
Abstract
Respiratory disorders in sulfur mustard (SM)-exposed and chronic obstructive pulmonary disease (COPD) patients are mostly associated with neutrophilic inflammation, severe airflow limitation, and oxidative stress. The objective of this study was to establish whether neutrophil (PMN) proteomes in these diseases were similar or differed. Blood neutrophil proteomes from healthy, SM-exposed, and COPD subjects were analyzed using two-dimensional gel electrophoresis followed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI TOF MS). Elastase activity was determined kinetically. The results showed that levels of S100 calcium-binding protein (CBP) A12, S100 CBP A8, glyceraldehyde-3-phosphate dehy-drogenase, superoxide dismutase, and protein disulfide isomerase proteins – as well as elastase activity – were significantly increased in PMN from ‘diseased’ hosts compared to in cells from healthy controls. In contrast, coactosin-like protein, RhoGDP dissociation inhibitor, and actin isoforms were significantly decreased in diseased subjects’ PMN compared to PMN of healthy controls. Moreover, serpin B1 and coronin-1A were expressed only in PMN of the healthy subjects. Lastly, S100 CBP A9, endoplasmic reticulum (ER)-60 protease, and glutathione-S-transferase isoforms were differentially expressed in the cells from the SM-exposed and COPD subjects. These results show that serpin B1, an efficient inhibitor of neutrophil serine proteases, was not detectable, and elastase activity significantly increased in PMN from both SM-exposed and COPD patients. It seems that, apart from inflammation and oxidative stress, a protease:anti-protease imbalance exists within PMN of both COPD and SM-exposed patients.
Introduction
Long-term pulmonary complications from sulfur mustard (SM) exposure have included chronic obstructive pulmonary disease (COPD), chronic bronchitis, bronchiolitis obliterans, bronchiectasis, airway hyper-reactivity, and lung fibrosis (Balali-Mood et al., Citation2005; Emad & Rezaian, Citation1997; Ghanei et al., Citation2008a,Citationb; Khateri et al., Citation2003). The SM-exposed patients suffer from pulmonary disorders due to previous documented exposure to a single high dose of SM gas they faced during the Iran–Iraq conflict in 1987. Late-onset complications from the gas exposure were found to appear almost 10 years after these exposures (Emad & Rezaian, 1997). Although many years have passed since the SM exposure, overactive inflammatory processes are still evident in many SM-exposed veterans (Weinberger et al., Citation2011; Yaraee et al., Citation2009). A systemic inflammation associated with pulmonary complications that is common among SM-exposed patients has been investigated (Attaran et al., Citation2009; Emad & Emad, Citation2007). The appearance of pulmonary inflammation in SM-exposed patients is often accompanied by changes in markers of oxidative stress in the lungs, e.g. increases in malondialdehyde (indicator of lipid peroxidation), and decreases in both general anti-oxidants and in glutathione (Jafari & Ghanei, Citation2010; Shohrati et al., Citation2008, Citation2010).
Many lung pathologies associated with SM exposure, such as systemic inflammation and increases in oxidative stress, are also a part of the pathophysiology of COPD (Heaney et al., Citation2007; MacNee, Citation2005; Wouters, Citation2005). As neutrophils (PMN) are a major cell type involved in COPD (Cowburn et al., Citation2008; Nathan, Citation2006; Oudijk et al., Citation2005; Quint & Wedzicha, Citation2007), this could imply PMN-dominated inflammatory processes also characterize many SM-related lung pathologies.
Systemic inflammatory responses are accompanied by changes in the phenotype of circulating PMN (i.e. resting, primed, or activated); these changes are induced by cytokines and chemokines and are likely characterized by epigenetic changes (Oudijk et al., Citation2005). Phenotype changes not only alter the PMN shape and permit infiltration of injured tissues, but also make the cells functionally hyper-active in response to stimuli (Pillay et al., Citation2010; Sheppard et al., Citation2005). Neutrophil serine proteases (NSP), i.e. elastase, cathepsin G, and proteinases-3, are high turnover enzymes present in/released by circulating PMN. These NSP can: degrade structural proteins; contribute to pathologies like bronchiolectasis and bronchiectasis; exacerbate airway dysfunction by increasing mucin release; propagate inflammation by inducing pro-inflammatory cytokines; and diminish anti-microbial defenses by cleaving phagocyte receptors, antibodies, and both complement and surfactant proteins (Cooley et al., Citation2011). Normally, released NSP are rapidly inactivated by protease inhibitors encountered in the plasma; however, in extravascular spaces, local conditions – such as a deficiency of inhibitors or an excess of the NSP – can extend the functional lifespan of these enzymes (Cooley et al., Citation2011). A detailed analysis of NSP expression profiles has shown that there is a tight transcriptional peak of expression in bone marrow pro-myelocytes that declines rapidly as the PMN mature (Theilgaard-Mönch et al., Citation2005).
Due to the role of PMN in many pulmonary pathologies and the critical nature of some of their products (i.e. NSP) in onset/progression of these adverse states, a study of PMN proteome profiles could provide key information about the role of these cells in respiratory disorders like COPD and SM-induced lung injury. In fact, through the use of proteomics, biomarkers in various diseases in which PMN play an important role have been identified (Langereis et al., Citation2011; Teles et al., Citation2012). Our own previous findings with SM-exposed patients revealed there were differing proteomic profiles between mild, moderate, and severe cases of exposure (Mehrani et al., Citation2009, Citation2011). Specifically, proteomic analyses of bronchoalveolar lavage (BAL) fluids from SM-exposed patients indicated that PMN and S100A8 protein expression (mostly produced by PMN) were significantly increased in an exposure dose-related manner (Mehrani et al., Citation2009). Moreover, proteomic analyses of plasma from these patients revealed a significant increase in serum amyloid A1 (marker of systemic inflammation) as well (Mehrani et al., Citation2011). However, there have been no studies of time-related changes in these profiles, in great part because the technologies available now did not exist in those earliest days after the SM exposures took place.
Nevertheless, building on those earlier findings, we hypothesize here that the altered protein expression levels observed in inflamed airways of COPD and SM-exposed subjects might be attributed in part to alterations in their formation/release by PMN that are/were recruited to the lungs during the disease progression. Specifically, in this study, two-dimensional gel electrophoresis (2-DE) followed by matrix assisted laser desorption ionization time-of-flight mass spectrometry (MALDI TOF MS) was used to investigate possible alterations in proteomes of PMN (in this case, peripheral blood PMN) isolated from COPD and SM-exposed patients.
Materials and methods
Chemicals
All chemicals used were analytical grade or equivalent and were obtained from Sigma (St. Louis, MO) unless otherwise stated. Milli-Q water was used in all experiments.
Patient population
This Baqiyatallah University of Medical Sciences Ethics Committee (Tehran, Iran) approved this study. For the actual study, two all male patient groups were defined, i.e. 10 SM-exposed and 10 COPD; a group of 10 healthy male individuals served as controls. The lung injuries in the study patients were defined as moderate according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) classification and based on spirometric and high resolution computed tomography findings (data not shown). Informed consent was obtained from all patients and healthy controls; all participants were free to leave the study if they wished.
The SM-exposed patients were suffering from pulmonary disorders due to previous documented exposure to a single high dose of SM gas during the Iran–Iraq conflict in 1987. All SM-exposed as well as the COPD patients had stable conditions without any exacerbation in the last 4 weeks prior to entry into the study. Applicants with other inflammatory conditions, chronic disease (diabetes, hypertension, heart disease, etc.), pneumonia, acute bronchitis, and/or who were undergoing lung surgical treatment or had consumed oral glucocorticoids were excluded.
Sample collection and preparation
All fasting venous blood samples were drawn in the morning (8–10 am). Blood samples (≈40 ml) from each participant were drawn into EDTA-coated tubes and placed on ice. Neutrophils (average PMN yield ∼4 × 107 cells) were isolated by density gradient centrifugation (Axis-Shield PoC AS, Oslo, Norway) according to the manufacturer instructions. Residual erythrocytes were lysed using isotonic ice-cold 0.84% [w/v] ammonium chloride solution. Samples were centrifuged at 500 × g and the PMN pellet washed twice with, and then suspended in, phosphate-buffered saline (PBS, pH 7.4) without Ca2+ and Mg2+ so as to prevent PMN activation. An aliquot was taken for cell counts (Neubauer chamber); cell viability was examined via trypan blue dye exclusion. Cell viability and purity was routinely >97%. Re-suspension of each pellet was performed slowly; vortex settings and centrifugation were kept in the low range so that cells did not become activated. To remove any contaminant plasma proteins, the PMN from each individual subject were then washed twice with a 0.34 M sucrose/1 mM EDTA/10 mM Tris solution. Thereafter, the cells were lysed in a pH 7.5 buffer containing 1% Triton X100, 20 mM Tris base, 1% 3-[(cholamidopropyl) dimethylamino]-1-propanesulfonate (CHAPS) and protease inhibitor cocktail (Sigma). Each set of cells was then solubilized by sonication (short bursts) while the samples were held on ice. The materials were then centrifuged (10,000 × g, 10 min, 4 °C); resultant supernatants were transferred to new tubes and stored at −80 °C for later analyses. Protein levels in aliquots of each sample were determined using a bicinchoninic acid (BCA) assay as previously described (Mehrani et al., Citation2009).
2-D gel electrophoresis (2-DE)
For 2-DE, a protein sample from each individual subject was incubated at room temperature for 30 min, and then a volume containing 500 µg protein was mixed with re-hydration buffer [7 M urea, 2 M thiourea, 4% [w/v] CHAPS, 0.5% immobilized pH gradient (IPG) buffer [matching the 24-cm linear IPG strips {pH 4-7}], 45 mM dithiothreitol (DTT), and 0.1% [w/v] bromophenol blue]. Thereafter, 2-DE analysis, protein staining, and spot analysis were done as described in Mehrani et al. (Citation2011).
Gel analysis and protein identification
For protein spot analysis, each gel generated for each individual patient and control was inspected. Only spots that were reproducibly and significantly different in intensity and those not matched in the patients versus the controls were selected for use in the mass analyses. Each protein spot was carefully inspected for inappropriate matching, staining artifacts, or bad-spot detection before any decision on its utility for further analysis was made.
Each protein spot of interest was aseptically picked and the gel plug processed as noted in Mehrani et al. (Citation2011). MALDI TOF MS was performed using a Bruker Autoflex III MALDI TOF/TOF instrument (Bruker, Kalkar, Denmark). An external peptide calibration standard containing bovine serum albumin (BSA) and transferrin was used for calibration. Spectra were acquired in a positive reflector mode. A list of peptides was obtained using a SNAP algorithm (Flexanalysis 3.0, Bruker). The most intense peaks were selected for tandem mass spectrometry (MS/MS) scans to obtain as many CID spectrums as possible. The mass spectrometry (MS) and MS/MS spectra were combined and used for database searches using MASCOT software (Matrix science, version 2.2.03) with the following selection criteria: Database search program = NCBInr (7930315 protein sequence); species of origin = Homo sapiens; peptide ion mass tolerance = 60 ppm; MS/MS tolerance = 0.2 Da; peptide cut-off = 25; and digestion by trypsin allowing for no more than one missed cleavage. The accuracy of mass detection was an MH+ (mass fragment plus proton) of 0.01, assuming a possibility of modification of cysteine by acrylamide and/or oxidation of methionine. Protein identifications were based on a combination of the peptide mass fingerprinting (PMF) with peptide masses and several MS/MS spectra of selected peptides in each MALDI MS spectrum. Positive protein identification was based on a probability-scoring algorithm (www.matrixscience.com); the 95% confidence level for positive identification was a score of 100.
PMN elastase activity assay
PMN elastase activity was assessed at 37 °C using a colorimetric assay employing the chromogenic peptide substrate N-methoxy-succinyl Ala-Ala-Pro-Val-p-nitroanilide (Sigma) (Oltmanns et al., Citation2005). In brief, 50 µl supernatant from the PMN lysate (without protease inhibitors) was incubated with HEPES buffer (0.1 M) containing 0.5 M NaCl, 10% dimethyl sulfoxide, and 2 mM substrate at pH 7.5. The amount of p-nitroanilide liberated was then measured at 405 nm in a Smart Spec Plus spectrophotometer (BioRad, Hercules, CA).
Statistical analysis
All data are representative of at least three independent experiments, with 10 individuals in each group. All data are expressed as mean ± SEM. Comparisons between groups were performed using a two-way analysis of variance (ANOVA) with post-hoc analysis using a Tukey’s range test in multiple groups of sample. The criterion for statistical significance was p < 0.05 for all comparisons.
Results
Patient data
Human PMN proteomics were performed for 30 participating male subjects, including 10 healthy controls, 10 patients suffering from SM-induced lung disease and 10 COPD patients. The mean (± SEM) age and clinical findings of the patients and controls are shown in .
Table 1. Clinical characteristics, spirometric, hematologic, andserologic findings in study subjects.
PMN proteome profiles
To investigate protein expression patterns in PMN from the participating subjects, 2-DE methods were applied. Initially, for the first dimension, 24-cm IPG strips (with pH value in 4–7 range) were used to obtain a greater resolution on protein separation. In the second dimension, proteins were resolved using 12% homogeneous gels. A typical 2-D gel representing the PMN of healthy subjects and the two test group subjects had ≈350 protein spots (as detected by colloidal Coomassie blue [CBB] staining). illustrates a representative protein pattern from PMN isolated from healthy, SM-exposed, and COPD subjects. Differences in PMN protein patterns were defined by comparing the gels generated using lysates from the PMN obtained from the subjects in each group. The analyses of protein patterns focused on those protein spots that indicated differences (comparing test patients against the controls) and that could be identified with sufficient reliability. These were compared with Image Master 2D software that indicated only protein results in all cases (100%) with the same condition. To identify the altered proteins, all differentially-expressed protein spots were subjected to MALDI TOF MS/MS analysis. The results indicating the identified proteins and their isoforms are presented in .
Figure 1. Representative 2-D protein patterns of PMN from study subjects. (A) Healthy control. (B) COPD patient. (C) SM-exposed subject. Sample preparation and 2-D analysis was performed as described in Materials and methods. Proteins (500 µg/subject) were separated first using linear IPG strips (pH 4–7) followed by 12% SDS-PAGE. Protein spots were then visualized by colloidal CBB staining.
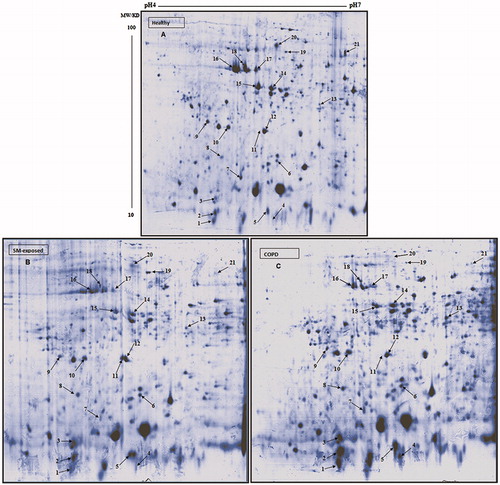
Table 2. Sequence coverage (in %) and MASCOT scores of proteins spots analyzed by MS/Ms.
Altered protein expression in PMN
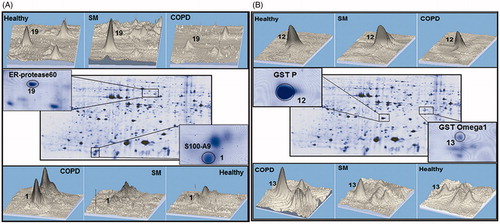
Analyses of proteome profiles of PMN from the different experimental groups showed there were significant differences in protein expression (, ). The data presented (each as mean ± SEM; n = 10/subject group) indicates the statistical analyses of the percentage spots volume on the gels for the diseased and healthy control subjects, with associated p values. Among the differentially-expressed proteins, the S100 family was predominant. Levels of S100 calcium binding protein A12 (S100A12) (spots 2 and 3) and S100 calcium binding protein A8 (S100A8) (spot 4) were significantly increased in PMN from COPD and SM-exposed patients compared to in cells from the healthy controls. Similarly, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (spot 5), superoxide dismutase (SOD) (spot 6), and protein disulfide isomerase (PDI) (spot 8) were each significantly increased in both disease groups compared to in PMN of healthy controls. In contrast to these same trends in changes among the cells from both test groups, the level of S100 calcium binding protein A9 (S100A9) (spot 1) was significantly increased only in PMN of COPD patients versus in the cells of the controls. Similarly, glutathione-S-transferase (GST) omega-1 (spot 13) expression was significantly increased versus in healthy controls’ cells again only in COPD PMN. On the other hand, GST P (spot 12) was significantly decreased only in these same PMN versus in control subject’s cells. ER-60 protease (spot 19) was significantly up-regulated in PMN from the SM-exposed patients, but significantly down-regulated in the PMN of the COPD patients.
Table 3. Up-/down-regulated proteins in PMN of SM-exposed and COPD patients versus PMN of healthy controls.
A representative 3-D view of some up- and down-regulated proteins, such as serpin B1, SOD, PDI, and S100A12, that were found in the COPD and SM-exposed subjects’ PMN are presented in . Coactosin-like protein (spot 7), Rho GDP-dissociation inhibitor (Rho GDPDI) (spot 9), Rho GDPDI 2 (spot 10), and actin isoforms (spots 14-17) were significantly decreased in the PMN from both disease groups. Further, serpin B1 (spot 20) and cronin-1A (spot 21) were present in the PMN of the healthy controls but not detectable in cells from SM-exposed or COPD patients. presents some of the differentially expressed proteins that significantly differed among the three groups of subjects assessed here.
Figure 2. Representative 3-D images of differentially-expressed proteins in PMN. Differentially-expressed proteins spots in lysate from PMN of the same control, SM-exposed, and COPD subject shown in . (A) PDI (Spot 8) and SOD (Spot 6). (B) SerpinB1 (Spot 20) and S100A12 (Spot 2). Data were analyzed using Image master 2-D platinum software.
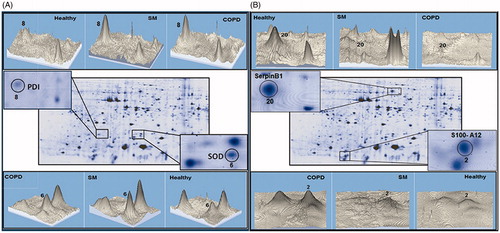
Figure 3. Differenially-expressed proteins in PMN from SM-exposed and COPD patients. Protein spots expressed differently in the PMN of the same SM-exposed and COPD patients shown in . (A) ER-proteses 60 (Spot 19) and S100A9 (spot 1). (B) GSTp (Spot 12) and GST omega1 (Spot 13). The 3-D images of differential spots between the patients were analyzed using ImageMaster 2D Platinum software.
Discussion
The present study sought to assess any differential proteomic profiles between blood PMN recovered from SM-exposed, COPD, and age-matched healthy control subjects. Recent genomic and proteomic studies showed that PMN isolated from COPD patients displayed altered protein profiles and mRNA expression patterns (Langereis et al., Citation2011; Oudijk et al., Citation2005; Pillay et al., Citation2010). The main finding in the present study’s non-control subjects was that the proteomes of their PMN had over-expression of S100A8, S100A12, PDI, GAPDH, and SOD, and under-expression of serpin B1, cronin 1A, actin isoforms, coactosin, and Rho GDPDI.
S100 proteins are an extremely diverse and highly specialized family of Ca2+-binding proteins with both intra- and inter-cellular functions. The S100 gene family is composed of at least 20 members that share a common structure defined, in part, by the Ca2+-binding EF-hand motif (similar to calmodulin). Expression of S100A8 and S100A12 were both significantly increased in the PMN from the SM-exposed and COPD hosts. This was in keeping with our previous study on BAL fluid from SM exposed patients that also showed significant increases in S100A8 protein (Mehrani et al., Citation2009). S100A12 is constitutively expressed in PMN (∼5% of cytosolic protein) and, as a calcium-, zinc-, and copper-binding protein, plays a prominent role in regulation of inflammatory processes and immune responses. Pro-inflammatory activity involves recruitment of leukocytes, promotion of cytokine synthesis/release, and regulation of leukocyte adhesion and migration. The best-known target of S100A12 is RAGE (receptor for advanced glycation end-products). Ligation of S100A12 with RAGE is followed by activation of intra-cellular signal cascade markers such as MAP-kinase and nuclear factor (NF)-κB, leading to production of pro-inflammatory cytokines like tumor necrosis factor (TNF)-α and interleukin (IL)-1β (Yang et al., Citation2007). The net effect of these responses is to mediate pro-inflammatory effects on lymphocytes, endothelial cells, PMN, and mononuclear phagocytes.
Protein disulfide isomerase (PDI) was significantly increased in the PMN from both the SM-exposed and the COPD subjects. PDI is a redox chaperone that classically responds to un-folded proteins; its up-regulation has been reported in the lungs of smokers (Kelsen et al., Citation2008). PDI also interacts with p47 phox one of the NADPH oxidase sub-unit in PMN and causes increased reactive oxygen species (ROS) production. PDI is also known as a cytoskeleton re-organizing protein (Chaudhuri et al., Citation2001; Fernandes et al., Citation2009; Laurindo et al., Citation2012). Hahm et al. (Citation2013) showed that PMN PDI was required for PMN adhesion and crawling during TNFα-induced vascular inflammation. Expression of the glycolytic enzyme GAPDH was also significantly increased in PMN from both SM-exposed and the COPD subjects. GAPDH, considered a simple ‘housekeeping’ protein, has been proven to be involved in many cellular processes in addition to glycolysis including: DNA repair, tRNA export, membrane fusion and transport, cytoskeleton dynamics, and cell death (Tristan et al., Citation2011). GAPDH is one of the first glycolytic enzymes known to interact with tubulin and actin, facilitating microtubule bundling and actin polymerization, respectively. Thus, the decrease in expression of the actin isoforms and the increase in GAPDH in the cells from SM-exposed and COPD subjects might reflect changes in PMN cytoskeleton dynamics (i.e. motility and activity) in these hosts’ lungs.
Apart from inflammatory reactions and oxidative stress, a shift toward a domination of activities of proteases over those of anti-proteases in the lungs is also an important factor in the pathology of COPD and in lung illnesses in SM-exposed patients (Barnes et al., Citation2003; Emad & Rezaian, Citation1997). Normally, serpin B1 – a prominent anti-protease among the various NSP inhibitors expressed by PMN – efficiently inhibits activities of NSP using a suicide mechanism (classical for serpins) that irreversibly traps/inactivates proteases (Benarafa et al., Citation2011). While most protease inhibitors can rapidly inactivate any released NSP, a deficiency of these inhibitors (or a concurrent/resultant excess of the NSP themselves) within extravascular spaces can extend the timespan and/or intensity of the destructive effects of the PMN; this process is known to occur during cystic fibrosis (Cooley et al., Citation2011). The lack of the serpin B1 in the PMN of both SM-exposed and COPD subjects suggests that many nascent NSP will be longer-lived, and any NSP-induced damage more intense, in these patients. This phenomenon could also help explain why there was a significant reduction in surfactant protein A (SPA) levels in the lungs of SM-exposed patients (Mehrani et al., Citation2009). SPA is apparently critical to maintaining lung integrity as an increase in its degradation by NSP has been reported in several inflammatory lung diseases (Rubio et al., Citation2004).
Human glutathione-S-transferases (GST) are a functionally diverse family of soluble detoxification enzymes that use reduced glutathione (GSH) during reduction reactions to eliminate many toxicants. Among the GST proteins whose expressions were assessed in the PMN from the SM-exposed and COPD subjects, GST-P was significantly decreased and GST-omega significantly increased only in cells from the COPD patients. GST-omega contains an N-terminal glutathione-binding domain, suggesting it has a role in the metabolism and maintenance of GSH levels in intact cells (Yin et al., Citation2001). Since GSH is a major airway anti-oxidant, it could be surmised that GST-omega might also partake in maintenance of GSH status in the extracellular space, and this process could be modulated by oxidative stress. Glutathione-S-transferase P1 (GSTP1) is widely expressed in normal tissues (Terrier et al., 1990) and is present to protect cells against adverse effects of toxins, carcinogens, and free radicals (Lo & Ali-Osman, Citation2007). Thus, in the case of COPD subjects, the significant increase in GST omega could reflect an increased need for GSH-dependent thiol-transferase reactions to compensate for the diminished presence of GSTP1, and reflect a pattern of overall reduction in ability to mitigate the impact of free radicals generated during local inflammatory events in the lungs. Conversely, the lack of change in GSTP1 and GST omega expression in PMN from the SM-exposed patients could mean that oxidative stresses in these patients were not as severe as in the lungs of COPD patients and, thus, there would be less demand upon GSH stores in the lungs of the SM subjects. Based on these disparate expression patterns, it would be reasonable to assume there should be differences in bronchoalveolar lavage levels of GSH between these two subject classes. Indeed, lavage levels of GSH in the lungs of these subjects have been shown to significantly differ (i.e. 5.7 [± 3.8] versus 2.4 [± 0.9] µmol/L in, respectively, SM-exposed and COPD patients) (Bridgeman et al. Citation1994; Jafari & Ghanei, Citation2010). The present results may now provide a basis for those earlier measures.
The down-regulation of cytoskeleton-related proteins, including actin isoforms and coactosin-like protein, seen in the PMN from the COPD and SM-exposed hosts may reflect changes that may have occurred with regard to cell re-modeling processes in the lungs (Sheppard et al., Citation2005). Coactosin-like protein, which belongs to the actin-depolymerizing factor family of actin-binding proteins, interacts with 5-lipoxygenase and filamentous actin; because extensive compositional re-organization is exhibited in PMN during initial stages of phenotypic conversion (Stie & Jesaitis, Citation2007), upon PMN activation, the assembly of polarized F-actin promotes exocytosis (Mitchell et al., Citation2008). Actin (de)polymerizetion is also important for the process of trans-endothelial migration, the means by which leukocytes exit the blood and enter tissues – a key step in inflammatory processes in organs like the lungs. As Stroka et al. (Citation2013) demonstrated that inhibition of actin (de)polymerization impedes trans-migration, a lesser presence of actin could result in fewer PMN exiting the bloodstream to enter the airways (after passing from alveolar capillaries/venules across the endothelium, extracellular matrix, and finally the alveolar epithelium to access the alveolar lumen).
Because neutrophilic inflammation is a key clinical presentation in the airway wall/lumen of COPD and SM-exposed patients, this suggested to us that the PMN in these patients were still able to perform the three crossings, even with a down-regulated presence of cytoskeleton-related proteins. As this would seem counterintuitive, further study is clearly needed to ascertain if the processes by which the PMN present in the airway lumen/walls are also impacted in COPD and SM-exposed patients. This is important to investigate in that if resolution of the neutrophilic inflammation (which occurs by apoptosis of PMN followed by removal by macrophages) does not occur properly in these hosts, their PMN would need instead to be removed via mucociliary transport. This, unfortunately, could result in release of proteases/chemoattractants that contribute to lung damage via a self-perpetuation of inflammation and localized necrosis (Hoenderdos & Condliffe, Citation2013; Quint & Wedzicha, Citation2007).
Individuals that were exposed to an acute single dose of SM gradually lost their lung function and slowly progressed from mild-to-moderate to severe lung damage over a nearly 25-year period. Such changes are comparable to what is seen during COPD exacerbation and the progressive deterioration of lung function that gradually occurs in smokers (or due to other etiologies). The overall lung function data presented in and the other results noted above indicated to us that COPD and exposure to SM gave rise to similar changes in lung function and, more importantly, in proteomic profiles of PMN. Nevertheless, there were still some key albeit minor differences between the groups with respect to expression of glutathione-S-transferase isoforms, S100A9 CBP, and ER60 proteases. Thus, while the clinical presentations and overall PMN proteomic findings appear similar among the test subjects here, how each of the two health states are ultimately related to the status of each host’s PMN (or other immune cell types) remains to be more fully determined in our ongoing studies.
Conclusions
The comparative profiling of human PMN performed here revealed overall proteome changes within PMN in hosts known to have undergone long periods of respiratory changes. The finding demonstrated that serpin B1 (a prominent anti-protease) and coronin-1A (a cytoskeleton modifier) were not detectable in PMN of SM-exposed or COPD hosts, while elastase activity was significantly increased in those cells as compared to in the PMN of healthy controls. These proteome alterations generally correlated with a protease:anti-protease imbalance in PMN. As such, these findings could be useful in the development of directed-therapies (i.e. against proteases) against COPD and against lingering health effects in individuals acutely exposed to SM decades earlier.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper. The Baqiyatallah University Research Branch and the Chemical Injuries Research Center supported this research.
Acknowledgements
We are grateful to Drs Thomas Kofoed and Ejvind Mørtz (Alphalyse, Denmark) for the mass analysis.
References
- Attaran, D., Lari, S. M., Khajehdaluee, M., et al. 2009. Highly sensitive C-reactive protein levels in Iranian patients with pulmonary complication of sulfur mustard poisoning and its correlation with severity of airway diseases. Human Exp. Toxicol. 28:739–745
- Balali-Mood, M., Hefazi, M., Mahmoudi, M., et al. 2005. Long-term complications of sulfur mustard poisoning in severely intoxicated Iranian veterans. Fundam. Clin. Pharmacol. 19:713–721
- Barnes, P., Shapiro, S., and Pauwels, R. 2003. Chronic obstructive pulmonary disease: Molecular and cellular mechanisms. Eur. Resp. J. 22:672–688
- Benarafa, C., Lecuyer, T. E., Baumann, M., et al. 2011. Serpin B1 protects the mature neutrophil reserve in the bone marrow. J. Leukocyte Biol. 90:21–29
- Bridgeman, M. M., Marsden, M., Selby, C., et al. 1994. Effect of N-acetyl cysteine on the concentrations of thiols in plasma, bronchoalveolar lavage fluid, and lung tissue. Thorax 49:670–675
- Chaudhuri, A. R., Khan, I. A., and Ludueña, R. F. 2001. Detection of disulfide bonds in bovine brain tubulin and their role in protein folding and microtubule assembly in vitro: A novel disulfide detection approach. Biochemistry 40:8834–8841
- Cooley, J., Sontag, M., Accurso, F., and Remold-O’Donnell, E. 2011. Serpin B1 in cystic fibrosis airway fluids: Quantity, molecular form and mechanism of elastase inhibition. Eur. Resp. J. 37:1083–1090
- Cowburn, A. S., Condliffe, A. M., Farahi, N., et al. 2008. Advances in neutrophil biology - clinical implications. Chest 134:606–612
- Emad, A., and Emad, Y. 2007. Levels of cytokine in bronchoalveolar lavage (BAL) fluid in patients with pulmonary fibrosis due to sulfur mustard gas inhalation. J. Interferon Cytokine Res. 27:38–43
- Emad, A., and Rezaian, G. R. 1997. The diversity of the effects of sulfur mustard gas inhalation on respiratory system 10 years after a single, heavy exposure. Analysis of 197 cases. Chest 112:734–738
- Fernandes, D. C., Manoel, A. H., Wosniak, J. Jr., and Laurindo, F. R. 2009. Protein disulfide isomerase over-expression in vascular smooth muscle cells induces spontaneous preemptive NADPH oxidase activation and Nox1 mRNA expression: Effects of nitrosothiol exposure. Arch. Biochem. Biophys. 484:197–204
- Ghanei, M., Shohrati, M., Jafari, M., et al. 2008a. N-Acetyl-cysteine improves clinical conditions of mustard gas-exposed patients with normal pulmonary function test. Basic Clin. Pharmacol. Toxicol. 103:428–432
- Ghanei, M., Tazelaar, H. D., Chilosi, M., et al. 2008b. An international collaborative pathologic study of surgical lung biopsies from mustard gas-exposed patients. Resp. Med. 102:825–830
- Hahm, E., Li, J., Kim, K., et al. 2013. Extracellular protein disulfide isomerase regulates ligand-binding activity of αMβ2 integrin and neutrophil recruitment during vascular inflammation. Blood 121:3789–3800
- Heaney, L. G., Lindsay, J. T., and McGarvey, L. P. 2007. Inflammation in chronic obstructive pulmonary disease: Implications for new treatment strategies. Curr. Med. Chem. 14:787–796
- Hoenderdos, K., and Condliffe, A. 2013. The neutrophil in COPD. Too little, too late, or too much, too soon? Am. J. Respir. Cell Mol. Biol. 48:531–539
- Jafari, M., and Ghanei, M. 2010. Evaluation of plasma, erythrocytes, and bronchoalveolar lavage fluid anti-oxidant defense system in sulfur mustard-injured patients. Clin. Toxicol. 48:184–192
- Kelsen, S. G., Duan, X., Ji, R., et al. 2008. Cigarette smoke induces an unfolded protein response in the human lung: A proteomic approach. Am. J. Respir. Cell Mol. Biol. 38:541–550
- Khateri, S., Ghanei, M., Keshavarz, S., et al. 2003. Incidence of lung, eye, and skin lesions as late complications in 34,000 Iranians with wartime exposure to mustard agent. J. Occup. Environ. Med. 45:1136–1143
- Langereis, J. D., Schweizer, R. C., Lammers, J. W., et al. 2011. A unique protein profile of peripheral neutrophils from COPD patients does not reflect cytokine-induced protein profiles of neutrophils in vitro. BMC Pulm. Med. 11:44
- Laurindo, F. R., Pescatore, L. A., and De Castro Fernandes, D. 2012. Protein disulfide isomerase in redox cell signaling and homeostasis. Free Rad. Biol. Med. 52:1954–1969
- Lo, H. W., and Ali-Osman, F. 2007. Genetic polymorphism and function of glutathione-S-transferases in tumor drug resistance. Curr. Opin. Pharmacol. 7:367–374
- MacNee, W. 2005. Pathogenesis of chronic obstructive pulmonary disease. Proc. Am. Thor. Soc. 2:258–266
- Mehrani, H., Ghanei, M., Aslani, J., and Golmanesh, L. 2009. Bronchoalveolar lavage fluid proteomic patterns of sulfur mustard-exposed patients. Proteomics - Clin. Applic. 3:1191–1200
- Mehrani, H., Ghanei, M., Aslani, J., and Tabatabaei, Z. 2011. Plasma proteomic profile of sulfur mustard-exposed lung diseases patients using 2-dimensional gel electrophoresis. Clin. Proteomics 8:2
- Mitchell, T., Lo, A., Logan, M. R., et al. 2008. Primary granule exocytosis in human neutrophils is regulated by Rac-dependent actin remodeling. Am. J. Physiol. 295:C1354–C1365
- Nathan, C. 2006. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 6:173–182
- Oltmanns, U., Sukkar, M. B., Xie, S., et al. 2005. Induction of human airway smooth muscle apoptosis by neutrophils and neutrophil elastase. Am. J. Respir. Cell Mol. Biol. 32:334–341
- Oudijk, E. D., Nijhuis, E., Zwank, M., et al. 2005. Systemic inflammation in COPD visualized by gene profiling in peripheral blood neutrophils. Thorax 60:538–544
- Pillay, J., Ramakers, B. P., Kamp, V. M., et al. 2010. Functional heterogeneity and differential priming of circulating neutrophils in human experimental endotoxemia. J. Leukocyte Biol. 88:211–220
- Quint, J. K., and Wedzicha, J. A. 2007. The neutrophil in chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 119:1065–1071
- Rubio, F., Cooley, J., Accurso, F., and Remold-O’Donnell, E. 2004. Linkage of neutrophil serine proteases and decreased surfactant protein-A (SP-A) levels in inflammatory lung disease. Thorax 59:318–323
- Sheppard, F. R., Kelher, M. R., Moore, E. E., et al. 2005. Structural organization of the neutrophil NADPH oxidase: Phosphorylation and translocation during priming and activation. J. Leukocyte Biol. 78:1025–1042
- Shohrati, M., Ghanei, M., Shamspour, N., et al. 2010. Glutathione and malondialdehyde levels in late pulmonary complications of sulfur mustard intoxication. Lung 188:77–83
- Shohrati, M., Ghanei, M., Shamspour, N., and Jafari, M. 2008. Activity and function in lung injuries due to sulfur mustard. Biomarkers 13:728–733
- Stie, J., and Jesaitis, A. J. 2007. Reorganization of the human neutrophil plasma membrane is associated with functional priming: Implications for neutrophil preparations. J. Leukocyte Biol. 81:672–685
- Stroka, K. M., Hayenga, H. N., and Aranda-Espinoza, H. 2013. Human neutrophil cytoskeletal dynamics and contractility actively contribute to trans-endothelial migration. Plos One 8:1–11
- Teles, L. M., Aquino, E. N., Neves, A. C., et al. 2012. Comparison of the neutrophil proteome in trauma patients and normal controls. Protein Peptide Lett. 19:663–672
- Terrier, P., Townsend, A. J., Coindre, J. M., et al. 1990. An immune-histological study of Pi class glutathione-S-transferase expression in normal human tissue. Am. J. Pathol. 137:845–853
- Theilgaard-Mönch, K., Jacobsen, L. C., Borup, R., et al. 2005. The transcriptional program of terminal granulocytic differentiation. Blood 105:1785–1796
- Tristan, C., Shahani, N., Sedlak, T. W., and Sawa, A. 2011. The diverse functions of GAPDH: Views from different subcellular compartments. Cell. Signal. 23:317–323
- Weinberger, B., Laskin, J. D., Sunil, V. R., et al. 2011. Sulfur mustard-induced pulmonary injury: Therapeutic approaches to mitigating toxicity. Pulm. Pharmacol. Ther. 24:92–99
- Wouters, E. F. 2005. Local and systemic inflammation in chronic obstructive pulmonary disease. Proc. Am. Thor. Soc. 2:26–33
- Yang, Z., Yan, W. X., Cai, H., et al. 2007. S100A12 provokes mast cell activation: A potential amplification pathway in asthma and innate immunity. J. Allergy Clin. Immunol. 119:106–114
- Yaraee, R., Ghazanfari, T., Faghihzadeh, S., et al. 2009. Alterations in serum levels of soluble L, P, and E-selectin 20 years after sulfur mustard exposure: Sardasht-Iran Cohort Study. Int. Immunopharmacol. 9:1477–1481
- Yin, Z. L., Dahlstrom, J. E., Le Couteur, D. G., and Board, P. G. 2001. Immunohistochemistry of omega class glutathione-S-transferase in human tissues. J. Histochem. Cytochem. 49:983–987