
Abstract
Acrolein (ACR), an α,β-unsaturated aldehyde and a major component of tobacco smoke, is a highly reactive electrophilic respiratory irritant implicated in asthma pathogenesis and severity. However, few studies have directly investigated the influence of ACR exposure on allergen sensitization and pulmonary inflammation. The present study was designed to examine the impact of ACR inhalation on allergic sensitization to the inhaled antigen ovalbumin (OVA), as well as pulmonary inflammation during subsequent OVA challenge. Adult male C57BL/6 mice were exposed to inhaled OVA (1%, 30 min/day, 4 days/week) and/or ACR (5 ppm, 4 h/day, 4 days/week) over 2 weeks and subsequently challenged with aerosolized OVA (1%, 30 min/day) over three consecutive days. Serum anti-OVA IgG1 levels were increased significantly in animals exposed to both OVA and ACR, compared to animals exposed to either OVA or ACR alone. In addition, differential cell counts and histological analysis revealed an increase in BAL neutrophils in animals exposed to both OVA and ACR. However, exposure to both OVA and ACR did not influence mRNA expression of the cytokines il5, il10, il13 or tnfa, but significantly increased mRNA expression of ccl20. Moreover, ACR exposure enhanced lung mRNA levels of il17f and tgfb1, suggesting development of enhanced inhalation tolerance to OVA. Overall, the findings indicate that ACR inhalation can promote airway-mediated sensitization to otherwise innocuous inhaled antigens, such as OVA, but also enhances immune tolerance, thereby favoring neutrophilic airway inflammation.
Introduction
Asthma is a chronic inflammatory disease of the airways associated with reoccurring symptoms that limit the flow of air to the lungs, such as wheezing, coughing, shortness of breath and chest tightness (Murdoch & Lloyd, Citation2010; Murphy & O’Byrne, Citation2010). The worldwide prevalence of asthma has steadily increased since the 1970s (Sears, Citation2014; To et al., Citation2012) and current estimates suggest that ∼300 million individuals are diagnosed with asthma globally (WHO, Citation2007). Individual pathogenesis of asthma arises from a complex inter-play between genetic predisposition and environmental exposures (Holgate et al., Citation2007; Mukherjee & Zhang, Citation2011). Yet, the continually rising prevalence of asthma, especially in heavily industrialized areas (Anandan et al., Citation2010; Wong et al., Citation2013), has prompted research to focus increasingly on the environmental aspect. To date, many studies have implicated various indoor and outdoor chemical air pollutants in asthma pathogenesis and asthma severity (Dick et al., Citation2014; Holgate et al., Citation2009; Strachan, Citation2000). One major environmental pollutant with constant worldwide human exposure (Oberg et al., Citation2011; WHO, Citation2008) and known association with pulmonary (CDC, Citation2004) and cardiovascular (Dunbar et al., Citation2013; Huxley & Woodward, Citation2011) diseases is cigarette smoke.
The relationship between environmental cigarette smoke exposure and asthma is not fully understood. Several epidemiologic studies have reported that exposure to either firsthand or secondhand cigarette smoke exacerbates asthma symptoms (Hersoug et al., Citation2010; Jung et al., Citation2012; Papaioannou et al., Citation2010; Stapleton et al., Citation2011) and increases the risk of developing asthma (Hunt et al., Citation2011; Mitchell et al., Citation2012; Tsai et al., Citation2010). However, other studies have reported no increased risk of asthma development in smokers (Mohammad et al., Citation2013; Siroux et al., Citation2000), while some have even reported a lowered risk of asthma in smokers compared to non-smokers and ex-smokers (Hjern et al., Citation2001; Troisi et al., Citation1995). A similar paradigm has been reported in studies using animal models of allergic asthma and cigarette smoke exposure (Lanckacker et al., Citation2013; Melgert et al., Citation2004; Robbins et al., Citation2005; Thatcher et al., Citation2008; Trimble et al., Citation2009). Biochemical interactions between cigarette smoke and lung tissue are highly complex, in part due to the chemical heterogeneity of cigarette smoke (Rodgman & Perfetti, Citation2013). Several lines of evidence (Rahman & MacNee, Citation1999) suggest that thiol-reactive components in cigarette smoke may be the main contributors of airway disease and that acrolein (ACR), an α,β-unsaturated aldehyde, is one of the most abundant thiol-reactive components in cigarette smoke (Faroon et al., Citation2008; Reddy et al., Citation2002). Therefore, examining ACR-induced alterations to airway inflammation can potentially provide clarification to the cigarette smoke-asthma dichotomy reported in the literature.
ACR is routinely produced by combustion of organic materials and has been identified as one of the non-cancerous air pollutants of greatest concern by the US Environmental Protection Agency (Leikauf, Citation2002). While a maximum exposure limit for ACR in ambient air has not been established, the permissible limit of indoor ACR exposure established by the US Occupational Safety and Health Administration is 0.1 ppm (0.25 mg/m3). Sources of ACR include wood smoke, automobile exhaust, power plant emissions and heated cooking oil (Bein & Leikauf, Citation2011; Faroon et al., Citation2008). However, the most common and one of the most concentrated sources of human exposure to ACR is cigarette smoke (Stevens & Maier, Citation2008). Cigarette smoke has been estimated to contain up to 220 μg ACR/cigarette and mainstream tobacco smoke contains ACR at levels up to 90 ppm (≈206 mg/m3). Epidemiologic evidence suggests that human exposure to ACR is associated with increased risk of developing asthma (Leikauf, Citation2002), although few reports have studied the influence of ACR exposure on asthma development and severity of pulmonary inflammation.
We recently reported that airway exposure to ACR in mice that were previously sensitized to ovalbumin (OVA) results in attenuated pulmonary inflammation in response to antigen challenge, due to the anti-inflammatory properties of this electrophile (Spiess et al., Citation2013). However, our previous study utilized a model of intraperitoneal antigen sensitization and did not address the potential impact of ACR inhalation on airway-dependent antigen sensitization, which would be more relevant to asthma development in humans. In the current study, we addressed whether ACR inhalation can enhance sensitization to inhaled antigen, as was previously reported with respect to environmental cigarette smoke exposure (Rumold et al., Citation2001; Trimble et al., Citation2009) and analogous to antigen sensitization during co-exposure to other environmental pollutants (Bevelander et al., Citation2007; Hollingsworth et al., Citation2010). Here, we report that co-exposure to ACR and aerosolized OVA indeed promotes antigen sensitization, indicated by increased antigen-specific IgG1, a marker of T-helper type 2 (TH2)-biased adaptive immune responses, although this was not accompanied by enhanced allergic inflammation, but rather by increased pulmonary neutrophilic inflammation.
Methods and materials
Animals
Eight-week-old male C57BL/6 mice were obtained from Charles River (Wilmington, MA). Animals were randomly assigned to one of four treatment conditions: naive (n = 5), ACR (n = 14), OVA (n = 14) or OVA/ACR (n = 14). All animals were housed in a University of Vermont animal facility maintained at 70 °C with a 25–30% relative humidity and a 12-h light:dark cycle. All procedures contributing to this work comply with the ethical standards of the National Institutes of Health on the care and use of laboratory animals and have been approved by the University of Vermont Institutional Animal Care and Use Committee.
ACR exposure
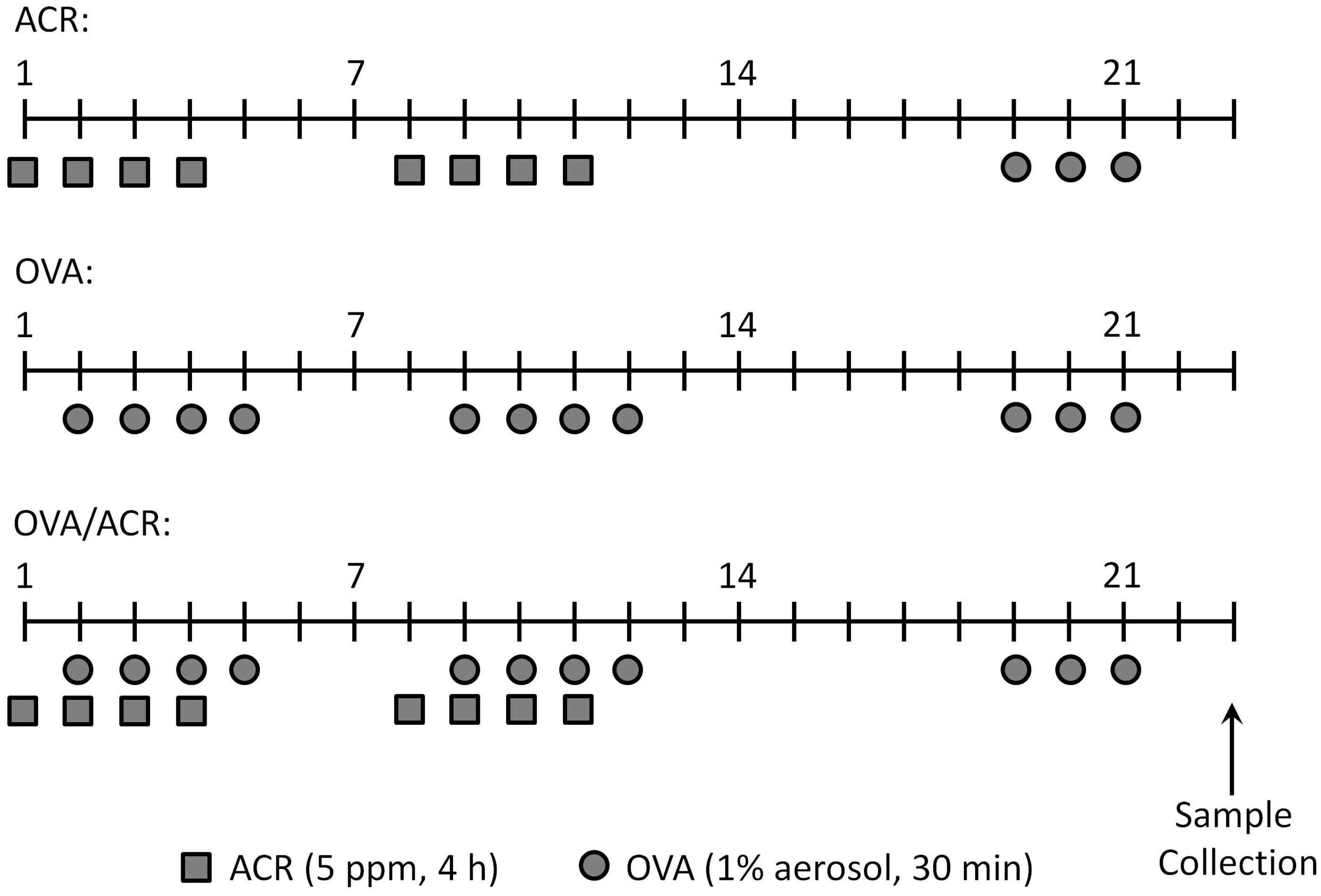
Mice assigned to the ACR and OVA/ACR treatment groups were exposed to vaporized ACR in an enclosed 2-L glass chamber with a continuous air flow rate of 2.7 L/min. ACR (≥95.5–99.0% pure stabilized with ∼0.2% hydrochinoine and ∼3% water; Sigma, St. Louis, MO or Chem Service, Inc., West Chester, PA) was vaporized into the constant flow of air and maintained at ≈5 ppm (≈11 mg/m3) for the duration of exposure via continuous monitoring by a 205B Series MIRAN SapphIRe (ThermoFisher Scientific, Waltham, MA) portable ambient air analyzer, as described in Kasahara et al. (Citation2008). Each ACR exposure lasted 4 h. Mice receiving ACR treatment were exposed 4 days/week for two consecutive weeks on Days 1–4 and 8–11 (). Our study uses environmentally-relevant ACR concentrations based on levels of indoor ACR in smoking areas that have been estimated to be as high as 1 ppm (Kasahara et al., Citation2008).
Figure 1. Schematic diagram of ACR and OVA administrations. On indicated days, mice were exposed to 5 ppm ACR for 4 h (squares) and/or 1% OVA in PBS for 30 min (circles). All animals treated with ACR only, OVA only or OVA and ACR were challenged with OVA on Days 19–21. Sample collection occurred on Day 23. Naive mice received no treatment.
Antigen sensitization and challenge
Animals assigned to the OVA and OVA/ACR treatment groups were subjected to repeated administrations of aerosolized 1% OVA (prepared in PBS and filter-sterilized) in individual whole-body chambers, as described previously (Spiess et al., Citation2013). During each administration, mice were exposed to aerosolized OVA for 30 min, which was repeated 4 days/week for two consecutive weeks on Days 2–5 and 9–12 (). OVA was delivered using an LC PLUS nebulizer powered by a Vios compressor that produced particles at a flow rate of 4.5 L/min (PARI Respiratory Equipment, Inc., Midlothian, VA). In subjects exposed to both ACR and OVA, mice were first exposed to ACR as described above and subsequently transferred to whole-body chambers for exposure to OVA, thus minimizing potential direct interactions between inhaled OVA and ACR. One week after the final OVA inhalation (Day 19), all animals were challenged with aerosolized 1% OVA for 30 min for three consecutive days (Days 19–21). Animals were euthanized by pentobarbital overdose 48 h after the last OVA administration (Day 23) for collection of bronchoalveolar lavage (BAL) fluids and lung tissues for histology (Spiess et al., Citation2013) and mRNA analyses.
Lung leukocyte recovery by bronchoalveolar lavage (BAL) and enumeration
Lungs of euthanized animals were cannulated through the trachea and lavaged 3 times with 500 μl of room temperature phosphate-buffered saline (PBS, pH 7.4; Invitrogen, Life Technologies, Grand Island, NY). Cells suspended in BAL fluid (BALF) were collected by centrifugation at 4 °C and re-suspended in 400 μl PBS. The total number of BALF cells was enumerated by counting on a hemocytometer. Differential counts were determined using a modified Wright-Giemsa stain (Differential Quik Stain, ThermoFisher Scientific) and counting a total of 400 cells in randomly chosen fields under a light microscope (100×). Two observers blinded to the experimental treatment groups counted the slides. The total number of each cell type was determined from percentage of each leukocyte subset and the total number of lung leukocytes in each sample.
Histology
Excised lungs were inflated with a solution of 4% paraformaldehyde (Sigma) and submerged overnight to fix the tissues. Samples were then embedded in paraffin, sectioned onto slides, de-paraffinized and then stained with hematoxylin and eosin (H&E). Representative images were then captured using a light microscope.
Analysis of OVA-specific IgG
Sera were collected at the time of dissection via arterial exsanguination and stored at −80 °C until analysis. The levels of anti-OVA IgG1 and IgG2a in sera were determined as previously described (Bevelander et al., Citation2007) using an enzyme immunoassay (EIA) that used OVA (Sigma) to capture anti-OVA IgG1 or IgG2a in sera and rat anti-mouse IgG1 and IgG2a antibodies (BD Biosciences, San Jose, CA) to detect anti-OVA IgG1 and IgG2a, respectively. Data are reported from identical serum dilutions from each treatment group (1:300 for OVA-specific IgG1 and 1:50 for OVA-specific IgG2a) and expressed as optical density (OD) values.
mRNA extraction and RT-PCR
Lung tissues collected at the time of dissection were incubated in RNAlater (Ambion, Life Technologies) for 24 h at 4 °C before being stored at −80 °C. Lung tissues were homogenized in TRIzol (Ambion) using a TissueLyser LT (Qiagen, Valencia, CA). mRNA extraction was completed by subsequent chloroform and ethanol treatment. Extracted mRNA was treated with DNase I (Qiagen) to remove contaminating DNA and then purified using a GeneJET RNA Purification Kit (ThermoFisher Scientific) according to manufacturer instructions. Complimentary DNA (cDNA) was prepared from 1 μg extracted mRNA using M-MLV Reverse Transcriptase (Invitrogen) and a dNTB Set (Invitrogen) according to manufacturer instructions. Quantitative PCR (qPCR) was performed on a CFX96 Real-Time Detection System (Bio-Rad, Hercules, CA) using iQ SYBR Green Supermix (Bio-Rad) and primers designed for specific mouse genes of interest (). Gapdh expression was measured as a housekeeping gene to normalize candidate gene expressions. Relative gene expression levels were calculated using the 2−ΔΔCT method to determine the relative quantifications (RQ) (Livak & Schmittgen, Citation2001).
Table 1. Primers used for qPCR of cytokine and chemokines in lung tissues.
Cytokine determinations
Levels of inflammatory cytokines (interleukin [IL]-5, IL-13, tumor necrosis factor [TNF]-α) or transforming growth factor (TGF)-β1 were measured in BALF using commercially available enzyme immunoassay (EIA) kits (DuoSet, R&D Systems, Minneapolis, MN) according to manufacturer instructions.
Statistical analyses
Data are expressed as mean ± SEM with an n = 4–14 individual subjects. Analyses were conducted in GraphPad Prism Software (GraphPad Software, Inc., La Jolla, CA) using a one-way analysis of variance with a post-hoc Bonferroni test for separation of the means. In all cases, a p value < 0.05 was considered statistically significant.
Results
Serum levels of anti-OVA IgG
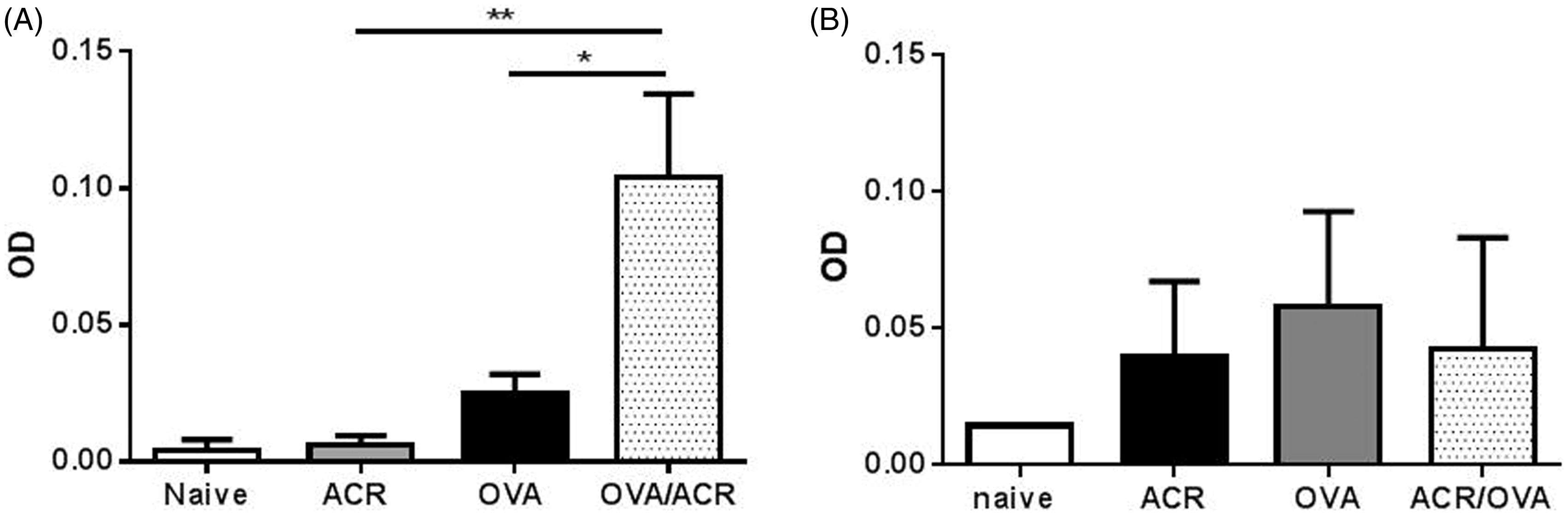
To assess systemic OVA sensitization in treated animals, sera were collected and analyzed for anti-OVA IgG1 and IgG2a. Serum IgG1 levels measured in the ACR and OVA treatment groups were similar to those of naive mice (ACR: p = 0.829, OVA: p = 0.311), but animals that received airway exposure to both OVA and ACR displayed a 3-fold increase in serum IgG1 levels compared to both ACR-treated (p = 0.004) and OVA-treated animals (p = 0.022), indicating that ACR inhalation can promote antigen sensitization via airway exposure (). It should be noted that the levels of serum anti-OVA IgG1 in the present studies were markedly lower than models using intraperitoneal (IP) antigen sensitization with aluminum hydroxide adjuvant (Spiess et al., Citation2013). In contrast to the observed increase in IgG1, serum IgG2a levels were not different between any of the treatment groups ().
Figure 2. (a) Anti-OVA IgG1 and (b) anti-OVA IgG2a levels in sera from naive, ACR-, OVA- and OVA/ACR-treated animals collected on Day 23. Bars represent mean ± SEM. *p < 0.05 and **p < 0.005 compared between indicated treatment groups. OD, Optical Density. Naive: n = 4; ACR, OVA and OVA/ACR: n = 10–14.
Leukocyte recruitment following OVA challenge
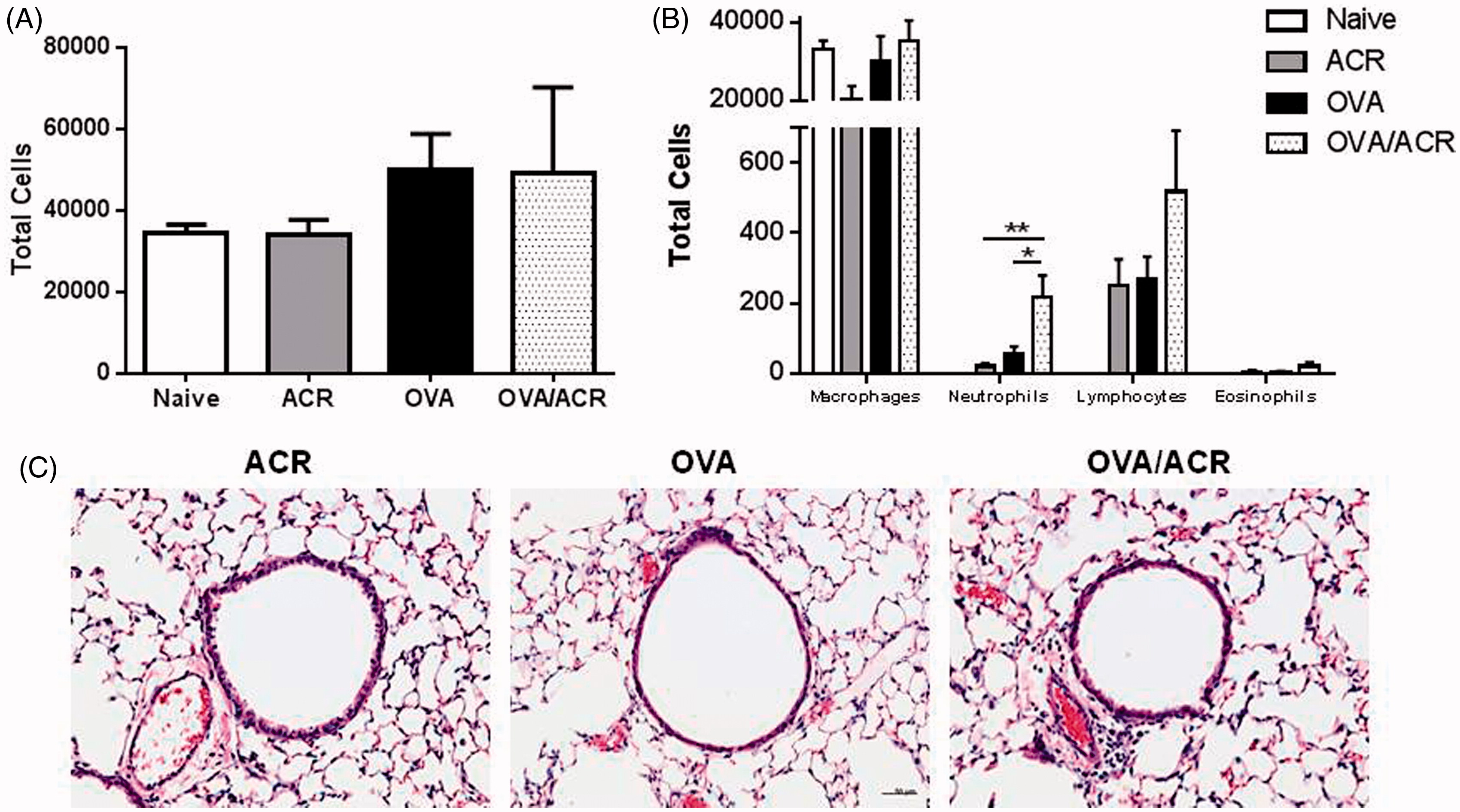
Following airway challenge with OVA, recruitment of leukocytes to the airways was examined in BALF (). No significant differences were observed in total BALF leukocyte numbers (p = 0.207) () or numbers of macrophages (p = 0.172), lymphocytes (p = 0.129) and eosinophils (p = 0.255) between any of the treatment groups (). However, significant, albeit modest, increases in the BALF neutrophils were observed in OVA/ACR animals compared to animals in the ACR (p = 0.007) and OVA (p = 0.029) treatment groups, whereas BALF neutrophil counts from animals in the ACR (p = 0.253) and OVA (p = 0.206) treatment groups did not differ from those of naive mice. The modest increase in lung inflammation in OVA/ACR-treated animals is also illustrated in H&E-stained lung tissue sections that indicate increased perivascular inflammation compared to that seen in ACR- or OVA-treated animals ().
Figure 3. Airway influx of (a) total leukocytes and (b) macrophages, neutrophils, lymphocytes and eosinophils in BALF of naive, ACR-, OVA- and OVA/ACR-treated animals collected on Day 23. Bars represent mean ± SEM. *p < 0.05 and **p < 0.01 compared between indicated treatment groups. Naive: n = 4; ACR, OVA and OVA/ACR: n = 14. (c) Representative lung sections from OVA-, ACR- and OVA/ACR-treated mice collected after subsequent OVA challenge (stained with H&E). Bar = 50 μm.
Effects of OVA/ACR on inflammatory cytokine production
To further evaluate lung tissue inflammation after OVA challenge, lung tissues were analyzed for inflammatory cytokine and chemokine mRNA expression (). As shown, lung tissue mRNA expression of the TH1 cytokine tnfa and the TH2 cytokines il5, il10 and il13 did not differ significantly between animals from any of the experimental groups. Furthermore, analyses of BALF by EIA did not reveal any differences in IL-5, IL-10, IL-13 or TNFα levels between each of the treatment groups (results not shown). Based on the observation of increased airway neutrophils in OVA/ACR-exposed mice, several cytokines and chemokines that are related to neutrophil recruitment were further evaluated (). Indeed, increased mRNA expression of ccl20, a neutrophil chemoattractant (Hieshima et al., Citation1997), was observed in lung tissues from OVA/ACR-treated mice compared to those from ACR-treated (p = 0.015) and naive (p = 0.029) mice; although, they did not differ significantly from OVA-treated mice (p = 0.109). Expression of the IL-17 family member il17f was also increased in both the ACR (p = 0.035) and OVA/ACR (p = 0.012) treatment groups, compared to in naive animals, whereas lung tissue il17f expression was not increased in OVA-treated animals (p = 0.125). Furthermore, animals in all exposure groups displayed significantly increased il1a mRNA expression compared to naive animals (ACR: p = 0.008, OVA: p = 0.022, OVA/ACR: p = 0.023), but il1a expression did not differ between treatment groups. Lastly, because of the potential role of TGFβ1 in immune regulation and inflammatory responses (Mantel & Schmidt-Weber, Citation2011), this study evaluated expression of tgfb1 and observed a significant increase in the ACR- (p = 0.001) and OVA/ACR- (p = 0.002) treated hosts compared to in the OVA-treated animals. These increases were associated with significant increases in TGFβ1 in the BALF from ACR and OVA/ACR mice ().
Figure 4. (a) mRNA expression of ccl20, il1a, il17f and tfgb1 in lung tissue and (b) TGFβ1 levels in BALF of naive, ACR-, OVA- and OVA/ACR-treated animals collected on Day 23. Bars represent mean ± SEM. *p < 0.05, **p < 0.01 and ***p < 0.001 compared to control (open bar) or between treatment groups indicated by line. RQ, relative quantifications. Naive: n = 2–5; ACR, OVA and OVA/ACR: n = 8–11.
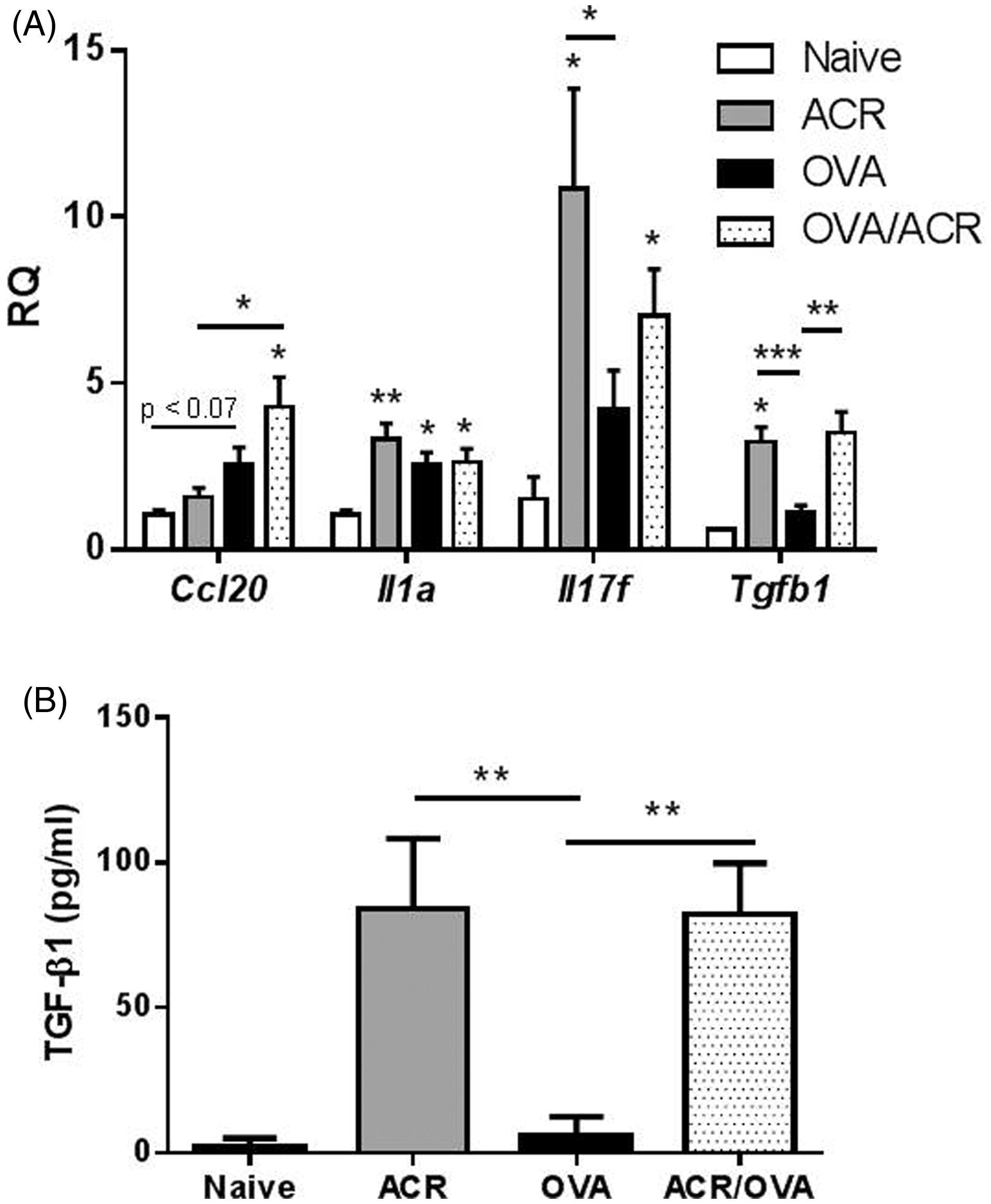
Table 2. Expression of cytokine and chemokine mRNA levels in lung tissues.
Discussion
Various lines of evidence indicate an important role for thiol-reactive electrophiles such as ACR in tobacco smoke-related injury and disease (Harris & Hansen, Citation2012; Myers et al., Citation2011); although, little is known regarding the potential contribution of ACR or other electrophilic aldehydes in smoking-related development and severity of allergic airway disease. We previously demonstrated that ACR exposure of previously sensitized mice can markedly suppress allergic airway inflammation in response to antigen challenge (Spiess et al., Citation2013). In the current study, we examined the ability of ACR exposure to enhance antigen sensitization to otherwise innocuous antigens (i.e. OVA) through the airway via potential adjuvant activity, analogous to similarly reported effects of environmental tobacco smoke (Moerloose et al., Citation2006; Rumold et al., Citation2001; Trimble et al., Citation2009) or other major pollutants (Bevelander et al., Citation2007; Hollingsworth et al., Citation2010). Indeed, our results indicate that ACR exposure is capable of promoting sensitization to inhaled OVA, as evidenced by enhanced OVA-specific IgG1 in sera of OVA/ACR-treated mice compared to mice exposed to OVA alone. Additionally, selective induction of OVA-specific IgG1 in the absence of changes in antigen-specific IgG2a levels would suggest that ACR exposure promotes a TH2-biased adaptive immune response. To our knowledge, the current study is the first to report that ACR (outside of context of cigarette smoke) can act as an adjuvant to enhance sensitization to OVA, although similar actions have been reported for other reactive aldehydes, such as formaldehyde (Riedel et al. Citation1996).
ACR-induced antigen sensitization in our studies was primarily associated with increased neutrophil recruitment, without significant increases in eosinophils. Moreover, in spite of apparent TH2-biased immune responses based on our findings with IgG1, we did not observe significant increases in expression of TH2 cytokines, such as IL-5, IL-10 or IL-13. Additional cytokine analyses did reveal an increase in lung tissue mRNA expression of ccl20 in the OVA/ACR group, possibly reflecting an effect on dendritic cell accumulation and antigen presentation (Botelho et al., Citation2012). CCL20 is also produced, in part, by neutrophils (Scapini et al., Citation2001) and can act as a mild chemoattractant for neutrophils (Hieshima et al., Citation1997). Additionally, the present study also revealed increased lung tissue mRNA levels of il17f in ACR and OVA/ACR treatment groups. IL-17F is a member of the IL-17 family that has been associated with enhanced neutrophilia in models of asthma (Kawaguchi et al., Citation2009). Our findings linking ACR exposure to antigen-induced neutrophil activation and recruitment may be relevant to the influence of smoking on asthma, which often is associated with increased neutrophilia and resistance to steroid treatment (Tamimi et al., Citation2012). Indeed, increased antigen sensitization by cigarette smoke exposure was also found to be associated with increased neutrophil recruitment (Rumold et al., Citation2001); although, other studies reported no change in neutrophil recruitment in a similar context (Moerloose et al., Citation2006).
While the findings here are analogous to previous studies demonstrating antigen sensitization and allergic inflammation by other environmental pollutants, such as nitrogen dioxide (Bevelander et al., Citation2007) or ozone (Hollingsworth et al., Citation2010), analysis of airway inflammation demonstrated a rather modest increase in neutrophilic inflammation in the OVA/ACR group and did not reveal significant increases in eosinophilic inflammation. In this regard, it should be noted that the exposure protocol here differed from those studies in that it involved repeated exposures to ACR and OVA for multiple days rather than a single pollutant exposure and antigen treatment to induce antigen sensitization. In fact, no significant antigen sensitization was observed when mice were exposed to OVA/ACR only on Days 1 and 7, followed by OVA challenge 2 weeks later (results not shown). Because repeated airway exposure to antigen is known to induce inhalation tolerance (Hoyne et al., Citation2000), our findings could also be interpreted as reflecting delayed inhalation tolerance due to ACR exposure, analogous to a recent report indicating similar actions of cigarette smoke (van Hove et al., Citation2008). The degree of allergic sensitization and subsequent inflammation in our studies was modest compared to previous studies of antigen sensitization by environmental pollutants (Bevelander et al., Citation2007; Rumold et al., Citation2001). This modest inflammation might also have been related to development of enhanced inhalation tolerance due to ACR exposure and to the fact that ACR has significant immuno-suppressive and anti-inflammatory properties due to its electrophilicity (Spiess et al., Citation2013).
With respect to the potential induction of inhalation tolerance by ACR, one potential mediator of such immune tolerance could be TGFβ which can be produced by regulatory T cells (Ostroukhova et al., Citation2004). Indeed, lung tissue analysis of mRNA of tgfb1 (the major isoform) demonstrated significantly enhanced tgfb1 mRNA in both ACR and OVA/ACR treatment groups, as well as enhanced TGFβ1 levels in the BALF, suggesting that induction of TGFβ1 is due to ACR exposure. Although ACR-induced TGFβ1 may originate from regulatory T-cells, it is more likely generated by structural lung cells as a result of persistent tissue injury. It is plausible that tissue injury may promote immune tolerance to minimize antigen sensitization and allergic inflammation (Ostroukhova et al., Citation2004). Intriguingly, since TGFβ1 can also act as a neutrophil chemoattractant and may promote TH17 differentiation (Mantel & Schmidt-Weber, 2011), induction of TGFβ1 by ACR may also help explain the observed increases in il17f mRNA and neutrophil recruitment in OVA/ACR mice. Thus, the findings here indicate that ACR exposure may promote antigen sensitization to OVA, but that subsequent inflammatory responses may be attenuated and skewed towards neutrophilic inflammation, in part due to a development of immune tolerance and pro-neutrophil responses by TGFβ1.
The different impacts of ACR exposure on allergen sensitization and tolerance demonstrated in the present study and on antigen-induced allergic airway inflammation in previously sensitized mice (Spiess et al. Citation2013) further highlight the complex relationship between smoking and asthma and emphasize the importance of exposure history. ACR can affect allergic airway disease in different ways dependent upon when exposure occurs during the course of disease pathogenesis; ACR exposure can promote allergic sensitization and asthma development, but can also suppress airway inflammation during ongoing asthma or during exacerbations. Our previous findings indicating that ACR exposure can dramatically suppress TH2 responses and eosinophilic inflammation in OVA-sensitized animals (Spiess et al., Citation2013), but not LPS-induced neutrophil recruitment (Kasahara et al., Citation2008), combined with our present findings, suggest that ACR exposure is primarily associated with neutrophilic inflammation, in spite of its immunosuppressive properties. In this regard, our findings are consistent with reports indicating that smoking is associated with a more prominent neutrophilic inflammatory response in asthmatics (Chalmers et al., Citation2001), which is often related to increased resistance to steroids and poor asthma management (Wenzel et al., Citation1997). Future studies to elucidate the molecular mechanism by which ACR exposure alters allergic inflammation will be useful in the development of improved therapeutics for managing asthma.
Conclusion
The current study has identified a novel association between ACR exposure, allergic sensitization and immune tolerance and indicates that ACR exposure can enhance antigen sensitization, but primarily promotes development of neutrophilic inflammation, whereas allergic inflammation may be suppressed by enhanced immune tolerance. Future studies will be required to address the molecular mechanisms by which ACR enhances antigen sensitization following ACR exposure, which may potentially involve epithelial injury and barrier disruption (Song et al., Citation2013), as well as altered inflammatory mechanisms that favor neutrophilia.
Declaration of interest
This research was funded by grants from the National Institutes of Environmental Health Sciences (ES021476, AvdV), the National Heart, Lung and Blood Institute (HL085646, AvdV) and the Flight Attendant Medical Research Institute (Clinical Investigator Award, AvdV). Support for PCS was provided by a Flight Attendant Medical Research Institute Young Clinical Scientist Award.
References
- Anandan, C., Nurmatov, U., van Schayck, O. C., and Sheikh, A. 2010. Is the prevalence of asthma declining? Systematic review of epidemiological studies. Allergy 65:152–167
- Bein, K., and Leikauf, G. D. 2011. Acrolein 0 A pulmonary hazard. Mol. Nutr. Food Res. 55:1342–1360
- Bevelander, M., Mayette, J., Whittaker, L. A., et al. 2007. Nitrogen dioxide promotes allergic sensitization to inhaled antigen. J. Immunol. 179:3680–3688
- Botelho, F. M., Nikota, J. K., Bauer, C. M., et al. 2012. Cigarette smoke-induced accumulation of lung dendritic cells is IL-1α-dependent in mice. Respir. Res. 13:81
- Centers for Disease Control and Prevention (CDC). 2004. The Health Consequences of Smoking: A Report of the Surgeon General. Atlanta, GA: CDC
- Chalmers, G. W., MacLeod, K. J., Thomson, L., et al. 2001. Smoking and airway inflammation in patients with mild asthma. Chest 120:1917–1922
- Dick, S., Doust, E., Cowie, H., et al. 2014. Associations between environmental exposures and asthma control and exacerbations in young children: A systematic review. BMJ Open 4:e003827
- Dunbar, A., Gotsis, W., and Frishman, W. 2013. Second-hand tobacco smoke and cardiovascular disease risk: An epidemiological review. Cardiol. Rev. 21:94–100
- Faroon, O., Roney, N., Taylor, J., et al. 2008. Acrolein environmental levels and potential for human exposure. Toxicol. Ind. Health 24:543–564
- Harris, C., and Hansen, J. M. 2012. Oxidative stress, thiols, and redox profiles. Meth. Mol. Biol. 889:325–346
- Hersoug, L. G., Husemoen, L. L., Sigsgaard, T., et al. 2010. Indoor exposure to environmental cigarette smoke, but not other inhaled particulates associates with respiratory symptoms and diminished lung function in adults. Respirology 15:993–1000
- Hieshima, K., Imai, T., Opdenakker, G., et al. 1997. Molecular cloning of a novel human CC chemokine liver and activation-regulated chemokine (LARC) expressed in liver. Chemotactic activity for lymphocytes and gene localization on chromosome 2. J. Biol. Chem. 272:5846–5853
- Hjern, A., Hedberg, A., Haglund, B., and Rosen, M. 2001. Does tobacco smoke prevent atopic disorders? A study of two generations of Swedish residents. Clin. Exp. Allergy 31:908–914
- Holgate, S. T., Davies, D. E., Powel, R. M., et al. 2007. Local genetic and environmental factors in asthma disease pathogenesis: Chronicity and persistence mechanisms. Eur. Respir. J. 29:793–803
- Holgate, S. T., Roberts, G., Arshad, H. S., et al. 2009. The role of airway epithelium and its interaction with environmental factors in asthma pathogenesis. Proc. Am. Thorac. Soc. 6:655–659
- Hollingsworth, J. W., Free, M. E., Li, Z., et al. 2010. Ozone activates pulmonary dendritic cells and promotes allergic sensitization through a Toll-like receptor 4-dependent mechanism. J. Allergy Clin. Immunol. 125:1167–11670
- Hoyne, G. F., Tan, K., Corsin-Jimenez, M., et al. 2000. Immunological tolerance to inhaled antigen. Am. J. Respir. Crit. Care Med. 162:S169–174
- Hunt, A., Crawford, J. A., Rosenbaum, P. F., and Abraham, J. L. 2011. Levels of household particulate matter and environmental tobacco smoke exposure in the first year of life for a cohort at risk for asthma in urban Syracuse, NY. Environ. Int. 37:1196–1205
- Huxley, R. R., and Woodward, M. 2011. Cigarette smoking as a risk factor for coronary heart disease in women compared with men: A systematic review and meta-analysis of prospective cohort studies. Lancet 378:1297–1305
- Jung, J. W., Ju, Y. S., and Kang, H. R. 2012. Association between parental smoking behavior and children’s respiratory morbidity: 5-year study in an urban city of South Korea. Pediatr. Pulmonol. 47:338–345
- Kasahara, D. I., Poynter, M. E., Othman, Z., et al. 2008. Acrolein inhalation suppresses lipopolysaccharide-induced inflammatory cytokine production but does not affect acute airway neutrophilia. J. Immunol. 181:736–745
- Kawaguchi, M., Kokubu, F., Fujita, J., et al. 2009. Role of IL-17F in asthma. Inflamm. Allergy Drug Targets 8:383–389
- Lanckacker, E. A., Tournoy, K. G., Hammad, H., et al. 2013. Short cigarette smoke exposure facilitates sensitization and asthma development in mice. Eur. Respir. J. 41:1189–1199
- Leikauf, G. D. 2002. Hazardous air pollutants and asthma. Environ. Health Perspect. 110:505–526
- Livak, K. J., and Schmittgen, T. D. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25:402–408
- Mantel, P. Y., and Schmidt-Weber, C. B. 2011. Transforming growth factor-β: Recent advances on its role in immune tolerance. Meth. Mol. Biol. 677:303–338
- Melgert, B. N., Postma, D. S., Geerlings, M., et al. 2004. Short-term smoke exposure attenuates ovalbumin-induced airway inflammation in allergic mice. Am. J. Respir. Cell. Mol. Biol. 30:880–885
- Mitchell, E. A., Beasley, R., Keil, U., et al; and the ISAAC Phase Three Study Group. 2012. The association between tobacco and the risk of asthma, rhino-conjunctivitis and eczema in children and adolescents: Analysis from Phase Three of the ISAAC Program. Thorax 67:941–949
- Moerloose, K. B., Robays, L. J., Maes, T., et al. 2006. Cigarette smoke exposure facilitates allergic sensitization in mice. Respir. Res. 7:49--57
- Mohammad, Y., Shaaban, R., Al-Zahad, B. A., et al. 2013. Impact of active and passive smoking as risk factors for asthma and COPD in women presenting to primary care in Syria: First report by the WHO-GARD survey group. Int. J. Chron. Obstruct. Pulmon. Dis. 8:473–482
- Mukherjee, A. B., and Zhang, Z. 2011. Allergic Asthma: Influence of genetic and environmental factors. J. Biol. Chem. 286:32883–32889
- Murdoch, J. R., and Lloyd, C. M. 2010. Chronic inflammation and asthma. Mutat. Res. 690:24–39
- Murphy, D. M., and O’Byrne, P. M. 2010. Recent advances in the pathophysiology of asthma. Chest 137:1417–1426
- Myers, C. R., Myers, J. M., Kufahl, T. D., et al. 2011. The effects of acrolein on the thioredoxin system: Implication for redox-sensitive signaling. Mol. Nutr. Food Res. 55:1361–1374
- Oberg, M., Jaakkola, M. S., Woodward, A., et al. 2011. Worldwide burden of disease from exposure to second-hand smoke: A retrospective analysis of data from 192 countries. Lancet 377:139–146
- Ostroukhova, M., Seguin-Devaux, C., Oriss, T. B., et al. 2004. Tolerance induced by inhaled antigen involves CD4+ T-cells expressing membrane-bound TGFβ and FOXP3. J. Clin. Invest. 114:28–38
- Papaioannou, A. I., Koutsokera, A., Tanou, K., et al. 2010. Acute effect of smoking in healthy and asthmatic smokers. Eur. J. Clin. Invest. 40:103–109
- Rahman, I., and MacNee, W. 1999. Lung glutathione and oxidative stress: Implications in cigarette smoke-induced airway disease. Am. J. Physiol. 277:L1067–1088
- Reddy, S., Finkelstein, E. I., Wong, P. S., et al. 2002. Identification of glutathione modifications by cigarette smoke. Free Radic. Biol. Med. 33:1490–1498
- Riedel, F., Hasenauer, E., Barth, P. J., et al. 1996. Formaldehyde exposure enhances inhalative allergic sensitization in the guinea pig. Allergy 51:94–99
- Robbins, C. S., Pouladi, M. A., Fattouh, R., et al. 2005. Mainstream cigarette smoke exposure attenuates airway immune inflammatory responses to surrogate and common environmental allergens in mice, despite evidence of increased systemic sensitization. J. Immunol. 175:2834–2842
- Rodgman, A., and Perfetti, T. A., (Eds.). 2013. The Chemical Components of Tobacco and Tobacco Smoke. Boca Raton, FL: CRC Press
- Rumold, R., Jyrala, M., and Diaz-Sanchez, D. 2001. Secondhand smoke induces allergic sensitization in mice. J. Immunol. 167:4765–4770
- Scapini, P., Laudanna, C., Pinardi, C., et al. 2001. Neutrophils produce biologically-active macrophage inflammatory protein-3α (MIP-3α)/CCL20 and MIP-3β/CCL19. Eur. J. Immunol. 31:1981–1988
- Sears, M. R. 2014. Trends in the prevalence of asthma. Chest 145:219–225
- Siroux, V., Pin, I., Oryszczyn, M. P., et al. 2000. Relationships of active smoking to asthma and asthma severity in the EGEA study. Epidemiological study on the Genetics and Environment of Asthma. Eur. Respir. J. 15:470–477
- Spiess, P. C., Kasahara, D., Habibovic, A., et al. 2013. Acrolein exposure suppresses antigen-induced pulmonary inflammation. Respir. Res. 14:107--120
- Song, J. J., Lee, J. D., Lee, B. D., et al. 2013. Effect of acrolein, a hazardous air pollutant in smoke, on human middle-ear epithelial cells. Int. J. Pediatr. Otorhino-laryngol. 77:1659–1664
- Stapleton, M., Howard-Thompson, A., George, C., et al. 2011. Smoking and asthma. J. Am. Board Fam. Med. 24:313–322
- Stevens, J. F., and Maier, C. S. 2008. Acrolein: Sources, metabolism, and biomolecular inter-actions relevant to human health and disease. Mol. Nutr. Food Res. 52:7–25
- Strachan, D. P. 2000. The role of environmental factors in asthma. Br. Med. Bull. 56:865–882
- Tamimi, A., Serdarevic, D., and Hanania, N. A. 2012. The effects of cigarette smoke on airway inflammation in asthma and COPD: Therapeutic implications. Respir. Med. 106:319–328
- Thatcher, T. H., Benson, R. P., Phipps, R. P., and Sime, P. J. 2008. High-dose but not low-dose mainstream cigarette smoke suppresses allergic airway inflammation by inhibiting T-cell function. Am. J. Physiol. 295:L412–L421
- To, T., Stanojevic, S., Moores, G., et al. 2012. Global asthma prevalence in adults: Findings from the cross-sectional world health survey. BMC Public Health 12:204–211
- Trimble, N. J., Botelho, F. M., Bauer, C. M., et al. 2009. Adjuvant and anti-inflammatory properties of cigarette smoke in murine allergic airway inflammation. Am. J. Respir. Cell. Mol. Biol. 40:38–46
- Troisi, R. J., Speizer, F. E., Rosner, B., et al. 1995. Cigarette smoking and incidence of chronic bronchitis and asthma in women. Chest 108:1557–1561
- Tsai, C. H., Huang, J. H., Hwang, B. F., and Lee, Y. L. 2010. Household environmental tobacco smoke and risks of asthma, wheeze and bronchitic symptoms among children in Taiwan. Respir. Res. 11:11--20
- van Hove, C. L., Moerloose, K., Maes, T., et al. 2008. Cigarette smoke enhances TH2-driven airway inflammation and delays inhalational tolerance. Respir. Res. 9:1–14
- Wenzel, S. E., Szefler, S. J., Leung, D. Y., et al. 1997. Bronchoscopic evaluation of severe asthma: Persistent inflammation associated with high-dose glucocorticoids. Am. J. Respir. Crit. Care Med. 156:737–742
- Wong, G. W., Leung, T. F., and Ko, F. W. 2013. Changing prevalence of allergic disease in the Asia-Pacific region. Allergy Asthma Immunol. Res. 5:251–257
- World Health Organization (WHO). 2007. Global Surveillance, Prevention, and Control of Chronic Respiratory Diseases: A Comprehensive Approach. Geneva: WHO
- World Health Organization (WHO). 2008. WHO Report on the Global Tobacco Epidemic, 2008: The MPOWER package. Geneva: WHO