
Abstract
Primary aromatic amine drugs are structural alerts in drug development because of their association with a high incidence of idiosyncratic drug reactions (IDRs). If biomarkers could be found that predict IDR risk, it would have a major impact on drug development. Previous attempts to do this through screening of hepatic gene expression profiles in rodents treated with aromatic amine drugs found limited changes. Of the drugs studied, aminoglutethimide (AMG) induced the most changes, and this led to a more comprehensive study of its effects on the liver. Brown Norway rats treated with AMG for up to 14 days showed only a transient elevation of glutamate dehydrogenase. Pathway-specific PCR arrays found few AMG-induced gene changes associated with an immune response and, of these changes, the majority were involved with innate immunity such as Tlr2, Ticam2, CD14, and C3. AMG treatment also led to significant changes in the apoptosis and mitochondrial panel of genes. It was recently found that AMG does induce significant changes in the bone marrow of rats, and agranulocytosis is a common IDR caused by AMG. In contrast, liver injury is not a common IDR associated with AMG. Therefore, the liver may be able to effectively deal with AMG reactive metabolites, and changes observed in this study may be involved in adaptation. Myeloperoxidase is also known to be able to oxidize aromatic amines to reactive metabolites, and these observations suggest that metabolism outside of the liver may be important for the mechanism of aromatic amine-induced IDRs.
Introduction
The primary aromatic amine functional group is a notorious structural alert in drug development because drugs with this group are associated with a high incidence of idiosyncratic drug reactions (IDRs; Uetrecht, Citation2002). This is presumably because they can be oxidized to reactive metabolites. The first step is oxidation to a hydroxylamine, which is not in itself chemically reactive, but the hydroxylamine is readily further oxidized to a nitroso metabolite that is reactive. In addition, these two metabolites readily redox cycle that has the potential to cause oxidative stress. Despite the potential for toxicity, many aromatic amine drugs remain pharmacologically very effective; therefore, understanding the mechanism of IDRs or determining biomarkers associated with these reactions could provide a major benefit to drug development. Although the types of IDRs vary from drug to drug, and one drug can cause more than one type of IDR, the toxicity of aromatic amine drugs is presumably due to their ability to form common reactive metabolites that could potentially induce cellular damage and lead to an adverse drug response (Ng & Uetrecht, Citation2013a).
Immune involvement has been implicated in the mechanisms of IDRs (Uetrecht & Naisbitt, Citation2013); however, our current understanding of IDRs is still far from complete. IDRs pose significant issues to drug development because of their lack of predictability, and the inherent risks posed to patients once the drug is out in the market. Ideally, the ability of drug candidates to induce IDRs would be detected early in pre-clinical studies to decrease the enormous financial risk of drug attrition and to ensure drug safety for patients. However, in reality, IDRs are much more difficult to predict, largely due to our lack of understanding of the underlying mechanisms, and, as a result, although there have been strong human leukocyte antigen (HLA) gene associations; there are currently no reliable tests to predict IDR risk. In addition, development of valid animal models of IDRs with clinical characteristics that closely mimic what occurs in humans has proven to be a daunting task, despite many attempts at various immune interventions (Ng et al., Citation2012). Analyzing changes in gene expression of drugs that induce IDRs has been proposed as a method of determining biomarkers to predict drugs pre-disposed to these reactions, and this has had some success in distinguishing model hepatotoxins in vitro (Waring et al., Citation2001). However, it is difficult to determine the relevancy of these gene changes and how well these signatures would apply to predicting IDR risk for new drug candidates.
In an attempt to understand the mechanisms underlying aromatic amine-induced IDRs, microarray studies were previously performed in both mice and rats to look for common hepatic gene changes induced acutely by aromatic amine drugs that could provide mechanistic clues or act as potential biomarkers to identify drugs pre-disposed to causing IDRs. The rationale for choosing early timepoints was that it is usually early cell injury – which may occur in most patients and animals – that presumably stimulates an immune response and later presents as an IDR in rare patients. It was also anticipated that the predictive value of these results would have significant implications for the assessment of pre-clinical drug safety. However, in mice treated with sulfamethoxazole, few gene changes were observed in the liver except for down-regulation of the expression of a few genes (Pacitto et al., Citation2007). Yet, it was uncertain whether these results were species- or drug-specific. Additional testing in rats with three different aromatic amine drugs – including sulfamethoxazole, dapsone, and aminoglutethimide – found few common gene changes and, again, the majority were down-regulated (Ng & Uetrecht, Citation2013a). Among these drugs, aminoglutethimide induced the most significant gene changes, and several were associated with the Keap-1-Nrf2-ARE pathway, which suggests that some cellular stress occurred. This is consistent with gene changes observed with other drugs associated with IDRs such as carbamazepine, phenytoin, tienilic acid, and D-penicillamine (Lu et al., Citation2008; Pacitto et al., Citation2007; Seguin et al., Citation2005). Given that the liver is the conventional target of drug metabolism studies because of its predominant function in drug metabolism, and aromatic amines are readily oxidized to reactive metabolites, the paucity of changes in gene expression was unexpected. Furthermore, given the postulates of the danger hypothesis, the liver would be expected to experience more adverse effects because it is assumed to be the site where most reactive metabolites would form and induce damage.
To ensure that any aromatic amine-induced changes in the liver were not overlooked, because of experimental limitations in the previous study that only focused on the differences between aromatic amine drugs at a single timepoint, a more comprehensive study combining physiological tests with gene arrays at successive timepoints was performed in the liver of rats to confirm whether aromatic amine drugs can affect the liver as a target organ. The aromatase inhibitor aminoglutethimide (AMG) was used in these studies as a prototypical aromatic amine drug because it induced the most changes in the previous microarray study and was more likely to result in observable effects. AMG is primarily indicated for breast and prostate cancer treatment and is associated with a variety of IDRs including cholestasis, agranulocytosis, lupus, and skin rashes (Cocconi, Citation1994). AMG is associated with a higher incidence of IDRs than sulfamethoxazole and dapsone, the other two aromatic amines that were studied previously, and, because of this, it is not currently used clinically. This higher incidence of IDRs with AMG may be a result of a higher electron density, which would facilitate the oxidation of the aromatic amine. The hydroxylamine metabolite of AMG has been found in the urine of humans on AMG therapy (Goss et al., Citation1985). The N-hydroxyl AMG metabolite has also been found in rats treated with AMG (Eweiss et al., Citation1983). The similarities in metabolic disposition between species suggest that the rat may provide a relevant species in which to study the effects of AMG. Thus, the changes induced by AMG on the liver in rats were tested, including pathway-specific gene expression, to gain a more detailed understanding of the mechanisms of aromatic amine-induced IDRs.
Materials and methods
Reagents
AMG was purchased from Toronto Research Chemical (North York, ON). Epitope Unmasking Solution and Antibody Dilution Buffer were acquired from ProHisto (Columbia, SC). 3,3′-diaminobenzidine (DAB) was obtained from Vector Labs (Burlingame, CA). Mouse anti-rat Ki-67 (clone: MIB-5), rabbit anti-mouse biotinylated IgG, and streptavidin-horseradish peroxidase (HRP) were purchased from Dako (Burlington, ON). Mouse anti-rat CD8 (clone: OX-8) and mouse anti-rat CD68 (clone: ED1) were obtained from Abcam (Cambridge, MA). Alanine aminotransferase (ALT) Liquid Stable Reagent was purchased from Thermo Scientific (Middletown, VA). Glutamate dehydrogenase (GLDH) assay kit was from Randox Laboratories Ltd. (Crumlin, UK). RNAlater RNA Stabilization Solution, Trizol Reagent, and RNase free water were from Life Technologies (Burlington, ON). RNeasy Mini Kit was purchased from Qiagen (Mississauga, ON). RT2 Profiler PCR Arrays and RT2 First Strand Kit were obtained from SABiosciences (Valencia, CA) and were supplied by Roche.
Animals
Male Brown Norway rats (≈9-weeks-old, weighing 200–250 g) were purchased from Charles River (Montreal, QC). Rats were housed at the Department of Comparative Medicine (University of Toronto) under standard care conditions (i.e. doubly-housed, 12:12 h light:dark cycle, and 22 °C room temperature). Standard rat chow and water were provided ad libitum. Rats were given 1 week to acclimatize prior to the start of experiments. The protocol was approved by the University of Toronto Faculties of Medicine and Pharmacy Animal Care Committee.
Treatments
AMG was suspended in 0.5% methylcellulose and administered by oral gavage at a dose of 125 mg/kg/day. This dosage is an increase from the 80 mg/kg AMG dosage that was used in our previous study (Ng & Uetrecht, Citation2013a) to ensure that maximal changes were induced, while maintaining the blood levels of AMG similar to what is found in humans taking AMG therapeutically (4.7–32.4 µg/ml AMG in blood of patients taking 1000 mg AMG daily for 12 weeks; Murray et al., Citation1979). Control rats were given 0.5% methylcellulose vehicle only. Although we did not specifically measure the plasma levels of AMG in this study, blood levels of AMG in rats had been measured in our previous studies and are within the range of AMG blood levels found in humans; a single dose of 80 or 160 mg/ml AMG in rats gave plasma levels of 15.0 (±2.0) and 18.4 (±2.1) µg/ml AMG 1 h after treatment, respectively (Ng & Uetrecht, Citation2013b). Four rats were tested for each treatment group unless otherwise stated. Body weight and general health of rats were monitored daily. At various timepoints (Days 0, 3, 7, and 14), blood samples were obtained for use in the protocols below. In addition, at Days 1, 7, and 14, sub-sets of rats were euthanized by CO2 asphyxiation, and liver (as well as a final blood) samples were isolated for use in the protocols below.
Histology
At necropsy, livers were excised and immediately immersed in 10% formalin solution. H&E-stained and unstained paraffin-embedded slides were prepared at the Division of Pathology at the Hospital for Sick Children (Toronto, ON). Immunohistochemistry was carried out using standard procedures. Briefly, slides were de-paraffinized with xylene and rehydrated with serial dilutions of ethanol. Heat-induced antigen retrieval was performed for 20 min (Ki-67 staining) or incubated at 71 °C overnight (for CD8 and CD68 staining) in Epitope Unmasking Solution. Endogenous peroxidases were blocked with 3% hydrogen peroxide in phosphate-buffered saline (PBS, pH 7.4) for 10 min. Washes were then performed using PBS-0.05% Tween-20 solution and antibodies were diluted in Antibody Dilution Buffer. Slides were incubated with primary antibodies against Ki-67, CD68, and CD8 at room temperature. For the mouse anti-rat Ki-67 antibody, a 1:25 dilution was used and incubation time was 30 min. For the mouse anti-rat CD68 and anti-rat CD8 antibodies, the dilution used was 1:100 and incubation time was 90 min. Rabbit anti-mouse biotinylated IgG was used as a secondary antibody (1:250 dilution) and incubated for 30–40 min at room temperature. Streptavidin-HRP (1:500) was then added and the slide incubated for 15 min, after which the signal was developed with DAB solution; hematoxylin was used as counterstain. Slides were dehydrated with ethanol and xylene before coverslips were attached with Cytoseal. Histological images were taken at the Microscopy Imaging Lab (Faculty of Medicine, University of Toronto) on a Zeiss fluorescence microscope with deconvolution. Immunohistochemical grading was performed by counting the number of cells/field of view; at least two slices of tissue (3–6 mm2) were mounted on glass slides. Five areas from each slice were manually counted under the microscope. All slides were read in a blinded manner.
Liver enzymes
Serial samples of tail vein blood were used to determine liver enzyme activities in the serum. Blood was processed within 1 h of collection, and ALT and GLDH activities determined using, respectively, ALT Liquid Stable Reagent and a GLDH Assay Kit, according to manufacturer protocols. For GLDH, minor modifications were made to the protocol previously described in Metushi et al. (Citation2012), wherein 200 µl Reagent 1 was pre-mixed with 8 µl Reagent 2, and 200 µl of this mixture was loaded into each well of a 96-well plate before serum samples were added. All reagents were reconstituted per manufacturer specification and absorbance values at 340 and 405 nm were monitored on a SpectraMax Plus384 (Molecular Devices, Sunnyvale, CA) plate-reader for at least 5 min at 25 °C.
RNA extraction
At necropsy, the livers were removed, rinsed in PBS, and immediately immersed in RNAlater RNA Stabilization Solution to preserve RNA for extraction. Liver samples were then homogenized with an IKA Ultra-Turrax T25 S1 rotor stator homogenizer (Janke and Kunel, Staufen, Germany), and RNA was extracted using Trizol Reagent as per manufacturer protocols. An additional RNA clean-up step was performed using an RNeasy Mini Kit. Ultimately, extracted RNA was eluted in 40 µl RNase-free water and stored at −80 °C. The quality and purity of the extracted RNA was measured using UV spectrophotometry based on the A260/A280 ratio.
PCR arrays
Extracted RNA was converted to cDNA using the RT2 First Strand Kit with 400 ng RNA per sample, following manufacturer protocols. Six different panels of genes were analyzed using the RT2 Profiler PCR Arrays, including: apoptosis (PARN-012), chemokines and receptors (PARN-022), common cytokines (PARN-021), mitochondria (PARN-087), toll-like receptor signalling (PARN-018), and Th17 and autoimmunity (PARN-073). However, limitations on the number of plates prevented analysis of every gene panel at all timepoints (1, 7, 14 days); specifically, only Day 14 data were collected for common cytokine, toll-like receptor signalling, and Th17 response pathways. Array studies were performed according to manufacturer protocols on 384-well plates that held four samples simultaneously. For each sample, cDNA converted from 400 ng total RNA was loaded on the plate, and each plate had samples staggered between AMG-treated and control animals. Arrays were run on an ABI7900HT real-time PCR instrument (Applied Biosystems, Grand Island, NY) according to the SABiosciences protocol. A threshold cycle value of 0.2 was assigned to each array plate for data normalization. Data was analyzed using RT2 Profiler PCR Array Data Analysis Template Version 3.2 (SABiosciences); lactate dehydrogenase A and ribosomal protein P1 were used as housekeeping genes. Pathway analyses were performed using Gene Network Central Pro software (SABiosciences).
Statistical analysis
Data were analyzed with GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA) using a Mann–Whitney U-test or two-way analysis of variance (ANOVA) depending on the constraints of the data being analyzed. PCR array data were analyzed with RT2 Profiler PCR Array Data Analysis Template v3.2 (SABiosciences) using Student’s t-test.
Results
Effects of AMG on the liver
Rats tolerated the 125 mg AMG/kg/day dose well for a period of up to 14 days without significant changes in body weight or evidence of distress. Histological analysis of the liver of AMG-treated rats found enlarged hepatocytes in Zone 3 as compared to control rats (). In the representative H&E staining of the liver, the noticeable presence of red blood cells in sinusoids of the control is likely an artifact from lack of perfusion prior to fixation rather than a treatment effect. An increase in the liver weight of AMG-treated rats was also found, and this was most significant after 7 days of treatment (). Although no data on liver weight was collected at the 14-day end-point, the histologic findings suggest the increase in liver weight with AMG was sustained. In addition, there appeared to be a slight increase in cell proliferation, as determined by the proliferation marker Ki-67, in the liver of AMG rats after 7 days compared to the untreated group (); however, this did not reach statistical significance.
Figure 1. Histological changes in livers of AMG-treated rats. Representative H&E staining of (A) control and (B) AMG-treated (125 mg/kg/day, 14 days) rats. Evidence of Zone 3 hepatocyte hypertrophy was found in rats treated with AMG as compared to controls; 20× magnification. (C) Rats treated with AMG had an increase in liver weight that was most significant on Day 7 of treatment. Values shown are mean (±SEM) for each group (n = 4). *p < 0.05 compared to control (Mann--Whitney U-test).
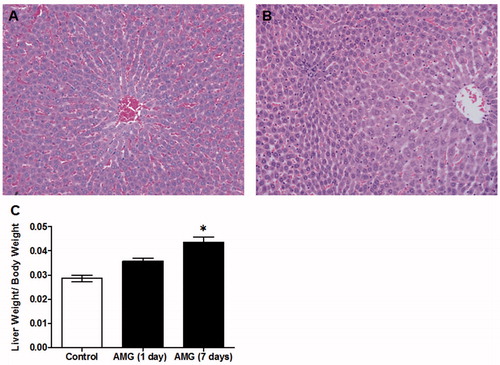
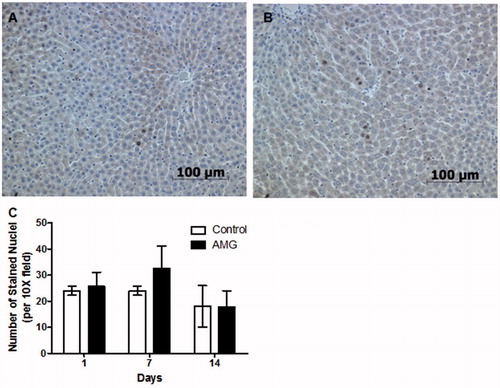
Figure 2. Comparison of hepatocyte proliferation in control and AMG-treated animals. Representative Ki-67 staining in tissue from (A) control and (B) AMG-treated (125 mg/kg/day, 14 days) rats; 20× magnification. (C) A possible increase in Ki-67 was observed at Day 7 of AMG treatment. Although some evidence of hepatocyte proliferation was found in the AMG-treated rats, the main effect was hypertrophy rather than hyperplasia. Open bars = control rats; solid bars = AMG-treated rats. Values shown are mean (±SEM) number of Ki-67-positively-stained nuclei per 10× field of view (n = 4). No results were statistically significant (two-way ANOVA).
Examination of H&E-stained slides did not reveal any evidence of liver injury (neither hepatocellular nor cholestatic) in AMG-treated rats after 14 days, nor were immune cell infiltrates detected in the liver. Staining for specific immune cells such as CD8 T-lymphocytes and CD68 macrophages in the liver found no differences using immunohistological grading between AMG-treated and control animals (data not shown).
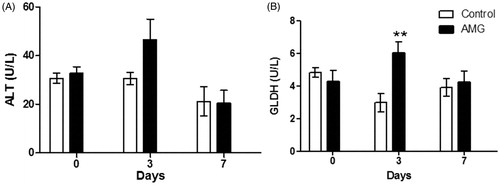
In serum, few changes were observed in enzymes associated with liver injury in AMG-treated rats. A subtle but transient increase in ALT was observed with AMG treatment at Day 3 (). A significant increase in GLDH was also observed with AMG at Day 3; however, these changes were transient and returned back to baseline/control levels at Day 7 (). No changes were observed in ALT or GLDH at Day 14 of AMG treatment (data not shown).
Figure 3. AMG-induced changes in serum levels of liver enzymes. (A) ALT and (B) GLDH in rats treated with AMG (125 mg/kg/day) compared to control rat values. Open bars = control group; solid bars = AMG-treated group. Values shown are mean (±SEM) (n = 4). **p < 0.01 as compared to control (two-way ANOVA).
AMG-induced hepatic gene changes
Hepatic gene expression was determined at various timepoints of AMG treatment using PCR arrays that were pathway-specific. The greatest number of AMG-induced differential changes in gene expression was primarily found within the apoptotic panel of genes, but both pro- and anti-apoptotic regulatory pathways were induced (). Although the majority of changed genes were involved with the induction of apoptosis, significant up-regulation of anti-apoptotic genes was also quite notable including Aven, Bag1, Bcl2l1, Birc3, and Nol3. At earlier timepoints, a general down-regulation of apoptotic genes was observed, whereas up-regulation of genes was mainly observed at the latter 14-day end-point. A pathway analysis of the up-regulated genes induced by AMG did not reveal any particular dominant signaling pathway, although most genes were associated with Nfkb1 (). In terms of downstream effects of the gene changes, TUNEL staining for apoptotic cells in the liver did not show any differences between AMG-treated and control rats (data not shown). This may be due to the activation of genes in opposing apoptosis pathways, which may act to modulate apoptotic events.
Figure 4. Pathway analysis of significantly changed hepatic genes in the apoptotic pathway in AMG-treated rats. Only genes with changes >2-fold (up-regulation) after 14 days of AMG treatment were included in analysis for trends in the apoptotic panel of genes. Arrows indicate known associations and do not represent how genes are regulated by AMG. Solid lines = up-regulation; dashed lines = down-regulation; dotted lines = physical interaction. Analysis was performed using Gene Network Central Pro software (SABiosciences).
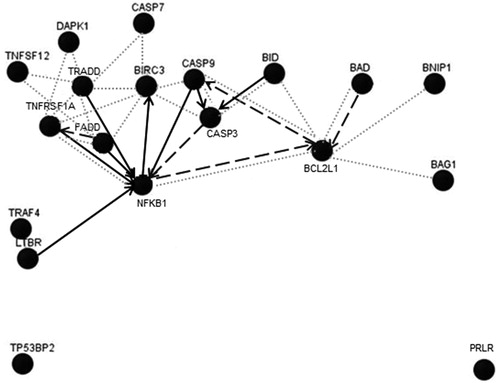
Table 1. Hepatic changes in gene expression induced by AMG in the apoptosis pathway.
Many mitochondria-associated genes were also significantly changed upon AMG treatment (). Again, down-regulation of genes was primarily observed at the earlier timepoints, whereas up-regulation was observed after 14 days of AMG treatment. Of these AMG-induced genes, many were associated with mitochondrial transportation such as the solute carrier family 25 (Slc25) and translocase (Tomm34, Tomm40, Tspo, Timm17b) genes. Mitochondria-associated apoptotic genes such as Aifm2, Bbc3, and Bid were also significantly up-regulated with AMG treatment; the function of these genes suggested induction of apoptosis via the intrinsic mitochondrial pathway.
Table 2. Mitochondrial gene expression changes in the livers of AMG-treated rats.
In terms of changes in immune-associated genes, AMG induced fewer significant changes than that observed for the other two pathways, and most of these changes were more suggestive of changes in innate immunity. In the chemokine and receptor pathways there were significant increases in C3, Ccl19, and Nfkb1 (); in contrast, a sustained down-regulation was observed for Cxcl13 upon AMG treatment. Increases in the toll-like receptor Tlr2 and the adaptor molecule Ticam2 genes were also observed with an increase in Cd14 and changes in the IL-1 pathway (Il1f5, IL1rn) upon AMG treatment (). Up-regulation of Cd4 was the predominant change associated with adaptive immunity.
Table 3. AMG-induced hepatic gene changes in chemokines and receptors.
Table 4. Hepatic gene changes induced by AMG in various immune pathways after 14 days of treatment.
Discussion
The current findings are consistent with our previous experiments in that aromatic amine drugs do not appear to cause significant liver injury even though AMG treatment resulted in more changes in hepatic mRNA expression that are consistent with liver injury than the other aromatic amines that were tested. In retrospect, although AMG is associated with a very high incidence of IDRs including cholestatic liver injury, the non-steroidal anti-inflammatory, bromfenac, is the only primary aromatic amine drug to be withdrawn from the market because of idiosyncratic hepatocellular liver injury (Goldkind & Laine, Citation2006). Sulfamethoxazole often causes idiosyncratic liver injury, but it is usually part of a more generalized hypersensitivity reaction than liver-specific toxicity. Furthermore, because the liver is so extensively exposed to xenobiotics that can be oxidized to reactive metabolites, the primary response of the liver to drug-modified proteins may be tolerance (Bowen et al., Citation2005); liver toxicity caused by aromatic amines may be more a consequence of a generalized hypersensitivity reaction rather than direct liver injury.
It has been proposed that primary activation of T-lymphocytes in the liver leads to immune suppression through CD8+ T-lymphocyte apoptosis – the liver has been referred to as a T-lymphocyte graveyard (Crispe et al., Citation2000) – whereas primary activation outside of the liver may lead to a robust immune response in the liver (Bowen et al., Citation2005). This may be an appropriate response for an organ with high xenobiotic exposure and reactive metabolite formation. Although inflammatory infiltrates were not observed in the liver histologically, the transient increase in GLDH, which is a marker of liver injury, may be a sign of mild injury followed by adaptation. Thus, if these AMG-induced changes in the liver are immune-mediated, then the response to AMG in the liver in the majority of patients may be immune tolerance.
Approximately 60% of AMG-treated patients experience elevations in γ-glutamyltransferase (Nagel et al., Citation1982), which may be an indicator of cholestatic liver injury. The more serious form of idiosyncratic liver injury is hepatocellular rather than cholestatic (Uetrecht & Naisbitt, Citation2013). Although enzymes of cholestatic liver injury were not measured, there was no histological evidence of cholestatic injury or bile accumulation in the AMG-treated rats. The hypertrophy and hyperplasia of hepatocytes observed in the AMG-treated rats is consistent with the ability of AMG to induce cytochrome P450 (Murray et al., Citation1993), and this is presumably the reason that the half-life of AMG decreases with sustained AMG therapy (Murray et al., Citation1979).
The largest number of AMG-induced gene changes was within the apoptotic panel of genes, and this suggests that apoptosis may play a role in AMG-induced IDRs. Apoptosis is a dynamic process; as cells undergo apoptotic cell death, they are cleared by phagocytic cells; if this process is not overwhelmed, then apoptotic changes may not be observed. However, in the context of IDRs, excessive apoptotic cell death that cannot be efficiently cleared by phagocytes may lead to secondary necrosis and the release of danger signals that activate the immune system (Kono & Rock, Citation2008).
Although, at later timepoints, AMG up-regulated more pro-apoptotic than anti-apoptotic genes, it was difficult to discern which of the pathways was dominant. Interestingly, a pathways analysis found many associations with Nfkb1, which is a part of the nuclear factor-κB (NF-κB) family of transcription factors. NF-κB is involved in immune responses and inflammation, which ranges from involvement in hematopoiesis and organogenesis to transcription of cytokines and chemokines (Hayden et al., Citation2006). Furthermore, NF-κB can regulate programmed cell death and has a predominant pro-survival effect because of its ability to transcribe anti-apoptotic genes such as inhibitor of apoptosis proteins and anti-apoptotic Bcl-2 proteins (Dutta et al., Citation2006). This suggests that these changes may modulate the potential effects of the pro-apoptotic gene expression. Consistent with this, TUNEL staining found no difference between AMG-treated rats and control rats in the number of apoptotic cells in the liver. Since NF-κB is regulated in the cytoplasm by the inhibitory protein IκB, there are limitations in the interpretation of changes in gene expression because the effect of NF-κB is dependent on cytoplasmic activation (Dutta et al., Citation2006). There is also evidence to suggest that NF-κB plays a role in immune tolerance since lack of NF-κB1 expression in dendritic cells activates CD8+ T-lymphocytes (Dissanayake et al., Citation2011), which could potentially be an additional factor contributing to the tolerogenic response in the liver. However, based on our results, it seems that AMG exposure in the liver induces modest gene expression in apoptotic pathways that is balanced by anti-apoptotic gene changes and prevents the induction of apoptosis.
Mitochondrial injury has been proposed to contribute to the mechanisms of IDRs as a danger signal (Uetrecht & Naisbitt, Citation2013). Given the importance of mitochondria in maintaining cellular energy and regulating programmed cell death, it is not surprising that AMG also induced a significant number of mitochondrial gene changes. Although the majority of mitochondrial gene changes are involved with solute carriers and transporters, all of the apoptotic genes that were up-regulated by AMG positively regulate the intrinsic apoptotic pathway, and these changes may compliment the observed changes in the apoptosis arrays.
AMG induced fewer changes in immune-related genes at these early timepoints, and the majority of these changes involved genes associated with the innate immune response. Although they provide some evidence for the up-regulation of immune signaling pathways, the only conclusive evidence for activation of an adaptive response was an increase in Cd4 gene expression. However, a histological increase in lymphocytes was not observed in the liver with AMG. Thus, activation of the innate immune response upon AMG treatment may be sufficient to deal with the drug insult without activating the adaptive immune response in the majority of patients. Moreover, the innate immune system has been proposed to be protective, such as in cases of hepatic inflammation induced by acetaminophen (Jaeschke et al., Citation2012).
Similar to previous gene array studies of aromatic amine drugs, successive timepoint testing found that, at least for aromatic amine drugs, the majority of genes are down-regulated at early timepoints. The true implications for these findings are still unknown; however, there may be some compensatory mechanisms in which a threshold needs to be crossed before gene expression is elicited. As seen with these studies, the mere presence of changes in gene expression does not necessarily correlate with pathology. Furthermore, we did not investigate downstream protein expression or signaling pathways because our main goal was only to detect early biomarkers to predict drug risk because, according to the danger hypothesis, it is early injury of the target cells, in this case the hepatocytes, that would determine the immune response.
Overall, the implications of this study are that the effects of AMG on the liver are small, and that the very high incidence of IDRs associated with this drug may originate from outside the liver. The finding that immune cells can also bioactivate aromatic amines may be the key to their ability to cause IDRs (Elsheikh et al., Citation2010). In a very recent study, we found that AMG caused marked effects on the bone marrow of rats, and agranulocytosis is a major IDR caused by AMG (Ng & Uetrecht, Citation2013b). This suggests that extra-hepatic metabolism may be more relevant to the IDRs caused by many drugs and makes the point that it is important to make links between investigations in animals and the type of IDR that a drug causes in patients. It also suggests that, in addition to screening for reactive metabolite formation in the liver, investigating bioactivation by immune cells, especially macrophages, may provide important biomarkers for detecting the potential of whether a drug candidate can cause IDRs. Nevertheless, the changes in gene expression induced by AMG in the liver may provide clues as to why significant liver injury did not occur in rats and why liver injury is not a significant target of IDRs in patients. Specifically, because the predominant immune changes were associated with innate immunity, it is quite possible that innate immune cells are able to effectively deal with injury caused by the drug and effectively down-regulate any potential adaptive immune response. We have performed high through-put arrays with multiple IDR-associated drugs and found differential changes suggestive of cellular stress and an immune response at early timepoints with most. The major exceptions are primary aromatic amine drugs; that emphasizes the uniqueness of this chemical moiety and the importance of consideration for all sources of drug metabolism in the induction of adverse effects.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper. This research was funded by the Canadian Institutes of Health Research. J.P.U. is the Canada Research Chair in Adverse Reactions.
| Abbreviations | ||
| IDRs, | = | idiosyncratic drug reactions; |
| AMG, | = | aminoglutethimide; |
| HLA, | = | human leukocyte antigen; |
| GLDH, | = | glutamate dehydrogenase; |
| ALT, | = | alanine aminotransferases; |
| HRP, | = | horseradish peroxidase; |
| H&E, | = | hematoxylin and eosin; |
| DAB, | = | 3′3 diaminobenzidine; |
| PBS, | = | phosphate buffered saline; |
| TUNEL, | = | terminal deoxynucleotidyl transferase dUTP nick end labeling. |
Acknowledgements
We thank Dr Ping Cai for his input into the experimental design of these studies. We also thank Roche for generously supplying the PCR array plates and reagents.
References
- Bowen, D. G., Mccaughan, G. W., and Bertolino, P. 2005. Intrahepatic immunity: A tale of two sites? Trends Immunol. 26:512–517
- Cocconi, G. 1994. First generation aromatase inhibitors-aminoglutethimide and testololactone. Breast Cancer Res.Treat. 30:57–80
- Crispe, I. N., Dao, T., Klugewitz, K., et al. 2000. The liver as a site of T-cell apoptosis: Graveyard or killing field? Immunol. Rev. 174:47–62
- Dissanayake, D., Hall, H., Berg-Brown, N., Elford, A. R., et al. 2011. Nuclear factor-κB1 controls the functional maturation of dendritic cells and prevents the activation of autoreactive T-cells. Nat. Med. 17:1663–1667
- Dutta, J., Fan, Y., Gupta, N., Fan, G., and Gelinas, C. 2006. Current insights into the regulation of programmed cell death by NF-κB. Oncogene 25:6800–6816
- Elsheikh, A., Lavergne, S. N., Castrejon, J. L., et al. 2010. Drug antigenicity, immunogenicity, and costimulatory signaling: Evidence for formation of a functional antigen through immune cell metabolism. J. Immunol. 185:6448–6460
- Eweiss, N., Nicholls, P. J., and Askam, V. 1983. Absorption and elimination of aminoglute-thimide in the rat and guinea pig. IRCS Med. Sci. Biochem. 11:843–844
- Goldkind, L., and Laine, L. 2006. A systematic review of NSAIDs withdrawn from the market due to hepatotoxicity: Lessons learned from the bromfenac experience. Pharmacoepidemiol. Drug Safety 15:213–220
- Goss, P. E., Jarman, M., and Griggs, L. J. 1985. Metabolism of aminoglutethimide in humans: Quantification and clinical relevance of induced metabolism. Br. J. Cancer 51:259–262
- Hayden, M. S., West, A. P., and Ghosh, S. 2006. NF-κB and the immune response. Oncogene. 25:6758–6780
- Jaeschke, H., Williams, C. D., Ramachandran, A., and Bajit, M. L. 2012. Acetaminophen hepatotoxicity and repair: The role of sterile inflammation and innate immunity. Liver Int. 32:8–20
- Kono, H., and Rock, K. L. 2008. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8:279–289
- Lu, W., Li, X., and Uetrecht, J. P. 2008. Changes in gene expression induced by carbamazepine and phenytoin: Testing the Danger Hypothesis. J. Immunotoxicol. 5:107–113
- Metushi, I. G., Nakagawa, T., and Uetrecht, J. 2012. Direct oxidation and covalent binding of isoniazid to rodent liver and human hepatic microsomes: Humans are more like mice than rats. Chem. Res. Toxicol. 25:2567–2576
- Murray, F. T., Santner, S., Samojlik, E., and Santen, R. J. 1979. Serum aminoglutethimide levels: Studies of serum half-life, clearance, and patient compliance. J. Clin. Pharmacol. 19:704–711
- Murray, M., Cantrill, E., and Farrell, G. C. 1993. Induction of cytochrome P450 2B1 in rat liver by the aromatase inhibitor aminoglutethimide. J. Pharmacol. Exp. Ther. 265:477–481
- Nagel, G. A., Wander, H. E., and Blossey, H. C. 1982. Phase II study of aminoglutethimide and medroxyprogesterone acetate in the treatment of patients with advanced breast cancer. Cancer Res. (Suppl.) 42:3442s–3444s
- Ng, W., and Uetrecht, J. 2013a. Changes in gene expression induced by aromatic amine drugs: Testing the danger hypothesis. J. Immunotox. 10:178–191
- Ng, W., and Uetrecht, J. 2013b. Effect of aminoglutethimide on neutrophils in rats - implications for idiosyncratic drug-induced blood dyscrasias. Chem. Res. Toxicol. 26:1271–1281
- Ng, W., Lobach, A. R. M., Zhu, X., et al. 2012. Animal models of idiosyncratic drug reactions. Adv. Pharmacol. 63:81–135
- Pacitto, S. R., Uetrecht, J. P., Boutros, P. C., and Popovic, M. 2007. Changes in gene expression induced by tienilic acid and sulfamethoxazole: Testing the danger hypothesis. J. Immunotoxicol. 4:253–266
- Seguin, B., Boutros, P. C., Li, X., et al. 2005. Gene expression profiling in a model of D-penicillamine-induced autoimmunity in the Brown Norway rat: Predictive value of early signs of danger. Chem. Res. Toxicol. 18:1193–1202
- Uetrecht, J. 2002. N-Oxidation of drugs associated with idiosyncratic drug reactions. Drug Metab. Rev. 34:651–665
- Uetrecht, J., and Naisbitt, D. J. 2013. Idiosyncratic adverse drug reactions: Current concepts. Pharmacol. Rev. 65:779–808
- Waring, J. F., Ciurlionis, R., Jolly, R. A., et al. 2001. Microarray analysis of hepatotoxins in vitro reveals a correlation between gene expression profiles and mechanisms of toxicity. Toxicol. Lett. 120:359–368