injury that occurs in patients. Earlier experiments performed in rats found that acetylhydrazine, a metabolite of INH, is responsible for the hepatotoxicity (Mitchell et al., Citation1976). However, this was an acute model of hepatotoxicity where rats were treated with very high doses of INH and developed liver injury within hours. In humans, INH-induced liver injury has almost always a delayed onset of at least a week and characteristics of fever, rash, and eosinophilic infiltrate into the liver (Black et al., Citation1975; Maddrey & Boitnott, Citation1973). It is unlikely that the rat model represents the same mechanism of INH-induced hepatotoxicity that occurs in humans because the characteristics are very different. Hydrazine has also been implicated as a potential hepatotoxic metabolite of INH (Blair et al., Citation1985; Gent et al., Citation1992; Noda et al., Citation1983; Sarich et al., Citation1996); however, given its potent hepatotoxic effects it is more likely that hydrazine would cause acute liver injury rather than liver injury with a delayed onset.
More recently we have shown that INH itself can be bioactivated and covalently bind to liver macromolecules in mice in vivo and to human liver microsomes in vitro; covalent binding also occurred in rats, but it was much less than in mice (Metushi et al., Citation2012). In particular, the metabolism of INH and covalent binding in human hepatic microsomes is closer to mice than rats (Metushi et al., Citation2012). The covalent binding of INH in the liver of mice appears to mimic human slow acetylators (Metushi et al., Citation2012), who are at increased risk of developing INH-induced liver injury (Huang et al., Citation2002).
There are multiple lines of evidence that INH-induced liver injury involves an immune response against INH-modified proteins. Specifically, T-cells from patients with mild INH-induced liver injury proliferate when incubated with INH-modified proteins, while T-cells from patients with severe INH-induced liver injury also proliferate when incubated with INH alone (Warrington et al., Citation1978, Citation1982). In addition, most patients with INH-induced liver injury have antibodies against either INH-modified proteins or the cytochromes P450 that bioactivate INH (Metushi et al., Citation2013). Taken together, these data suggest that INH-induced liver injury is immune-mediated, as reviewed elsewhere (Metushi et al., Citation2011), and mice are more likely to be a model for INH-induced liver injury in humans than rats. Therefore, we set out to develop an animal model of INH-induced liver injury in mice.
Materials and methods
Animals
All mice were between 6–8 weeks-of-age. Inbred mice (Balb/c AnNCrl, C57BL/6NCrl, or C3H/HeNCrl) were purchased from Charles River Laboratories (Montreal, QC, Canada). All mice were housed in a specific pathogen-free environment maintained at 22 °C with a 30–70% relative humidity and a 12-hour light/dark cycle. All mice had ad libitum access to filtered water. Upon arrival, all mice were allowed to acclimatize for 1 week before treatment. All animal protocols used in this study were approved by the University of Toronto Animal Care Committee (and conducted in an animal facility accredited by the Canadian Council on Animal Care).
Cbl-b−/− mice are on a C57BL/6 background and were bred in house with the permission of the developer, Dr J. Penninger, at the Institute of Molecular Biotechnology of the Austrian Academy of Science, Vienna. PD1−/− mice are also on a C57BL/6 background and were bred in house with the permission of the developer, Dr H. Honjo at the Kyoto University Graduate School of Medicine. T- and B-cell immunodeficient Rag−/− (Rag1tm1Mom/J) mice were purchased from Jackson Laboratories (Bar Harbor, ME).
Treatment
INH (Sigma; Oakville, ON) was ground to fine powder, thoroughly mixed with food, and given to rodents at a dose of 0.1%, 0.15%, 0.2%, or 0.4% by weight (w/w) in food. Food was provided to the animals (four mice/cage) in small jars ad libitum, and the average amount consumed was measured. This resulted in an INH dose of 150–450 mg/kg/day depending on the percentage dose of INH and the amount of daily food consumption by mice (). The most common dose of INH in food was 0.2% [w/w] and this resulted in blood levels anywhere from 2–6 µg/µl that is similar to the Cmax of INH in humans (Fukino et al., Citation2008). Pyridoxine hydrochloride (MP Biomedicals, Solon, OH) was also mixed in food; given at 0.05% by weight in food, this resulted in a dose of ≈50–90 mg/kg/day. Rifampicin (RMP; Sigma) was suspended in saline and given by gavage to female Cbl-b−/− mice at 50 mg/kg/day; the vehicle, saline, was given to control mice.
Table 1. Food consumption by mice.
Attempts to deplete T-regulatory (Treg) cells were carried out as previously described (Onizuka et al., Citation1999; Quintana et al., Citation2008). Briefly, Cbl-b−/− mice were injected intraperitoneally with 1 µg/mouse of 6 formylindolo-(3,2-b)carbazole (FICZ; Enzo Life Sciences, Brockville, ON) or with 0.25 mg/mouse anti-CD25 antibody (BioXcell, West Lebanon, NH). INH was started 2 days after the first injection of FICZ or anti-CD25 antibody; one additional subsequent injection with FICZ or anti-CD25 antibody were repeated 1 week after the first injection.
Biochemical measures of liver injury
Liver enzyme activities were measured by collecting blood from the saphenous vein at baseline and every week during the course of the exposures, unless otherwise stated. As biomarkers of liver injury, the activity of alanine aminotransferase (ALT, Thermo Scientific, Middletown, VA) and sorbitol dehydrogenase (SDH, Catachem, Oxford, CT) were measured as described by the manufacturer. Given the limited amount of blood that can be drawn from mice at each timepoint, the method for glutamate dehydrogenase (GLDH, Randox, Crumlin, UK) was slightly modified wherein 200 µl Reagent 1 was pre-mixed with 8 µl Reagent 2, and 200 µl of this mixture was loaded into wells of a 96-well plate; to this was added either 10 or 20 µl serum. All reagents were reconstituted as per manufacturer specification, and the absorbance was monitored for at least 5 min at 25 °C as described in the kit.
Correlation of GLDH/SDH assay with ALT
Because INH reacts with and depletes pyridoxal 5′-phosphate (a co-factor in ALT assay) and given the limited amount of blood that can be drawn from mice, GLDH and SDH assays were used and optimized to decrease the amount of serum required for assessing liver injury. Amodiaquine hydrochloride (AQ) was provided from IPCA Laboratories (Mumbai, India) and was also used as a positive control of liver injury and given to female Cbl-b−/− mice (18–20 g of weight) at 0.15% or 0.20% [w/w] in food. This resulted in an amodiaquine dose of 200–300 mg/kg/day. Upon AQ treatment the activities for ALT, SDH, and GLDH were increased compared to controls (Supplemental Figures S1A–C); this was also consistent with H&E staining, which showed infiltration of lymphocytes and foamy hepatocytes (Supplemental Figures S1D and E). By using 10 µl serum, SDH correlated well with ALT levels, and by modifying the GLDH assay, a good correlation with ALT was also achieved (R2 = 0.74 and 0.82, respectively; Supplemental Figures S1F and G). Correlation of SDH with GLDH gave an R2 = 0.71. As expected, the modification of the GLDH and SDH assays also correlated well with ALT when 20 µl serum was used (Supplemental Figure S2). In addition, a comparison of the fold-difference in enzyme activities between control and mice treated with amodiaquine found the highest fold difference was for ALT (5.6-fold higher than control) followed by GLDH (2.1-fold) and SDH (1.7-fold).
Blood level measurements of INH
Eighty microliters of methanol containing 4-dimethylaminoantipyrene (Sigma) was added to 10 µl plasma as the internal standard to precipitate the protein. The mixture was vortexed and placed at −20 °C for 30 min; after centrifugation (11,000 x g, 10 min), the supernatant was dried under a N2 stream. The samples were then reconstituted with the initial mobile phase, and 20 µl of sample was injected onto a 150 × 2 mm Gemini 5-µm C18 110 Å column (Phenomenex, Torrance, CA) with an isocratic mobile phase containing 10% methanol in water-ammonium acetate (10 mM, pH 4.0) at a flow of 0.2 ml/min. Total run time was 5 min. The outlet from the HPLC was connected to an API3000 mass spectrometer (PE Sciex, Concord, ON). Metabolites were detected using an LC-MS system (Shimadzu, Columbia, MD); optimizations for multi-reaction monitoring were performed using synthetic standards (Sigma) for INH (Q1/Q3: 138/79) and 4-dimethylaminoantipyrene (Q1/Q3: 232.12/56.1).
Western blotting
Western blotting to detect covalent binding of INH was performed using isolated liver samples. Mice were euthanized by CO2 asphyxiation at the various timepoints (as specified in and and Supplemental Figure S10). Thereafter, one portion of the liver was isolated for Western analysis; another portion was isolated for histologic analysis (see below). Liver S9 fraction (liver homogenate after 9000 × g centrifugation) was prepared in the presence of protease inhibitors (Sigma) as previously described (Metushi et al., Citation2012). The concentration of protein in each sample was then measured using a bicinchoninic acid (BCA) kit (Fisher Scientific, Ottawa, ON) and 20 µg protein/lane was loaded into an 8% SDS-PAGE gel. SuperSignal enhanced molecular weight marker (Fisher Scientific) was loaded onto one of the lanes. The proteins were then resolved by electrophoresis and electrotransferred onto a nitrocellulose membrane (BioRad, Mississauga, ON). Each Western blot was repeated at least twice. Rabbit anti-INH primary antibody was developed as previously described (Metushi et al., Citation2012), detected by goat anti-rabbit IgG-peroxidase (Sigma). Bound peroxidase was visualized by using Supersignal West Pico Chemiluminescent Substrate (Fisher Scientific). Mouse monoclonal anti-GAPDH (Sigma) was used as a loading control and detected with goat anti-mouse IgG-peroxidase (Jackson ImmunoResearch; West Grove, PA).
Figure 1. GLDH activities, body weights, and INH covalent binding in INH-treated Balb/c mice. (a) GLDH in male mice treated at 0.10 or 0.15% INH [w/w] in food for 5 weeks. (b) GLDH in female mice treated at 0.10 or 0.15% of INH [w/w] in food for 3 weeks or treated with 0.2% of INH + 0.05% pyridoxine•HCl (Pyr) [w/w] in food for 2 weeks. (c, d) Body weights of treated mice. (e, f) Covalent binding of INH in livers of male and female mice. Values represent mean (±SE) of four mice/group. Analyzed for statistical significance by a Mann-Whitney U test; *p < 0.05.
![Figure 1. GLDH activities, body weights, and INH covalent binding in INH-treated Balb/c mice. (a) GLDH in male mice treated at 0.10 or 0.15% INH [w/w] in food for 5 weeks. (b) GLDH in female mice treated at 0.10 or 0.15% of INH [w/w] in food for 3 weeks or treated with 0.2% of INH + 0.05% pyridoxine•HCl (Pyr) [w/w] in food for 2 weeks. (c, d) Body weights of treated mice. (e, f) Covalent binding of INH in livers of male and female mice. Values represent mean (±SE) of four mice/group. Analyzed for statistical significance by a Mann-Whitney U test; *p < 0.05.](/cms/asset/fea7a25f-ea24-48b4-8622-d700688c907f/iimt_a_860644_f0001_b.jpg)
Figure 2. Comparison of INH covalent binding in livers of different mice. (A) Male Cbl-b−/− mice treated with INH (0.2% or 0.2% + 0.05% pyridoxine•HCl [w/w] in food, n = 3). (B) Female (n = 4) and male (n = 3) Cbl-b−/− mice treated with 0.2% INH [w/w] in food. (C) Female C57BL/6 and Cbl-b−/− mice treated with 0.2% INH [w/w] in food (n = 4). (D) Female Cbl-b−/− control (n = 2), Cbl-b−/− (n = 4), or C3H mice (n = 3) treated with 0.2% INH [w/w] in food.
![Figure 2. Comparison of INH covalent binding in livers of different mice. (A) Male Cbl-b−/− mice treated with INH (0.2% or 0.2% + 0.05% pyridoxine•HCl [w/w] in food, n = 3). (B) Female (n = 4) and male (n = 3) Cbl-b−/− mice treated with 0.2% INH [w/w] in food. (C) Female C57BL/6 and Cbl-b−/− mice treated with 0.2% INH [w/w] in food (n = 4). (D) Female Cbl-b−/− control (n = 2), Cbl-b−/− (n = 4), or C3H mice (n = 3) treated with 0.2% INH [w/w] in food.](/cms/asset/41680d35-b21e-4562-903d-039a9d79aaf4/iimt_a_860644_f0002_b.jpg)
Histology
Isolated liver samples (see above) were perfused, extracted, placed in 10% neutral buffered formalin solution (Sigma) overnight, and then embedded in paraffin. For preparation of frozen sections, the liver tissue was placed in OCT medium (VWR International; Radnor, PA) and immediately frozen using liquid N2. Thereafter, the paraffin-embedded and frozen materials were sectioned and stained (H&E or as outlined below) by the Department of Pathology at the Hospital for Sick Children (University of Toronto).
Immunohistochemical analysis
Rat monoclonal primary antibodies against mouse CD11b (clone M1/70), F4/80 (clone CI:A3-1), and CD45R (clone RA3-6B2) were purchased from Abcam (Cambridge, MA). Rabbit polyclonal antibody against mouse KI67 was also from Abcam. Monoclonal antibodies against mouse CD4 (clone GK 1.5) and CD8 (clone YTS169) were donated by Pamela Ohashi’s laboratory (Princess Margaret Hospital, University of Toronto). The proliferating cell nuclear antigen (PCNA) kit was purchased from Invitrogen (Camarillo, CA). Polyclonal rabbit secondary antibody anti-rat IgG-biotinylated and streptavidin-peroxidase were purchased from Dako (Burlington, ON). Goat anti-rabbit IgG-peroxidase was purchased from Sigma. Each experiment was repeated at least twice, and the signal was developed using 3,3′-diamino-benzidine for paraffin-embedded slides or NovaRed for frozen slides (Vector; Burlington, ON), and Mayer’s hematoxylin (Sigma) was used as the counterstain. Paraffin-embedded slides were stained with antibodies against F4/80, CD45R, or KI-67. Antibodies against CD11b, CD4, and CD8 were used to stain frozen sections. Immunohistochemical grading was blinded and done by counting the number of cells per field of view under a microscope; at least two slices (3–6 mm2) were mounted on glass slides and five areas from each slice were counted under the microscope.
Serum cytokines
Female Cbl-b−/− mice were treated with INH (0.2% by w/w in food) for 5 weeks. At the end of this period, mice were euthanized by CO2 asphyxiation; blood was collected by cardiac puncture and allowed to clot for 30 min at room temperature before being centrifuged (2000 x g, 10 min, 4 °C) to isolate the serum. Levels of IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-17A, eotaxin, GCSF, GMCSF, IFNγ, KC, MCP-1 (MCAF), MIP-1α, MIP-1β, RANTES, and TNFα in 25 µl serum/mouse were measured using a BioRad BioPlex Pro Mouse Cytokine 23plex.
Statistical analysis
Statistical analyses were performed using GraphPad Prism (GraphPad Software, San Diego, CA). Data were analyzed using a two-way analysis of variance (ANOVA), one-way ANOVA (Kruskal-Wallis test), or Mann-Whitney U test. A p value <0.05 was considered significant (*p < 0.05; **p < 0.01; ***p < 0.001).
Results
Treatment of Balb/c mice with INH
We recently reported that treatment of Balb/c mice with 0.2% of INH [w/w] in food resulted in mild increases in GLDH activity (Metushi et al., Citation2012); however, the mice could not be treated for longer than 3 weeks because of weight loss. To develop an animal model of chronic liver injury similar to that observed in humans, Balb/c mice were treated with lower doses of INH in order to keep treating them for longer and prevent severe weight loss. Treatment of male Balb/c mice with 0.1% or 0.15% of INH [w/w] in food for up to 5 weeks produced mild increases in GLDH without severe weight loss (). Covalent binding appeared to be a little greater in male Balb/c mice that received the higher dose (0.15% of INH; ; ). However, liver histology (H&E) of male Balb/c mice at the end of 5 weeks did not reveal any pathology (data not shown). Treatment of female Balb/c mice with 0.1% and 0.15% of INH by weight in food also resulted in slight elevations in GLDH levels (). Pyridoxine hydrochloride was added to the group of mice treated with the highest dose of INH (0.2% INH [w/w] in food) as a supplement to protect from the depletion of Vitamin B6 and to prevent weight loss. This approach is similar to what was used to prevent extensive weight loss in rabbits (Whitehouse et al., Citation1983), and it is consistent with the B6 supplement that is given to patients undergoing INH therapy. However, despite administration of pyridoxine, mice treated with 0.2% INH lost significant body weight and could not be maintained for more than 2 weeks (). Covalent binding of INH in female Balb/c mice revealed no difference between the groups (), presumably because, although there was an increase in concentration of INH in the food, these mice also ate less food, making the total INH intake similar between the groups (). Liver histology (H&E) of female Balb/c mice was normal (data not shown).
Treatment of Cbl-b−/−, PD1−/−, and Rag−/− mice with INH
Treatment of C57BL/6 mice with INH did not cause liver injury (Metushi et al., Citation2012). If the lack of injury is due to immune tolerance, it is possible that treatment of Cbl-b−/− mice that lack an E3 ubiquitin ligase, which is a molecule important for regulating immune tolerance (Venuprasad, Citation2010), would be more susceptible to liver injury. Male Cbl-b−/− mice treated with 0.2% INH [w/w] in food had mild elevations in GLDH, which returned to normal despite continued treatment (). We tried to prevent the increase in GLDH by also adding pyridoxine hydrochloride, but no significant difference from the INH-only group was observed. Male Cbl-b−/− mice did not eat as much food as the controls (), but their body weight was stable for 5 weeks (). Female Cbl-b−/− mice also had a mild increase in GLDH activity and could be maintained for up to 5 weeks when treated with 0.2% INH [w/w] in their food (). The blood level of INH appeared slightly higher in female mice than males, but the difference was not statistically significant (Supplementary Figure S3), and it is similar to the peak therapeutic concentration of INH in humans (Fukino et al., Citation2008). Food consumption in female Cbl-b−/− mice was also less than in the control animals, but it was sufficient to maintain stable body weight ().
Figure 3. GLDH activities and body weights in INH-treated Cbl-b−/− mice. (A) GLDH in male mice treated with 0.2% INH [w/w] in food or 0.2% INH + 0.05% pyridoxine•HCl (INH + Pyr) [w/w] in food for 5 weeks. (B) GLDH in female mice treated with INH at 0.2% [w/w] in food for 5 weeks. (C, D) Body weights of male and female INH-treated mice. Values represent mean (±SE) from four mice/group. Analyzed for statistical significance by two-way ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001.
![Figure 3. GLDH activities and body weights in INH-treated Cbl-b−/− mice. (A) GLDH in male mice treated with 0.2% INH [w/w] in food or 0.2% INH + 0.05% pyridoxine•HCl (INH + Pyr) [w/w] in food for 5 weeks. (B) GLDH in female mice treated with INH at 0.2% [w/w] in food for 5 weeks. (C, D) Body weights of male and female INH-treated mice. Values represent mean (±SE) from four mice/group. Analyzed for statistical significance by two-way ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001.](/cms/asset/69cfd049-38cc-4531-b6e7-b7e74dad858a/iimt_a_860644_f0003_b.jpg)
The liver histology of one female Cbl-b−/− mouse showed an infiltration of lymphocytes, focal necrosis, steatosis, and cholestasis, as shown in . This mouse also had an increase in cells staining positive for F4/80, CD45R, and PCNA (), which implies an immune response and liver cell regeneration. An increase in cells staining positive for CD45R and F4/80 was also observed in the spleen of the mouse that had abnormal liver histology (Supplementary Figure S4), but not in the other three mice treated with INH. Staining for F4/80 in the livers and spleens of C57BL/6 mice treated with 0.2% [w/w] INH in food showed no changes compared to controls (data not shown). A decrease in serum IL-12 (p70) and IL-1α was observed in female Cbl-b−/− mice treated with INH at the end of the 5 weeks (), other serum cytokines did not significantly change (Supplementary Figure S5). Treatment of C3H female mice with INH also produced a slight increase in GLDH, which returned to normal (Supplementary Figure S6); however, no abnormal histology (H&E) was observed (data not shown). Comparison of INH covalent binding revealed no difference between INH− and INH+ pyridoxine (INH + Pyr)-treated male Cbl-b−/− mice (). There was no difference in covalent binding between male and female Cbl-b−/− mice treated with INH (); this is similar to our previous results in C57BL/6 mice (Metushi et al., Citation2012). Also, no difference in covalent binding was observed between female Cbl-b−/− mice and female C57BL/6 or C3H mice (), suggesting that covalent binding is not the only determinant of liver injury.
Figure 4. H&E and immunohistochemical staining for anti-CD45R, F4/80, and PCNA in livers of Cbl-b−/− mice treated with INH (0.2% w/w in food) for 5 weeks. Control = untreated mice (n = 4); INH = mice treated with INH that did not develop abnormal liver histology (n = 3); INH steatosis = one mouse treated with INH that developed significant abnormal liver histology (n = 1). Red arrow = lymphocyte infiltration, yellow arrow = microvesicular steatosis, green arrow = macrovesicular steatosis, blue arrow = focal necrosis, orange arrow = cholestasis. 40× magnification for H&E; 20× magnification for CD45R, F4/80, and PCNA.
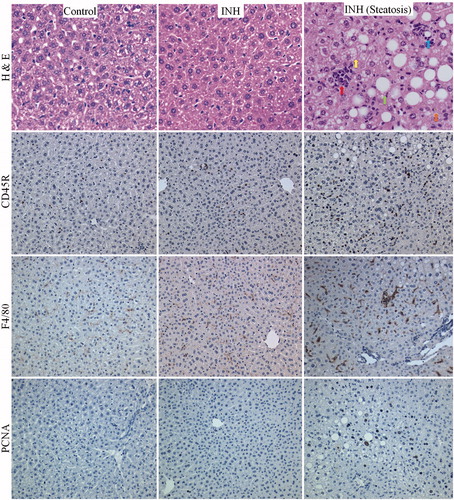
Figure 5. Serum concentrations of IL-1α and IL-12 (p70) in Cbl-b−/− control or mice treated with INH for 5 weeks. Values represent mean (±SE) from four mice/group. INH was given at 0.2% INH [w/w] in food. Analyzed for statistical significance by a Mann-Whitney U test; *p < 0.05.
![Figure 5. Serum concentrations of IL-1α and IL-12 (p70) in Cbl-b−/− control or mice treated with INH for 5 weeks. Values represent mean (±SE) from four mice/group. INH was given at 0.2% INH [w/w] in food. Analyzed for statistical significance by a Mann-Whitney U test; *p < 0.05.](/cms/asset/1e8fff1f-0140-4e55-a5fa-5f4012ef56a9/iimt_a_860644_f0005_b.jpg)
Treatment of female PD1−/− mice with INH also produced mild increases in GLDH at Week 5 (). However, these mice had significant weight loss after 1 week of treatment, and the dose of INH in food had to be reduced for 2 weeks (Supplementary Figure S7). We looked for an immune response in the liver by staining a variety of immune cell through immunohistochemistry. However, immunohistochemical staining in the livers of control or INH treated PD1−/− mice showed no difference in cells staining positive for KI67, F4/80, CD11b, CD4, CD8, or CD45R (Supplementary Figure S8). Only in the spleen was there a decrease in cell proliferation as determined by KI67 staining (Supplementary Figure S9). Treatment of Rag−/− mice, which are B- and T-cell deficient, for up to 12 weeks, resulted in consistently higher GLDH levels, but this injury did not become worse with continued treatment ().
Figure 6. GLDH activity in female PD−/− and Rag−/− mice treated with INH. (A) INH was given at 0.2% [w/w] in food until Week 2 wherein the dose was decreased to 0.1% [w/w] in food for 1 week (until Week 3) because of weight loss. Mice were then put back on 0.2% INH [w/w] from Week 3–5. (B) Female C57BL/6 control or Rag−/− mice were treated with INH at 0.2% [w/w] for up to 12 weeks. Values represent mean (±SE). Analyzed for statistical significance by two-way ANOVA. A p value <0.05 was considered significant (*p < 0.05; **p < 0.01; ***p < 0.001).
![Figure 6. GLDH activity in female PD−/− and Rag−/− mice treated with INH. (A) INH was given at 0.2% [w/w] in food until Week 2 wherein the dose was decreased to 0.1% [w/w] in food for 1 week (until Week 3) because of weight loss. Mice were then put back on 0.2% INH [w/w] from Week 3–5. (B) Female C57BL/6 control or Rag−/− mice were treated with INH at 0.2% [w/w] for up to 12 weeks. Values represent mean (±SE). Analyzed for statistical significance by two-way ANOVA. A p value <0.05 was considered significant (*p < 0.05; **p < 0.01; ***p < 0.001).](/cms/asset/234cb001-8a95-4551-b1bc-c1bdf04d84e2/iimt_a_860644_f0006_b.jpg)
Treatment of Cbl-b−/− mice with INH/RMP and depletion of Treg cells
Female Cbl-b−/− mice appeared to be the most promising strain in which to induce liver injury; therefore, we tried addition of rifampicin (RMP), which is an inducer of P450 in humans, to the INH treatment in order to increase hepatic covalent binding of INH. Treatment of female Cbl-b−/− mice with INH/RMP for up to 2 weeks resulted in slight elevations in GLDH and SDH, which seemed to be higher in the INH + RMP group, but the difference was not significant (Supplementary Figures S10a and b). Covalent binding was greater in the INH + RMP group (Supplementary Figure S10c), suggesting induction of drug metabolism, but the mice that were co-treated with INH + RMP, on average, also ate more INH-containing food (2.9 [±0.32] for the INH group vs 3.8 [±0.41] for the INH + RMP group; values represented as mean [±SE]; p > 0.05). Despite the increased covalent binding, the H&E slides of the liver indicated no evidence of damage in either treatment group.
Another strategy to break immune tolerance was to deplete Treg cells by treating Cbl-b−/− mice with anti-CD25 antibodies or FICZ. We repeated an injection schedule reported to deplete Treg cells, but no changes in liver histopathology were seen between control and the INH + FICZ or anti-CD25 antibodies co-treated group (data not shown). We also tried to give a higher INH dose to Cbl-b−/− mice (0.4% of INH [w/w] in food), but the drug had to be discontinued at the end of the second week because of extensive weight loss.
Discussion
The mechanism of idiosyncratic INH-induced liver injury in humans remains uncertain. As discussed in the Introduction, there are multiple lines of evidence that INH-induced liver injury in patients is immune-mediated, and a reactive metabolite of the parent drug is responsible (Metushi et al., Citation2011, Citation2012, Citation2013). Mice represent a better model for the metabolism of INH in humans than rats because blood levels and covalent binding of INH were higher in mice vs rats (Metushi et al., Citation2012); therefore, we attempted to develop an animal model of INH-induced liver injury in mice. In a previous study we did not find evidence of liver injury in male or female C57BL/6 mice; a small increase in GLDH was observed in male and female Balb/c mice, but at the dose utilized (300–350 mg/kg/day of INH given in food) the animals lost significant weight, and the treatment could not be sustained (Metushi et al., Citation2012). In this study, male Balb/c mice were treated with a lower dose of INH, which allowed mice to be treated with INH for longer. Although there was a significant increase in GLDH, which was greater in female mice (), there was no histological evidence of severe liver injury. With various treatments, the amount of covalent binding did not correlate with the increase in GLDH/liver injury. In particular, female Balb/c mice which were treated with 0.2% INH w/w in food had higher GLDH levels than female Balb/c mice treated with 0.1% INH w/w (), but the amount of covalent binding was the same (). Similarly, female Cbl-b−/− mice had similar INH covalent binding to male Cbl-b−/− mice, female C57BL/6 mice, and C3H mice (); yet, female Cbl-b−/− mice seemed to have the highest incidence and severity of liver injury ( and ). This data indicates a clear disconnect between INH covalent binding and liver pathology, which suggests that covalent binding of INH in the liver may be necessary in order to develop liver injury, but it is not the only factor determining liver pathology.
We have hypothesized that the most likely barrier to developing a valid animal model of idiosyncratic liver injury is the fact that the dominant immune response in the liver is immune tolerance; in particular, the liver has been referred to as the ‘lymphocyte graveyard’ (Crispe et al., Citation2000). The liver is the major site of xenobiotic metabolism, and the formation of chemically reactive metabolites from food constituents is common; if this led to an immune response against the modified hepatic proteins it could cause extensive damage. This also suggests that the default response of the liver to covalent binding is immune tolerance. This is in contrast to skin where there is little bioactivation, but, if covalent binding does occur, it is likely to lead to an immune response (e.g. contact hypersensitivity).
There are many redundant systems involved in immune tolerance. One is the ubiquitin ligase pathway, including Cbl-b. Although Cbl-b−/− mice are generally healthy, they have been shown to have a hyper-proliferative T-cell response and are resistant to anergy induction compared to the wild-type C57BL/6 mice from which they were developed (Venuprasad, Citation2010). Treatment of Cbl-b−/− mice with INH resulted in a small increase in GLDH activity, which appeared to return towards normal despite continued treatment (). One female Cbl-b−/− mouse developed abnormal liver histology that featured lymphocyte infiltration, focal necrosis, cholestasis, and steatosis. This mouse also had an increase in the number of macrophages and B-cells in the liver and spleen implying an immune response ( and Supplemental Figure S4). However, paradoxically, the serum levels of IL-1α and IL-12 were decreased by INH treatment in female Cbl-b−/− mice (). IL-1α and IL-12 are two key inflammatory cytokines that modulate the induction of an immune response (Naisbitt et al., Citation2000); the fact that INH down-regulated these cytokines suggests that INH has immunosuppressive effects. Overall, Cbl-b−/− mice tolerated INH quite well despite the increase in GLDH, but there was still a lack of serious liver injury with the exception of one mouse. INH also led to an increase in GLDH in PD-1−/− mice, which also have impaired immune tolerance (Pardoll, Citation2012), but they did not tolerate the treatment as well as Cbl-b−/− mice. Isolated animals in different treatment groups had more serious liver injury, but it is not clear what the basis for this difference was; this may be evidence of the idiosyncratic nature of the response. As with the Cbl-b−/− mice, the PD-1−/− mice were developed from C57BL/6 mice in which we saw no evidence of INH-induced liver injury. The observation that Cbl-b−/− and PD-1−/− mice are more sensitive than the wild-type implies that these pathways are important for adaptation of the liver to INH treatment, but the fact that Rag−/− mice, which lack competent T- and B-cells, also had an increase in GLDH that appeared more persistent than in other strains suggests that the initial immune response may be mediated by the innate immune system, and that T- and/or B-cells are involved in modulating this response. Although we utilized mouse strains that have somewhat impaired immune responses, there are several redundant mechanisms of immune tolerance (Laskin, Citation2009; Pardoll, Citation2012; Tiegs & Lohse, Citation2010); therefore these animals are by no means totally unable to develop immune tolerance.
Attempts to increase the liver injury in Cbl-b−/− mice by depleting T-regulatory (Treg) cells were not successful. Treg cells (classically defined as CD4+CD25+FoxP3+) are known to suppress inflammation and lead to maintenance of immune tolerance through production of suppressive cytokines, such as IL-10 (Soyer et al., Citation2013). Specifically, the use of FICZ has been reported to interfere with Treg cell development and to increase the severity of an experimental autoimmune model of encephalomyelitis in mice (Quintana et al., Citation2008). Likewise, although Treg cells are not the only cells that express CD25, administration of anti-CD25 monoclonal antibody was reported to reduce the number of CD4+CD25+ cells in peripheral lymphoid tissues, and this was associated with regression of tumors that grew in syngeneic mice (Onizuka et al., Citation1999). However, the agents were not effective in worsening INH-induced liver injury. In addition, co-treatment with RMP, which appears to increase the risk of INH-induced liver injury in humans, did not exacerbate liver injury. As we have previously proposed, this data is in agreement with the fact that rodents may not be a good model to study effects of RMP on INH toxicity because RMP appears to be a human-specific pregnane X receptor activator, which is required for induction of cytochrome P450s (Jones et al., Citation2000; Lehmann et al., Citation1998).
So what do these data mean and why is it so difficult to develop an animal model of idiosyncratic drug-induced liver injury? Given that severe liver injury is idiosyncratic in humans, it should not be surprising that it is difficult to reproduce this injury in animals. If it is immune-mediated as argued above, it is possible that there is a requirement for a specific MHC/T-cell receptor combination in order to produce an immune response that leads to severe liver injury. There is one report of an HLA association, but it does not appear to be a strong association (Sharma et al., Citation2002). In addition, given that INH covalently binds to a very large number of hepatic proteins (Metushi et al., Citation2012), which would lead to an even greater number of drug-modified peptides, it seems likely that at least one MHC/T-cell receptor pair would have the necessary fit, although it is possible that the affinity would not be sufficient to produce a strong response. It is possible that some environmental exposure, such as an infection, might be required to stimulate a strong immune response; however, patients with active tuberculosis do not appear to be a significantly increased risk, and it does not appear that infections or other agents are required to precipitate INH-induced liver failure. This also appears to be true of other idiosyncratic drug-induced liver failure; specifically, pre-existing liver disease does not appear to be a major risk factor (Zimmerman, Citation1999). Therefore, immune tolerance/immunosuppression is still a reasonable hypothesis for the reason that most patients and animals do not develop severe INH-induced liver injury.
The immune response to INH in humans is complex. It commonly induces anti-nuclear antibodies and sometimes induces an autoimmune syndrome resembling lupus (Salazar-Paramo et al., Citation1992). It also commonly causes a fever and rash (Maddrey & Boitnott, Citation1973). On the other hand, there are observations that suggest that INH can lead to immune suppression, which include the fact that administration of INH together with intra-vesical bacillus Calmette-Guérin (BCG) therapy for superficial bladder cancer appeared to: reduce the induction of mononuclear cell infiltrate in the bladder wall, inhibit enlargement of regional lymph nodes, inhibit the increase in MHC II expression of lymph node cells and diminish systemic immunity that was induced by BCG administration (de Boer et al., Citation1992). Signs of immune impairment were seen even if INH was administered later on during the course of BCG therapy, or if the dose of BCG was increased. Similarly, in humans, the absolute number of granulocytes and the concentration of IgG antibodies after BCG instillation were significantly suppressed by INH administration (Stassar et al., Citation1994). This immunosuppression may be part of a response that prevents an immune response to covalent binding of INH and protects the liver.
In short, we developed a mouse model of INH-induced liver injury in which the immune system appears to play a both protective and pathogenic role, but we were unable to develop a model of INH-induced liver failure. The two most likely explanations for this failure are that there are many redundant mechanisms of immune tolerance and/or that the mice simply did not have the requisite MHC/T-cell receptor repertoire to develop a strong immune response.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Supplementary Material
Download PDF (1.1 MB)Acknowledgements
Jack Uetrecht holds the Canada Research Chair in Adverse Drug Reactions. Imir G. Metushi is a trainee of the Drug Safety and Effectiveness Cross Disciplinary Training Program that is funded by the Canadian Institutes for Health Research. The Canadian Institutes for Health Research also supplied operating funds for this research. We would like to thank Dr Pamela Ohashi for giving us breeding pairs of PD1−/− and Cbl−/− mice, as well as for providing us with anti-CD4/CD8 antibody. We thank Dr Tony Hayes for interpretation of the liver H&E slides.
References
- Black, M., Mitchell, J. R., Zimmerman, H. J., et al. 1975. Isoniazid-associated hepatitis in 114 patients. Gastroenterology 69:289–302
- Blair, I. A., Mansilla Tinoco, R., Brodie, M. J., et al. 1985. Plasma hydrazine concentrations in man after isoniazid and hydralazine administration. Human Toxicol. 4:195–202
- Centers for Disease Control and Prevention (CDC). 2010. Severe isoniazid-associated liver injuries among persons being treated for latent tuberculosis infection - United States, 2004–2008. MMWR 59:224–229
- Crispe, I. N., Dao, T., Klugewitz, K., et al. 2000. The liver as a site of T-cell apoptosis: Graveyard, or killing field? Immunol. Rev. 174:47–62
- de Boer, L. C., Steerenberg, P. A., van der Meijden, P. M., et al. 1992. Impaired immune response by isoniazid treatment during intra-vesical BCG administration in the guinea pig. J. Urol. 148:1577–1582
- Fukino, K., Sasaki, Y., Hirai, S., et al. 2008. Effects of N-acetyltransferase 2 (NAT2), CYP2E1, and glutathione-S-transferase (GST) genotypes on serum concentrations of isoniazid and metabolites in tuberculosis patients. Toxicol. Sci. 33:187–195
- Gent, W. L., Seifart, H. I., Parkin, D. P., et al. 1992. Factors in hydrazine formation from isoniazid by paediatric and adult tuberculosis patients. Eur. J. Clin. Pharmacol. 43:131–136
- Huang, Y. S., Chern, H. D., Su, W. J., et al. 2002. Polymorphism of the N-acetyltransferase 2 gene as a susceptibility risk factor for anti-tuberculosis drug-induced hepatitis. Hepatology 35:883–889
- Jones, S. A., Moore, L. B., Shenk, J. L., et al. 2000. The pregnane X receptor: A promiscuous xenobiotic receptor that has diverged during evolution. Mol. Endocrinol. 14:27–39
- Laskin, D. L. 2009. Macrophages and inflammatory mediators in chemical toxicity: A battle of forces. Chem. Res. Toxicol. 22:1376–1385
- Lehmann, J. M., McKee, D. D., Watson, M. A., et al. 1998. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Invest. 102:1016–1023
- Maddrey, W. C., and Boitnott, J. K. 1973. Isoniazid hepatitis. Ann. Int. Med. 79:1–12
- Metushi, I. G., Cai, P., Zhu, X., et al. 2011. A fresh look at the mechanism of isoniazid-induced hepatotoxicity. Clin. Pharmacol. Ther. 89:911–914
- Metushi, I. G., Nakagawa, T., and Uetrecht, J. 2012. Direct oxidation and covalent binding of isoniazid to rodent liver and human hepatic microsomes: Humans are more like mice than rats. Chem. Res. Toxicol. 25:2567–2576
- Metushi, I. G., Sanders, C., Lee, W. M., and Uetrecht, J. 2013. Detection of anti-isoniazid and anti-cyp antibodies in patients with isoniazid-induced liver failure. Hepatology (Epub ahead of print)
- Mitchell, J. R., Zimmerman, H. J., Ishak, K. G., et al 1976. Isoniazid liver injury: Clinical spectrum, pathology, and probable pathogenesis. Ann. Int. Med. 84:181–192
- Naisbitt, D. J., Gordon, S. F., Pirmohamed, M., and Park, B. K. 2000. Immunological principles of adverse drug reactions: The initiation and propagation of immune responses elicited by drug treatment. Drug Safety 23:483–507
- Noda, A., Hsu, K. Y., Noda, H., et al. 1983. Is isoniazid-hepatotoxicity induced by the metabolite, hydrazine? J. UOEH 5:183–190
- Onizuka, S., Tawara, I., Shimizu, J., et al. 1999. Tumor rejection by in vivo administration of anti-CD25 (IL-2Rα) monoclonal antibody. Cancer Res. 59:3128–3133
- Pardoll, D. M. 2012. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12:252–264
- Quintana, F. J., Basso, A. S., Iglesias, A. H., et al. 2008. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 453:65–71
- Salazar-Paramo, M., Rubin, R. L., and Garcia de la Torre, I. 1992. Systemic lupus erythematosus induced by isoniazid. Ann. Rheum. Dis. 51:1085–1087
- Sarich, T. C., Youseffi, M., Zhou, T., et al. 1996. Role of hydrazine in the mechanism of isoniazid hepatotoxicity in rabbits. Arch. Toxicol. 70:835–840
- Sharma, S. K., Balamurugan, A., Saha, P. K., et al. 2002. Evaluation of clinical and immunogenetic risk factors for the development of hepatotoxicity during anti-tuber-culosis treatment. Am. J. Resp. Crit. Care Med. 166:916–919
- Soyer, O. U., Akdis, M., Ring, J., et al. 2013. Mechanisms of peripheral tolerance to allergens. Allergy 68:161–170
- Stassar, M. J., Vegt, P. D., Steerenberg, P. A., et al. 1994. Effects of isoniazid (INH) on the BCG-induced local immune response after intra-vesical BCG therapy for superficial bladder cancer. Urol. Res. 22:177–184
- Tiegs, G., and Lohse, A. W. 2010. Immune tolerance: What is unique about the liver? J. Autoimmun. 34:1–6
- Venuprasad, K. 2010. Cbl-b and itch: Key regulators of peripheral T-cell tolerance. Cancer Res. 70:3009–3012
- Warrington, R. J., McPhillips-Feener, S., and Rutherford, W. J. 1982. The predictive value of the lymphocyte transformation test in isoniazid-associated hepatitis. Clin. Allergy 12:217–222
- Warrington, R. J., Tse, K. S., Gorski, B. A., et al. 1978. Evaluation of isoniazid-associated hepatitis by immunological tests. Clin. Exp. Immunol. 32:97–104
- Whitehouse, L. W., Tryphonas, L., Paul, C. J., et al. 1983. Isoniazid-induced hepatic steatosis in rabbits: An explanation for susceptibility and its antagonism by pyridoxine hydrochloride. Can. J. Physiol. Pharmacol. 61:478–487
- Zimmerman, H. J. (Ed.). 1999. Hepatotoxicity: The Adverse Effects of Drugs and Other Chemicals on the Liver. Philadelphia, PA: Lippincott Williams & Wilkins